Involvement of Sphingolipids in Ethanol Neurotoxicity in the Developing Brain

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ethanol-Induced Neuronal Apoptosis in the Developing Brain
2.1. Neuronal Apoptosis Triggered by Ethanol during the Period of Synaptogenesis
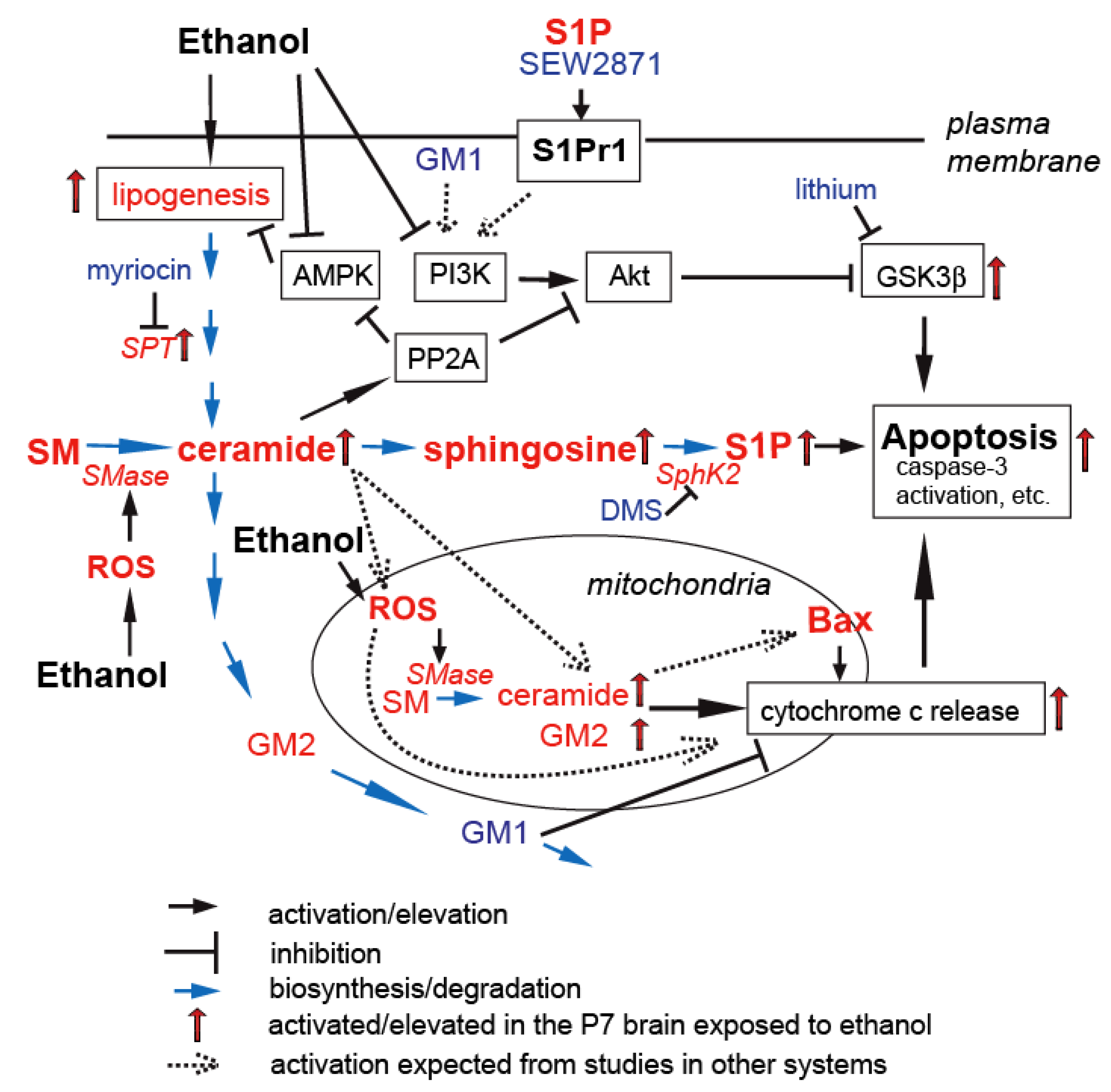
2.2. Mechanisms behind Ethanol-Induced Neuronal Apoptosis
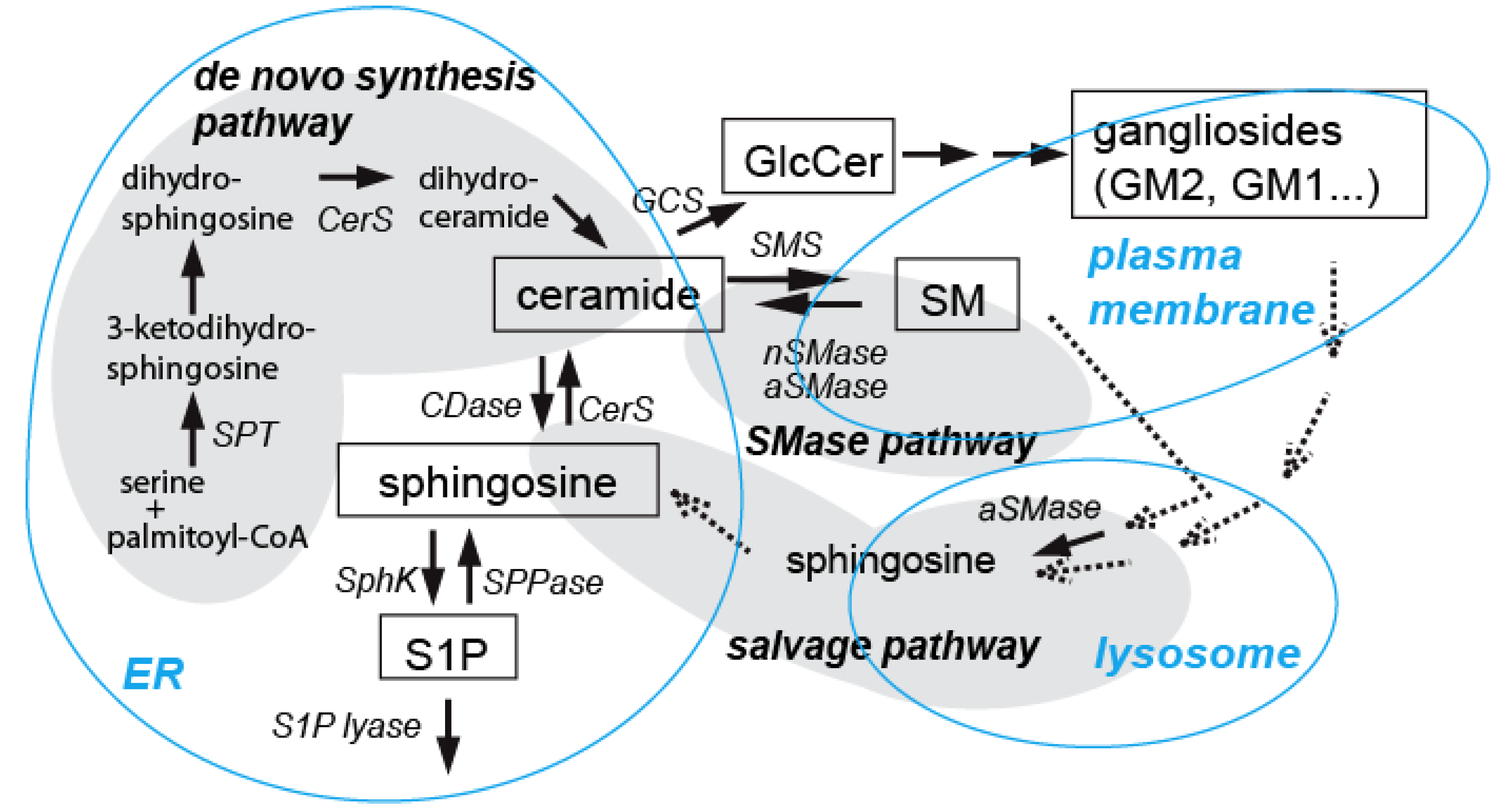
3. Ceramide Involvement in Ethanol-Induced Neuronal Apoptosis in the Developing Brain
3.1. Involvement of Ceramide in Apoptosis in the Developing Brain

3.2. Ceramide in Ethanol-Induced Apoptosis in the Developing Brain
4. S1P in Ethanol-Induced Apoptosis in the Developing Brain

5. Gangliosides in Ethanol-Induced Neuronal Apoptosis in the Developing Brain
5.1. Pro- and Anti-Apoptotic Effects of Gangliosides in the Brain
5.2. Gangliosides in Ethanol-Induced Apoptosis in the Developing Brain
6. Conclusion
Acknowledgments
Conflict of Interest
References
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Futerman, A.H.; Hannun, Y.A. The complex life of simple sphingolipids. EMBO Rep. 2004, 5, 777–782. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 2002, 1585, 153–162. [Google Scholar] [CrossRef]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Arboleda, G.; Morales, L.C.; Benitez, B.; Arboleda, H. Regulation of ceramide-induced neuronal death: Cell metabolism meets neurodegeneration. Brain Res. Rev. 2009, 59, 333–346. [Google Scholar] [CrossRef]
- Colombaioni, L.; Garcia-Gil, M. Sphingolipid metabolites in neural signalling and function. Brain Res. Brain Res. Rev. 2004, 46, 328–355. [Google Scholar] [CrossRef]
- Goswami, R.; Dawson, G. Does ceramide play a role in neural cell apoptosis? J. Neurosci. Res. 2000, 60, 141–149. [Google Scholar] [CrossRef]
- Jana, A.; Hogan, E.L.; Pahan, K. Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. J. Neurol. Sci. 2009, 278, 5–15. [Google Scholar] [CrossRef]
- Posse de Chaves, E.I. Sphingolipids in apoptosis, survival and regeneration in the nervous system. Biochim. Biophys. Acta 2006, 1758, 1995–2015. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2010, 12, e27. [Google Scholar] [CrossRef]
- Bieberich, E.; MacKinnon, S.; Silva, J.; Yu, R.K. Regulation of apoptosis during neuronal differentiation by ceramide and b-series complex gangliosides. J. Biol. Chem. 2001, 276, 44396–44404. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Chudakova, D.A.; Wheeler, B.W.; Bielawski, J.; Kindy, M.S.; Obeid, L.M.; Gudz, T.I. Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J. Biol. Chem. 2011, 286, 4644–4658. [Google Scholar] [CrossRef]
- Rotstein, N.P.; Miranda, G.E.; Abrahan, C.E.; German, O.L. Regulating survival and development in the retina: Key roles for simple sphingolipids. J. Lipid Res. 2010, 51, 1247–1262. [Google Scholar] [CrossRef]
- Schwarz, A.; Futerman, A.H. Distinct roles for ceramide and glucosylceramide at different stages of neuronal growth. J. Neurosci. 1997, 17, 2929–2938. [Google Scholar]
- Chen, H.; Tran, J.T.; Brush, R.S.; Saadi, A.; Rahman, A.K.; Yu, M.; Yasumura, D.; Matthes, M.T.; Ahern, K.; Yang, H.; et al. Ceramide signaling in retinal degeneration. Adv. Exp. Med. Biol. 2012, 723, 553–558. [Google Scholar]
- De La Monte, S.M. Triangulated mal-signaling in Alzheimer’s disease: Roles of neurotoxic ceramides, ER stress, and insulin resistance reviewed. J. Alzheimers Dis. 2012, 30 (Suppl. 2), S231–S249. [Google Scholar]
- Horres, C.R.; Hannun, Y.A. The roles of neutral sphingomyelinases in neurological pathologies. Neurochem. Res. 2012, 37, 1137–1149. [Google Scholar] [CrossRef]
- Ferrari, G.; Greene, L.A. Promotion of neuronal survival by GM1 ganglioside. Phenomenology and mechanism of action. Ann. N. Y. Acad. Sci. 1998, 845, 263–273. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. In search of a solution to the sphinx-like riddle of GM1. Neurochem. Res. 2010, 35, 1867–1874. [Google Scholar] [CrossRef]
- Mocchetti, I. Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell. Mol. Life Sci. 2005, 62, 2283–2294. [Google Scholar] [CrossRef]
- Ohmi, Y.; Ohkawa, Y.; Yamauchi, Y.; Tajima, O.; Furukawa, K.; Furukawa, K. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochem. Res. 2012, 37, 1185–1191. [Google Scholar] [CrossRef]
- Schengrund, C.L. The role(s) of gangliosides in neural differentiation and repair: A perspective. Brain Res. Bull. 1990, 24, 131–141. [Google Scholar] [CrossRef]
- Skaper, S.D.; Leon, A. Monosialogangliosides, neuroprotection, and neuronal repair processes. J. Neurotrauma 1992, 9 (Suppl. 2), S507–S516. [Google Scholar]
- Amacher, D.E. Strategies for the early detection of drug-induced hepatic steatosis in preclinical drug safety evaluation studies. Toxicology 2011, 279, 10–18. [Google Scholar] [CrossRef]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef]
- Sozio, M.; Crabb, D.W. Alcohol and lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E10–E16. [Google Scholar] [CrossRef]
- Longato, L.; Ripp, K.; Setshedi, M.; Dostalek, M.; Akhlaghi, F.; Branda, M.; Wands, J.R.; de la Monte, S.M. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef]
- Supakul, R.; Liangpunsakul, S. Alcoholic-induced hepatic steatosis-role of ceramide and protein phosphatase 2A. Transl. Res. 2011, 158, 77–81. [Google Scholar] [CrossRef]
- Guerri, C.; Bazinet, A.; Riley, E.P. Foetal Alcohol Spectrum Disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009, 44, 108–114. [Google Scholar] [CrossRef]
- Cudd, T.A. Animal model systems for the study of alcohol teratology. Exp. Biol. Med. 2005, 230, 389–393. [Google Scholar]
- Guerri, C. Neuroanatomical and neurophysiological mechanisms involved in central nervous system dysfunctions induced by prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 1998, 22, 304–312. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- Farber, N.B.; Creeley, C.E.; Olney, J.W. Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol. Dis. 2010, 40, 200–206. [Google Scholar] [CrossRef]
- Wilson, D.A.; Peterson, J.; Basavaraj, B.S.; Saito, M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2011, 35, 1974–1984. [Google Scholar] [CrossRef]
- Han, J.Y.; Jeong, E.Y.; Kim, Y.S.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S. C-jun N-terminal kinase regulates the interaction between 14-3-3 and Bad in ethanol-induced cell death. J. Neurosci. Res. 2008, 86, 3221–3229. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Madorsky, I.; Siler-Marsiglio, K.; Shaw, G. Effect of bax deletion on ethanol sensitivity in the neonatal rat cerebellum. J. Neurobiol. 2006, 66, 95–101. [Google Scholar] [CrossRef]
- Nowoslawski, L.; Klocke, B.J.; Roth, K.A. Molecular regulation of acute ethanol-induced neuron apoptosis. J. Neuropathol. Exp. Neurol. 2005, 64, 490–497. [Google Scholar]
- Young, C.; Klocke, B.J.; Tenkova, T.; Choi, J.; Labruyere, J.; Qin, Y.Q.; Holtzman, D.M.; Roth, K.A.; Olney, J.W. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003, 10, 1148–1155. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Hegde, M.; Ohsie, J.; Paik, S.M.; Vadasz, C.; Saito, M. Involvement of ceramide in ethanol-induced apoptotic neurodegeneration in the neonatal mouse brain. J. Neurochem. 2010, 115, 168–177. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Mao, R.F.; Wang, R.; Cooper, T.B.; Vadasz, C.; Saito, M. Ethanol alters lipid profiles and phosphorylation status of AMP-activated protein kinase in the neonatal mouse brain. J. Neurochem. 2007, 103, 1208–1218. [Google Scholar] [CrossRef]
- Saito, M.; Saito, M.; Cooper, T.B.; Vadasz, C. Ethanol-induced changes in the content of triglycerides, ceramides, and glucosylceramides in cultured neurons. Alcohol. Clin. Exp. Res. 2005, 29, 1374–1383. [Google Scholar] [CrossRef]
- Duffy, O.; Menez, J.F.; Floch, H.H.; Leonard, B.E. Changes in whole brain membranes of rats following pre- and post-natal exposure to ethanol. Alcohol Alcohol. 1991, 26, 605–613. [Google Scholar]
- Kim, H.Y. Biochemical and biological functions of docosahexaenoic acid in the nervous system: Modulation by ethanol. Chem. Phys. Lipids 2008, 153, 34–46. [Google Scholar] [CrossRef]
- Omodeo-Salè, F.; Pitto, M.; Masserini, M.; Palestini, P. Effects of chronic ethanol exposure on cultured cerebellar granule cells. Mol. Chem. Neuropathol. 1995, 26, 159–169. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Shah, R.; Mao, R.F.; Kumar, A.; Yang, D.S.; Dobrenis, K.; Saito, M. Elevation of GM2 ganglioside during ethanol-induced apoptotic neurodegeneration in the developing mouse brain. J. Neurochem. 2012, 121, 649–661. [Google Scholar] [CrossRef]
- Ravasi, D.; Ferraretto, A.; Omodeo-Salè, M.F.; Tettamanti, G.; Pitto, M.; Masserini, M. Ethanol-induced increase of sphingosine recycling for ganglioside biosynthesis: A study performed on cerebellar granule cells in culture. J. Neurosci. Res. 2002, 69, 80–85. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Bonthius, N.E.; Napper, R.M.; West, J.R. Early postnatal alcohol exposure acutely and permanently reduces the number of granule cells and mitral cells in the rat olfactory bulb: A stereological study. Comp. Neurol. 1992, 324, 557–566. [Google Scholar] [CrossRef]
- Green, J.T.; Tran, T.; Steinmetz, J.E.; Goodlett, C.R. Neonatal ethanol produces cerebellar deep nuclear cell loss and correlated disruption of eyeblink conditioning in adult rats. Brain Res. 2002, 956, 302–311. [Google Scholar] [CrossRef]
- Napper, R.M.; West, J.R. Permanent neuronal cell loss in the inferior olive of adult rats exposed to alcohol during the brain growth spurt: A stereological investigation. Alcohol. Clin. Exp. Res. 1995, 19, 1321–1326. [Google Scholar] [CrossRef]
- Pierce, D.R.; Goodlett, C.R.; West, J.R. Differential neuronal loss following early postnatal alcohol exposure. Teratology 1989, 40, 113–126. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Johnson, T.B. Neonatal binge ethanol exposure using intubation: Timing and dose effects on place learning. Neurotoxicol. Teratol. 1997, 19, 435–446. [Google Scholar] [CrossRef]
- Johnson, T.B.; Goodlett, C.R. Selective and enduring deficits in spatial learning after limited neonatal binge alcohol exposure in male rats. Alcohol. Clin. Exp. Res. 2002, 26, 83–93. [Google Scholar] [CrossRef]
- Woolfrey, K.M.; Hunt, P.S.; Burk, J.A. Postnatal ethanol exposure disrupts signal detection in adult rats. Neurotoxicol. Teratol. 2005, 27, 815–823. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Madorsky, I.; Mayer, J.; Moore, D.B. Effects of ethanol on neurotrophic factors, apoptosis-related proteins, endogenous antioxidants, and reactive oxygen species in neonatal striatum: Relationship to periods of vulnerability. Dev. Brain Res. 2003, 140, 237–252. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Madorsky, I.; Shaw, G. Ethanol effects on neonatal rat cortex: Comparative analyses of neurotrophic factors, apoptosis-related proteins, and oxidative processes during vulnerable and resistant periods. Dev. Brain Res. 2003, 145, 249–262. [Google Scholar]
- Moore, D.B.; Walker, D.W.; Heaton, M.B. Neonatal ethanol exposure alters bcl-2 family mRNA levels in the rat cerebellar vermis. Alcohol. Clin. Exp. Res. 1999, 23, 1251–1261. [Google Scholar] [CrossRef]
- Young, C.; Olney, J.W. Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiol. Dis. 2006, 22, 548–554. [Google Scholar] [CrossRef]
- Furumiya, J.; Hashimoto, Y. Effects of ethanol exposure on spatial learning in mice during synaptogenesis. Nihon Arukoru Yakubutsu Igakkai Zasshi 2011, 46, 250–259. [Google Scholar]
- Ieraci, A.; Herrera, D.G. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006, 3, e101. [Google Scholar] [CrossRef]
- Sadrian, B.; Subbanna, S.; Wilson, D.A.; Basavarajappa, B.S.; Saito, M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience 2012, 206, 122–135. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Hartman, R.E.; Boyle, M.P.; Vogt, S.K.; Brooks, A.R.; Tenkova, T.; Young, C.; Olney, J.W.; Muglia, L.J. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 2004, 17, 403–414. [Google Scholar] [CrossRef]
- Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Nemmers, B.; Olney, J.W. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Brain Res. Dev. Brain Res. 2005, 155, 1–13. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Eilers, A.T. Alcohol-induced Purkinje cell loss with a single binge exposure in neonatal rats: A stereological study of temporal windows of vulnerability. Alcohol. Clin. Exp. Res. 1997, 21, 738–744. [Google Scholar]
- Goodlett, C.R.; Marcussen, B.L.; West, J.R. A single day of alcohol exposure during the brain growth spurt induces brain weight restriction and cerebellar Purkinje cell loss. Alcohol 1990, 7, 107–114. [Google Scholar] [CrossRef]
- Heaton, M.B.; Moore, D.B.; Paiva, M.; Madorsky, I.; Mayer, J.; Shaw, G. The role of neurotrophic factors, apoptosis-related proteins, and endogenous antioxidants in the differential temporal vulnerability of neonatal cerebellum to ethanol. Alcohol. Clin. Exp. Res. 2003, 27, 657–669. [Google Scholar] [CrossRef]
- Siler-Marsiglio, K.I.; Paiva, M.; Madorsky, I.; Pan, Q.; Shaw, G.; Heaton, M.B. Functional mechanisms of apoptosis-related proteins in neonatal rat cerebellum are differentially influenced by ethanol at postnatal days 4 and 7. J. Neurosci. Res. 2005, 81, 632–643. [Google Scholar] [CrossRef]
- Luo, J. Mechanisms of ethanol-induced death of cerebellar granule cells. Cerebellum 2012, 11, 145–154. [Google Scholar] [CrossRef]
- Climent, E.; Pascual, M.; Renau-Piqueras, J.; Guerri, C. Ethanol exposure enhances cell death in the developing cerebral cortex: Role of brain-derived neurotrophic factor and its signaling pathways. J. Neurosci. Res. 2002, 68, 213–225. [Google Scholar] [CrossRef]
- Guerri, C.; Renau-Piqueras, J. Alcohol, astroglia, and brain development. Mol. Neurobiol. 1997, 15, 65–81. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Leo, J.T.; O’Callaghan, J.P.; Mahoney, J.C.; West, J.R. Transient cortical astrogliosis induced by alcohol exposure during the neonatal brain growth spurt in rats. Brain Res. Dev. Brain Res. 1993, 72, 85–97. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Mao, R.F.; Paik, S.M.; Vadasz, C.; Saito, M. Tau phosphorylation and cleavage in ethanol-induced neurodegeneration in the developing mouse brain. Neurochem. Res. 2010, 35, 651–659. [Google Scholar] [CrossRef]
- Chen, C.P.; Kuhn, P.; Chaturvedi, K.; Boyadjieva, N.; Sarkar, D.K. Ethanol induces apoptotic death of developing beta-endorphin neurons via suppression of cyclic adenosine monophosphate production and activation of transforming growth factor-beta1-linked apoptotic signaling. Mol. Pharmacol. 2006, 69, 706–717. [Google Scholar]
- Cherian, P.P.; Schenker, S.; Henderson, G.I. Ethanol-mediated DNA damage and PARP-1 apoptotic responses in cultured fetal cortical neurons. Alcohol. Clin. Exp. Res. 2008, 32, 1884–1892. [Google Scholar]
- De, A.; Boyadjieva, N.I.; Pastorcic, M.; Reddy, B.V.; Sarkar, D.K. Cyclic AMP and ethanol interact to control apoptosis and differentiation in hypothalamic beta-endorphin neurons. J. Biol. Chem. 1994, 269, 26697–26705. [Google Scholar]
- Druse, M.J.; Tajuddin, N.F.; Gillespie, R.A.; Dickson, E.; Atieh, M.; Pietrzak, C.A.; Le, P.T. The serotonin-1A agonist ipsapirone prevents ethanol-associated death of total rhombencephalic neurons and prevents the reduction of fetal serotonin neurons. Brain Res. Dev. Brain Res. 2004, 150, 79–88. [Google Scholar]
- Naseer, M.I.; Ullah, N.; Ullah, I.; Koh, P.O.; Lee, H.Y.; Park, M.S.; Kim, M.O. Vitamin C protects against ethanol and PTZ-induced apoptotic neurodegeneration in prenatal rat hippocampal neurons. Synapse 2011, 65, 562–571. [Google Scholar] [CrossRef]
- Light, K.E.; Brown, D.P.; Newton, B.W.; Belcher, S.M.; Kane, C.J. Ethanol-induced alterations of neurotrophin receptor expression on Purkinje cells in the neonatal rat cerebellum. Brain Res. 2002, 924, 71–81. [Google Scholar] [CrossRef]
- Chakraborty, G.; Saito, M.; Mao, R.F.; Wang, R.; Vadasz, C.; Saito, M. Lithium blocks ethanol-induced modulation of protein kinases in the developing brain. Biochem. Biophys. Res. Commun. 2008, 367, 597–602. [Google Scholar] [CrossRef]
- Han, J.Y.; Jeong, J.Y; Lee, Y.K.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S. Suppression of survival kinases and activation of JNK mediate ethanol-induced cell death in the developing rat brain. Neurosci. Lett. 2006, 398, 113–117. [Google Scholar] [CrossRef]
- Young, C.; Straiko, M.M.; Johnson, S.A.; Creeley, C.; Olney, J.W. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol. Dis. 2008, 31, 355–360. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Yao, W.; Lee, W. Lithium protects ethanol-induced neuronal apoptosis. Biochem. Biophys. Res. Commun. 2006, 350, 905–910. [Google Scholar] [CrossRef]
- Luo, J. Lithium-mediated protection against ethanol neurotoxicity. Front. Neurosci. 2010, 4, 41. [Google Scholar]
- Heaton, M.B.; Paiva, M.; Kubovic, S.; Kotler, A.; Rogozinski, J.; Swanson, E.; Madorsky, V.; Posados, M. Differential effects of ethanol on c-jun N-terminal kinase, 14-3-3 proteins, and Bax in postnatal day 4 and postnatal day 7 rat cerebellum. Brain Res. 2012, 1432, 15–27. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Mayer, J.; Miller, R. Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci. Lett. 2002, 334, 83–86. [Google Scholar] [CrossRef]
- Ke, Z.; Liu, Y.; Wang, X.; Fan, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Ou, X.; Shi, X.; Luo, J. Cyanidin-3-glucoside ameliorates ethanol neurotoxicity in the developing brain. J. Neurosci. Res. 2011, 89, 1676–1684. [Google Scholar] [CrossRef]
- Heaton, M.B.; Mitchell, J.; Paiva, M. Amelioration of ethanol-induced neurotoxicity in the neonatal rat central nervous system by antioxidant therapy. Alcohol. Clin. Exp. Res. 2000, 24, 512–518. [Google Scholar] [CrossRef]
- Marino, M.D.; Aksenov, M.Y.; Kelly, S.J. Vitamin E protects against alcohol-induced cell loss and oxidative stress in the neonatal rat hippocampus. Int. J. Dev. Neurosci. 2004, 22, 363–377. [Google Scholar] [CrossRef]
- Wang, X.; Ke, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Zhang, Z.; Shi, X.; Luo, J. Cdc42-dependent activation of NADPH oxidase is involved in ethanol-induced neuronal oxidative stress. PLoS One 2012, 7, e38075. [Google Scholar]
- Ke, Z.; Wang, X.; Liu, Y.; Fan, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Li, M.; Fang, S.; et al. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcohol. Clin. Exp. Res. 2011, 35, 1574–1583. [Google Scholar]
- Heaton, M.B.; Paiva, M.; Siler-Marsiglio, K. Ethanol influences on Bax translocation, mitochondrial membrane potential, and reactive oxygen species generation are modulated by vitamin E and brain-derived neurotrophic factor. Alcohol. Clin. Exp. Res. 2011, 35, 1122–1133. [Google Scholar] [CrossRef]
- Ullah, I.; Ullah, N.; Naseer, M.I.; Lee, H.Y.; Kim, M.O. Neuroprotection with metformin and thymoquinone against ethanol-induced apoptotic neurodegeneration in prenatal rat cortical neurons. BMC Neurosci. 2012, 13, 11. [Google Scholar] [CrossRef]
- Bhave, S.V.; Hoffman, P.L. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J. Neurochem. 1997, 68, 578–586. [Google Scholar] [CrossRef]
- Zhang, F.X.; Rubin, R.; Rooney, T.A. Ethanol induces apoptosis in cerebellar granule neurons by inhibiting insulin-like growth factor 1 signaling. J. Neurochem. 1998, 71, 196–204. [Google Scholar] [CrossRef]
- Bhave, S.V.; Ghoda, L.; Hoffman, P.L. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: Signal transduction cascades and site of ethanol action. J. Neurosci. 1999, 19, 3277–3286. [Google Scholar]
- Jiang, X.; Zhu, D.; Okazaki, P.; Lipsky, R.; Wu, X.; Banaudha, K.; Mearow, K.; Strauss, K.I.; Marini, A.M. N-methyl-d-aspartate and TrkB receptor activation in cerebellar granule cells: An in vitro model of preconditioning to stimulate intrinsic survival pathways in neurons. Ann. N. Y. Acad. Sci. 2003, 993, 134–145. [Google Scholar] [CrossRef]
- Yao, R.; Cooper, G.M. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [Google Scholar]
- Liu, L.; Cao, J.X.; Sun, B.; Li, H.L.; Xia, Y.; Wu, Z.; Tang, C.L.; Hu, J. Mesenchymal stem cells inhibition of chronic ethanol-induced oxidative damage via upregulation of phosphatidylinositol-3-kinase/Akt and modulation of extracellular signal-regulated kinase 1/2 activation in PC12 cells and neurons. Neuroscience 2010, 167, 1115–1124. [Google Scholar] [CrossRef]
- Zhou, H.; Li, X.M.; Meinkoth, J.; Pittman, R.N. Akt regulates cell survival and apoptosis at a postmitochondrial level. J. Cell Biol. 2000, 151, 483–494. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.; Ma, C.; Bower, K.A.; Xu, M.; Fan, Z.; Shi, X.; Ke, Z.J.; Luo, J. Overexpression of glycogen synthase kinase 3 beta sensitizes neuronal cells to ethanol toxicity. J. Neurosci. Res. 2009, 87, 2793–2802. [Google Scholar] [CrossRef]
- Takadera, T.; Ohyashiki, T. Glycogen synthase kinase-3 inhibitors prevent caspase-dependent apoptosis induced by ethanol in cultured rat cortical neurons. Eur. J. Pharmacol. 2004, 499, 239–245. [Google Scholar] [CrossRef]
- McAlhany, R.E., Jr.; West, J.R.; Miranda, R.C. Glial-derived neurotrophic factor (GDNF) prevents ethanol-induced apoptosis and JUN kinase phosphorylation. Brain Res. Dev. Brain Res. 2000, 119, 209–216. [Google Scholar] [CrossRef]
- Pascual, M.; Valles, S.L.; Renau-Piqueras, J.; Guerri, C. Ceramide pathways modulate ethanol-induced cell death in astrocytes. J. Neurochem. 2003, 87, 1535–1545. [Google Scholar] [CrossRef]
- Vallés, S.; Blanco, A.M.; Pascual, M.; Guerri, C. Chronic ethanol treatment enhances inflammatory mediators and cell death in the brain and in astrocytes. Brain Pathol. 2004, 14, 365–371. [Google Scholar]
- Villegas, S.N.; Njaine, B.; Linden, R.; Carri, N.G. Glial-derived neurotrophic factor (GDNF) prevents ethanol (EtOH) induced B92 glial cell death by both PI3K/AKT and MEK/ERK signaling pathways. Brain Res. Bull. 2006, 71, 116–126. [Google Scholar] [CrossRef]
- Antonio, A.M.; Druse, M.J. Antioxidants prevent ethanol-associated apoptosis in fetal rhombencephalic neurons. Brain Res. 2008, 1204, 16–23. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef]
- Ramachandran, V.; Watts, L.T.; Maffi, S.K.; Chen, J.; Schenker, S.; Henderson, G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003, 74, 577–588. [Google Scholar] [CrossRef]
- Sheth, D.S.; Tajuddin, N.F.; Druse, M.J. Antioxidant neuroprotection against ethanol-induced apoptosis in HN2-5 cells. Brain Res. 2009, 1285, 14–21. [Google Scholar] [CrossRef]
- Chu, J.; Tong, M.; de la Monte, S.M. Chronic ethanol exposure causes mitochondria dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol. 2007, 113, 659–673. [Google Scholar] [CrossRef]
- Sun, A.Y.; Chen, Y.M.; James-Kracke, M.; Wixom, P.; Cheng, Y. Ethanol-induced cell death by lipid peroxidation in PC12 cells. Neurochem. Res. 1997, 22, 1187–1192. [Google Scholar] [CrossRef]
- Maffi, S.K.; Rathinam, M.L.; Cherian, P.P.; Pate, W.; Hamby-Mason, R.; Schenker, S.; Henderson, G.I. Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis. J. Neurosci. Res. 2008, 86, 1064–1076. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. The Ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar] [CrossRef]
- Buccoliero, R.; Futerman, A.H. The roles of ceramide and complex sphingolipids in neuronal cell function. Pharmacol. Res. 2003, 47, 409–419. [Google Scholar] [CrossRef]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef]
- Falluel-Morel, A.; Aubert, N.; Vaudry, D.; Desfeux, A.; Allais, A.; Burel, D.; Basille, M.; Vaudry, H.; Laudenbach, V.; Gonzalez, B.J. Interactions of PACAP and ceramides in the control of granule cell apoptosis during cerebellar development. J. Mol. Neurosci. 2008, 36, 8–15. [Google Scholar] [CrossRef]
- Toman, R.E.; Spiegel, S.; Faden, A.I. Role of ceramide in neuronal cell death and differentiation. J. Neurotrauma 2000, 17, 891–898. [Google Scholar] [CrossRef]
- Wiegmann, K.; Schütze, S.; Machleidt, T.; Witte, D.; Krönke, M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 1994, 78, 1005–1015. [Google Scholar] [CrossRef]
- Qin, J.; Berdyshev, E.; Goya, J.; Natarajan, V.; Dawson, G. Neurons and oligodendrocytes recycle sphingosine 1-phosphate to ceramide: Significance for apoptosis and multiple sclerosis. J. Biol. Chem. 2010, 285, 14134–14143. [Google Scholar]
- Ginkel, C.; Hartmann, D.; vom Dorp, K.; Zlomuzica, A.; Farwanah, H.; Eckhardt, M.; Sandhoff, R.; Degen, J.; Rabionet, M.; Dere, E.; et al. Ablation of neuronal ceramide synthase 1 in mice decreases ganglioside levels and expression of myelin-associated glycoprotein in oligodendrocytes. J. Biol. Chem. 2012, 287, 41888–41902. [Google Scholar] [CrossRef]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef]
- Jin, J.; Hou, Q.; Mullen, T.D.; Zeidan, Y.H.; Bielawski, J.; Kraveka, J.M.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A.; Hsu, Y.T. Ceramide generated by sphingomyelin hydrolysis and the salvage pathway is involved in hypoxia/reoxygenation-induced Bax redistribution to mitochondria in NT-2 cells. J. Biol. Chem. 2008, 283, 26509–26517. [Google Scholar] [CrossRef]
- Yu, J.; Novgorodov, S.A.; Chudakova, D.; Zhu, H.; Bielawska, A.; Bielawski, J.; Obeid, L.M.; Kindy, M.S.; Gudz, T.I. JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J. Biol. Chem. 2007, 282, 25940–25949. [Google Scholar] [CrossRef]
- Aflaki, E.; Doddapattar, P.; Radović, B.; Povoden, S.; Kolb, D.; Vujić, N.; Wegscheider, M.; Koefeler, H.; Hornemann, T.; Graier, W.F.; et al. C16 ceramide is crucial for triacylglycerol-induced apoptosis in macrophages. Cell Death Dis. 2012, 3, e280. [Google Scholar] [CrossRef] [Green Version]
- Kroesen, B.J.; Pettus, B.; Luberto, C.; Busman, M.; Sietsma, H.; de Leij, L.; Hannun, Y.A. Induction of apoptosis through B-cell receptor cross-linking occurs via de novo generated C16-ceramide and involves mitochondria. J. Biol. Chem. 2001, 276, 13606–13614. [Google Scholar]
- Seumois, G.; Fillet, M.; Gillet, L.; Faccinetto, C.; Desmet, C.; François, C.; Dewals, B.; Oury, C.; Vanderplasschen, A.; Lekeux, P.; Bureau, F. De novo C16- and C24-ceramide generation contributes to spontaneous neutrophil apoptosis. J. Leukoc. Biol. 2007, 81, 1477–1486. [Google Scholar] [CrossRef]
- Wang, G.; Silva, J.; Dasgupta, S.; Bieberich, E. Long-chain ceramide is elevated in presenilin 1 (PS1M146V) mouse brain and induces apoptosis in pS1 astrocytes. Glia 2008, 56, 449–456. [Google Scholar] [CrossRef]
- Stoica, B.A.; Movsesyan, V.A.; Knoblach, S.M.; Faden, A.I. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol. Cell. Neurosci. 2005, 29, 355–371. [Google Scholar] [CrossRef]
- Willaime, S.; Vanhoutte, P.; Caboche, J.; Lemaigre-Dubreuil, Y.; Mariani, J.; Brugg, B. Ceramide-induced apoptosis in cortical neurons is mediated by an increase in p38 phosphorylation and not by the decrease in ERK phosphorylation. Eur. J. Neurosci. 2001, 13, 2037–2046. [Google Scholar] [CrossRef]
- Arboleda, G.; Cárdenas, Y.; Rodríguez, Y.; Morales, L.C.; Matheus, L.; Arboleda, H. Differential regulation of AKT, MAPK, and GSK3β during C2-ceramide induced neuronal death. Neurotoxicology 2010, 31, 687–693. [Google Scholar] [CrossRef]
- Goswami, R.; Kilkus, J.; Dawson, S.A.; Dawson, G. Overexpression of Akt (protein kinase B) confers protection against apoptosis and prevents formation of ceramide in response to pro-apoptotic stimuli. J. Neurosci. Res. 1999, 57, 884–893. [Google Scholar] [CrossRef]
- Dobrowsky, R.T.; Kamibayashi, C.; Mumby, M.C.; Hannun, Y.A. Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 1993, 268, 15523–15530. [Google Scholar]
- Lin, C.F.; Chen, C.L.; Chiang, C.W.; Jan, M.S.; Huang, W.C.; Lin, Y.S. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J. Cell Sci. 2007, 120, 2935–2943. [Google Scholar] [CrossRef]
- Junttila, M.R.; Li, S.P.; Westermarck, J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008, 22, 954–965. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Andrieu-Abadie, N.; Gouazé, V.; Salvayre, R.; Levade, T. Ceramide in apoptosis signaling: Relationship with oxidative stress. Free Radic. Biol. Med. 2001, 31, 717–728. [Google Scholar] [CrossRef]
- Won, J.S.; Singh, I. Sphingolipid signaling and redox regulation. Free Radic. Biol. Med. 2006, 40, 1875–1888. [Google Scholar]
- Barth, B.M.; Gustafson, S.J.; Kuhn, T.B. Neutral sphingomyelinase activation precedes NADPH oxidase dependent damage in neurons exposed to the proinflammatory cytokine tumor necrosis factor-α. J. Neurosci. Res. 2012, 90, 229–242. [Google Scholar] [CrossRef]
- Young, M.M.; Kester, M.; Wang, H.G. Sphingolipids: Regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013, 54, 5–19. [Google Scholar] [CrossRef]
- Chipuk, J.E.; McStay, G.P.; Bharti, A.; Kuwana, T.; Clarke, C.J.; Siskind, I.J.; Obeid, L.M.; Green, D.R. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012, 148, 988–1000. [Google Scholar] [CrossRef]
- Ganesan, V.; Perera, M.N.; Colombinini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef]
- Kashkar, H.; Wiegmann, K.; Yazdanpanah, B.; Haubert, D.; Krönke, M. Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J. Biol. Chem. 2005, 280, 20804–20813. [Google Scholar]
- Lee, H.; Rotolo, J.A.; Mesicek, J.; Penate-Medina, T.; Rimner, A.; Liao, W.C.; Yin, X.; Ragupathi, G.; Ehleiter, D.; Gulbins, E.; et al. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One 2011, 6, e19783. [Google Scholar] [CrossRef]
- Rego, A.; Costa, M.; Chaves, S.R.; Matmati, N.; Pereira, H.; Sousa, M.J.; Moradas-Ferreira, P.; Hannun, Y.A.; Costa, V.; Côrte-Real, M. Modulation of mitochondrial outer membrane permeabilization and apoptosis by ceramide metabolism. PLoS One 2012, 7, e48571. [Google Scholar]
- Kiebish, M.A.; Han, X.; Cheng, H.; Lunceford, A.; Clarke, C.F.; Moon, H.; Chuang, J.H.; Seyfried, T.N. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 2008, 106, 299–312. [Google Scholar] [CrossRef]
- Wu, B.X.; Rajagopalan, V.; Roddy, P.L.; Clarke, C.J.; Hannun, Y.A. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 2010, 285, 17993–18002. [Google Scholar]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar]
- Novgorodov, S.A.; Gudz, T.I. Ceramide and mitochondria in ischemic brain injury. Int. J. Biochem. Mol. Biol. 2011, 2, 347–361. [Google Scholar]
- Sanvicens, N.; Cotter, T.G. Ceramide is the key mediator of oxidative stress-induced apoptosis in retinal photoreceptor cells. J. Neurochem. 2006, 98, 1432–1444. [Google Scholar]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef]
- Uchida, Y.; Murata, S.; Schmuth, M.; Behne, M.J.; Lee, J.D.; Ichikawa, S.; Elias, P.M.; Hirabayshi, Y.; Holleran, W.M. Glucosylceramide synthesis and synthase expression protect against ceramide-induced stress. J. Lipid Res. 2002, 43, 1293–1302. [Google Scholar]
- Sietsma, H.; Veldman, R.J.; Kok, J.W. The Involvement of sphingolipids in multidrug resistance. J. Membr. Biol. 2001, 181, 153–162. [Google Scholar]
- Chakraborty, G.; Saito, M.; Mao, R.F.; Wang, R.; Vadasz, C.; Saito, M. Involvement of sphingomyelinase and the AKT-pathway in ethanol-induced neurodegeneration in the neonatal mouse brain. Alcohol. Clin. Exp. Res. 2007, 31 (Suppl. 2), 40A. [Google Scholar]
- Saito, M.; Chakraborty, G.; Shah, R.; Mao, R.F.; Saito, M. Nathan Kline Institute for Psychiatric Research: New York, NY, USA, Unpublished work. 2011.
- Ramirez, T.; Longato, L.; Dostalek, M.; Tong, M.; Wands, J.R.; de la Monte, S.M. Insulin resistance, ceramide accumulation and endoplasmic reticulum stress in experimental chronic alcohol-induced steatohepatitis. Alcohol Alcohol. 2013, 48, 39–52. [Google Scholar] [CrossRef]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 246–251. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Rahmini, Y.; Ross, R.A.; Zhao, Z.; Xu, Y.; Crabb, D.W. Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G515–G523. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Sozio, M.S.; Shin, E.; Zhao, Z.; Xu, Y.; Ross, R.A.; Zeng, Y.; Crabb, D.W. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G1004–G1012. [Google Scholar] [CrossRef]
- Helfer, J.L.; Calizo, L.H.; Dong, W.K.; Goodlett, C.R.; Greenough, W.T.; Klintsova, A.Y. Binge-like postnatal alcohol exposure triggers cortical gliogenesis in adolescent rats. J. Comp. Neurol. 2009, 514, 259–271. [Google Scholar] [CrossRef]
- Tiwari, V.; Chopra, K. Resveratrol prevents alcohol-induced cognitive deficits and brain damage by blocking inflammatory signaling and cell death cascade in neonatal rat brain. J. Neurochem. 2011, 117, 678–690. [Google Scholar]
- Dasgupta, S.; Adams, J.A.; Hogan, E.L. Maternal alcohol consumption increases sphingosine levels in the brains of progeny mice. Neurochem. Res. 2007, 32, 2217–2224. [Google Scholar] [CrossRef]
- Wang, G.; Bieberich, E. Prenatal alcohol exposure triggers ceramide-induced apoptosis in neural crest-derived tissues concurrent with defective cranial development. Cell Death Dis. 2010, 1, e46. [Google Scholar] [CrossRef]
- Schatter, B.; Jin, S.; Löffelholz, K.; Klein, J. Cross-talk between phosphatidic acid and ceramide during ethanol-induced apoptosis in astrocytes. BMC Pharmacol. 2005, 5, 3. [Google Scholar]
- Saito, M.; Chakraborty, G.; Mao, R.F.; Wang, R.; Shah, R.; Saito, M. Ceramide and GM2 increased in mitochondria in the developing brain exposed to ethanol. Alcohol. Clin. Exp. Res. 2011, 35 (Suppl. 1), 44A. [Google Scholar]
- Heaton, M.B.; Siler-Marsiglio, K.; Paiva, M.; Kotler, A.; Rogozinski, J.; Kubovec, S.; Coursen, M.; Madorsky, V. Ethanol influences on bax associations with mitochondrial membrane proteins in neonatal rat cerebellum. J. Neurobiol. 2013, 66, 95–101. [Google Scholar]
- Adams, S.M.; de Rivero Vaccari, J.C.; Corriveau, R.A. Pronounced cell death in the absence of NMDA receptors in the developing somatosensory thalamus. J. Neurosci. 2004, 24, 9441–9450. [Google Scholar] [CrossRef]
- Fredriksson, A.; Archer, T.; Alm, H.; Gordh, T.; Eriksson, P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav. Brain Res. 2004, 153, 367–376. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Wang, C.; Sadovova, N.; Hotchkiss, C.; Fu, X.; Scallet, A.C.; Patterson, T.A.; Hanig, J.; Paule, M.G.; Slikker, W., Jr. Blockade of N-methyl-d-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicol. Sci. 2006, 91, 192–201. [Google Scholar] [CrossRef]
- Blomgren, K.; Leist, M.; Groc, L. Pathological apoptosis in the developing brain. Apoptosis 2007, 12, 993–1010. [Google Scholar] [CrossRef]
- Allgaier, C. Ethanol sensitivity of NMDA receptors. Neurochem. Int. 2002, 41, 377–382. [Google Scholar] [CrossRef]
- Krystal, J.H.; Petrakis, I.L.; Krupitsky, E.; Schutz, C.; Trevisan, L.; D’Souza, D.C. NMDA receptor antagonism and the ethanol intoxication signal: From alcoholism risk to pharmacotherapy. Ann. N. Y. Acad. Sci. 2003, 1003, 176–184. [Google Scholar] [CrossRef]
- Ota, K.; Yakovlev, A.G.; Itaya, A.; Kameoka, M.; Tanaka, Y.; Yoshihara, K. Alteration of apoptotic protease-activating factor-1 (APAF-1)-dependent apoptotic pathway during development of rat brain and liver. J. Biochem. 2002, 131, 131–135. [Google Scholar] [CrossRef]
- Vekrellis, K.; McCarthy, M.J.; Watson, A.; Whitfield, J.; Rubin, L.L.; Ham, J. Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development 1997, 124, 1239–1249. [Google Scholar]
- Chakraborty, G.; Saito, M.; Shah, R.; Mao, R.F.; Vadasz, C.; Saito, M. Ethanol triggers sphingosine 1-phosphate elevation along with neuroapoptosis in the developing mouse brain. J. Neurochem. 2012, 121, 806–817. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Mao, R.F.; Vadasz, C.; Saito, M. Developmental profiles of lipogenic enzymes and their regulators in the neonatal mouse brain. Neurochem. Res. 2009, 34, 1945–1954. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Longato, L.; Tong, M.; DeNucci, S.; Wands, J.R. The liver-brain axis of alcohol-mediated neurodegeneration: Role of toxic lipids. Int. J. Environ. Res. Public Health 2009, 6, 2055–2075. [Google Scholar] [CrossRef]
- Okada, T.; Kajimoto, T.; Jahangeer, S.; Nakamura, S. Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal. 2009, 21, 7–13. [Google Scholar] [CrossRef]
- Singh, I.N.; Hall, E.D. Multifaceted roles of sphingosine-1-phosphate: How does this bioactive sphingolipid fit with acute neurological injury? J. Neurosci. Res. 2008, 86, 1419–1433. [Google Scholar] [CrossRef]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A house divided: Ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 2006, 1758, 2027–2036. [Google Scholar] [CrossRef]
- Snider, A.; Orr Gandy, K.A.; Obeid, L.M. Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 2010, 92, 707–715. [Google Scholar] [CrossRef]
- Davaille, J.; Li, L.; Mallat, A.; Lotersztajn, S. Sphingosine 1-phosphate triggers both apoptotic and survival signals for human hepatic myofibroblasts. J. Biol. Chem. 2002, 277, 37323–37330. [Google Scholar]
- Nakahara, T.; Iwase, A.; Nakamura, T.; Kondo, M.; Bayasula; Kobayashi, H.; Takikawa, S.; Manabe, S.; Goto, M.; Kotani, T.; Kikkawa, F. Sphingosine-1-phosphate inhibits H2O2-induced granulosa cell apoptosis via the PI3K/Akt signaling pathway. Fertil. Steril. 2012, 98, 1001–1008. [Google Scholar] [CrossRef]
- Nieuwenhuis, B.; Lüth, A.; Kleuser, B. Dexamethasone protects human fibroblasts from apoptosis via an S1P3-receptor subtype dependent activation of PKB/Akt and Bcl XL. Pharmacol. Res. 2010, 61, 449–459. [Google Scholar] [CrossRef]
- Zhang, J.; Honbo, N.; Goetz, E.J.; Chatterjee, K.; Karliner, J.S.; Gray, M. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3150–H3158. [Google Scholar] [CrossRef]
- Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 2003, 278, 46832–46839. [Google Scholar]
- Hagen, N.; Van Veldhoven, P.P.; Proia, R.L.; Park, H.; Merrill, A.H., Jr.; van Echten-Deckert, G. Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 2009, 284, 11346–11353. [Google Scholar]
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; Milstien, S.; Spiegel, S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef]
- Moore, A.N.; Kampfl, A.W.; Zhao, X.; Hayes, R.L.; Dash, P.K. Sphingosine-1-phosphate induces apoptosis of cultured hippocampal neurons that requires protein phosphatases and activator protein-1 complexes. Neuroscience 1999, 94, 405–415. [Google Scholar] [CrossRef]
- Agudo-López, A.; Miguel, B.G.; Fernández, I.; Martínez, A.M. Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia. Neurosci. Lett. 2010, 470, 130–133. [Google Scholar] [CrossRef]
- Shinpo, K.; Kikuchi, S.; Moriwaka, F.; Tashiro, K. Protective effects of the TNF-ceramide pathway against glutamate neurotoxicity on cultured mesencephalic neurons. Brain Res. 1999, 819, 170–173. [Google Scholar] [CrossRef]
- Lee, D.H.; Jeon, B.T.; Jeong, E.A.; Kim, J.S.; Cho, Y.W.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Altered expression of sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 in mouse hippocampus after kainic acid treatment. Biochem. Biophys. Res. Commun. 2010, 393, 476–480. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Muraki, T.; Ishikawa, K.; Kuwabara, A.; Okajima, F. Assessment of the role of sphingosine 1-phosphate and its receptors in high-density lipoprotein-induced stimulation of astroglial cell function. Biochem. J. 2003, 370, 817–827. [Google Scholar] [CrossRef]
- Pébay, A.; Toutant, M.; Prémont, J.; Calvo, C.F.; Venance, L.; Cordier, J.; Glowinski, J.; Tencé, M. Sphingosine-1-phosphate induces proliferation of astrocytes: Regulation by intracellular signalling cascades. Eur. J. Neurosci. 2001, 13, 2067–2076. [Google Scholar] [CrossRef]
- Sorensen, S.D.; Nicole, O.; Peavy, R.D.; Montoya, L.M.; Lee, C.J.; Murphy, T.J.; Traynelis, S.F.; Hepler, J.R. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol. Pharmacol. 2003, 64, 1199–1209. [Google Scholar] [CrossRef]
- Yamagata, K.; Tagami, M.; Torii, Y.; Takenaga, F.; Tsumagari, S.; Itoh, S.; Yamori, Y.; Nara, Y. Sphingosine 1-phosphate induces the production of glial cell line-derived neurotrophic factor and cellular proliferation in astrocytes. Glia 2003, 41, 199–206. [Google Scholar] [CrossRef]
- Nayak, D.; Huo, Y.; Kwang, W.X.; Pushparaj, P.N.; Kumar, S.D.; Ling, E.A.; Dheen, S.T. Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 2010, 166, 132–144. [Google Scholar] [CrossRef]
- Blondeau, N.; Lai, Y.; Tyndall, S.; Popolo, M.; Topalkara, K.; Pru, J.K.; Zhang, L.; Kim, H.; Liao, J.K.; Ding, K.; Waeber, C. Distribution of sphingosine kinase activity and mRNA in rodent brain. J. Neurochem. 2007, 103, 509–517. [Google Scholar] [CrossRef]
- Johnson, K.R.; Becker, K.P.; Faccinetti, M.M.; Hannun, Y.A.; Obeid, L.M. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA). J. Biol. Chem. 2002, 277, 35257–35262. [Google Scholar]
- Pitson, S.M.; Xia, P.; Leclercq, T.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Wattenberg, B.W.; Vadas, M.A. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J. Exp. Med. 2005, 201, 49–54. [Google Scholar]
- Hasegawa, Y.; Suzuki, H.; Sozen, T.; Rolland, W.; Zhang, J.H. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 2010, 41, 368–374. [Google Scholar] [CrossRef]
- Pfeilschifter, W.; Czech-Zechmeister, B.; Sujak, M.; Mirceska, A.; Koch, A.; Rami, A.; Steinmetz, H.; Foerch, C.; Huwiler, A.; Pfeilschifter, J. Activation of sphingosine kinase 2 is an endogenous protective mechanism in cerebral ischemia. Biochem. Biophys. Res. Commun. 2011, 413, 212–217. [Google Scholar] [CrossRef]
- Wacker, B.K.; Park, T.S.; Gidday, J.M. Hypoxic preconditioning-induced cerebral ischemic tolerance: Role for microvascular sphingosine kinase 2. Stroke 2009, 40, 3342–3348. [Google Scholar] [CrossRef]
- Yung, L.M.; Wei, Y.; Qin, T.; Wang, Y.; Smith, C.D.; Waeber, C. Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke 2012, 43, 199–204. [Google Scholar] [CrossRef]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef]
- Gomez, L.; Paillard, M.; Price, M.; Chen, Q.; Teixeira, G.; Spiegel, S.; Lesnefsky, E.J. A novel role for mitochondrial sphingosine-1-phosphate produced by sphingosine kinase-2 in PTP-mediated cell survival during cardioprotection. Basic Res. Cardiol. 2011, 106, 1341–1353. [Google Scholar] [CrossRef]
- Zheng, D.M.; Kitamura, T.; Ikejima, K.; Enomoto, N.; Yamashina, S.; Suzuki, S.; Takei, Y.; Sato, N. Sphingosine 1-phosphate protects rat liver sinusoidal endothelial cells from ethanol-induced apoptosis: Role of intracellular calcium and nitric oxide. Hepatology 2006, 44, 1278–1287. [Google Scholar] [CrossRef]
- Fournie, P.; Galiacy, S.; Ranty, M.L.; Rico-Lattes, I.; Malecaze, F.; Quintyn, J.C. Sphingosine-1 phosphate prevents ethanol-induced corneal epithelial apoptosis. Indian J. Ophthalmol. 2012, 60, 115–118. [Google Scholar] [CrossRef]
- Huang, J.S.; Mukherjee, J.J.; Kiss, Z. Ethanol potentiates the mitogenic effects of sphingosine 1-phosphate by a zinc- and calcium-dependent mechanism in fibroblasts. Arch. Biochem. Biophys. 1999, 366, 131–138. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Shah, R.; Saito, M. Nathan Kline Institute for Psychiatric Research: New York, NY, USA, Unpublished work. 2012.
- Brinkmann, V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [Google Scholar] [CrossRef]
- Chen, G.; Ke, Z.; Xu, M.; Liao, M.; Wang, X.; Qi, Y.; Zhang, T.; Frank, J.A.; Bower, K.A.; Shi, X.; Luo, J. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012, 8, 1577–1589. [Google Scholar]
- Prock, T.L.; Miranda, R.C. Embryonic cerebral cortical progenitors are resistant to apoptosis, but increase expression of suicide receptor DISC-complex genes and suppress autophagy following ethanol exposure. Alcohol. Clin. Exp. Res. 2007, 31, 694–703. [Google Scholar]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Yin, X.M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy 2011, 7, 248–249. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Thomes, P.G.; Ding, W.X.; Lemasters, J.J.; Donohue, T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcohol. Clin. Exp. Res. 2012, 36, 1301–1308. [Google Scholar] [CrossRef]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Charlie Dong, X.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013. [Google Scholar] [CrossRef]
- Thomes, P.G.; Ehlers, R.A.; Trambly, C.S.; Clemens, D.L.; Fox, H.S.; Tuma, D.J.; Donohue, T.M. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy 2013, 9, 63–73. [Google Scholar]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Is autophagy the key mechanism by which the sphingolipid rheostat controls the cell fate decision? Autophagy 2007, 3, 45–47. [Google Scholar]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar] [CrossRef]
- Sims, K.; Haynes, C.A.; Kelly, S.; Allegood, J.C.; Wang, E.; Momin, A.; Leipelt, M.; Reichart, D.; Glass, C.K.; Sullards, M.C.; Merrill, A.H., Jr. Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J. Biol. Chem. 2010, 285, 38568–38579. [Google Scholar] [CrossRef]
- Yamagata, M.; Obara, K.; Kihara, A. Sphingolipid synthesis is involved in autophagy in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2011, 410, 786–791. [Google Scholar] [CrossRef]
- Pattingre, S.; Bauvy, C.; Levade, T.; Levine, B.; Codogno, P. Ceramide-induced autophagy: To junk or to protect cells? Autophagy 2009, 5, 558–560. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Taniguchi, M.; Kitatani, K.; Kondo, T.; Hashimoto-Nishimura, M.; Asano, S.; Hayashi, A.; Mitsutake, S.; Igarashi, Y.; Umehara, H.; Takeya, H.; et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: Reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J. Biol. Chem. 2012, 87, 39898–39910. [Google Scholar]
- Lavieu, G.; Scarlatti, F.; Sala, G.; Carpentier, S.; Levade, T.; Ghidoni, R.; Botti, J.; Codogno, P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 2006, 281, 8518–8527. [Google Scholar]
- Ledeen, R.W.; Wu, G. Ganglioside function in calcium homeostasis and signaling. Neurochem. Res. 2002, 27, 637–647. [Google Scholar] [CrossRef]
- Hakomori, S. Structure, organization, and function of glycosphingolipids in membrane. Curr. Opin. Hematol. 2003, 10, 16–24. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—an overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef]
- Castiglione, M.; Spinsanti, P.; Iacovelli, L.; Lenti, L.; Martini, F.; Gradini, R.; Di Giorgi Gerevini, V.; Caricasole, A.; Caruso, A.; de Maria, R.; et al. Activation of Fas receptor is required for the increased formation of the disialoganglioside GD3 in cultured cerebellar granule cells committed to apoptotic death. Neuroscience 2004, 126, 889–898. [Google Scholar]
- Copani, A.; Melchiorri, D.; Caricasole, A.; Martini, F.; Sale, P.; Carnevale, R.; Gradini, R.; Sortino, M.A.; Lenti, L.; de Maia, R.; Nicoletti, F. Beta-Amyloid-induced synthesis of the ganglioside GD3 is a requisite for cell cycle reactivation and apoptosis in neurons. J. Neurosci. 2002, 22, 3963–3968. [Google Scholar]
- Hasegawa, T.; Sugeno, N.; Takeda, A.; Matsuzaki-Kobayashi, M.; Kikuchi, A.; Furukawa, K.; Miyagi, T.; Itoyama, Y. Role of Neu4L sialidase and its substrate ganglioside GD3 in neuronal apoptosis induced by catechol metabolites. FEBS Lett. 2007, 581, 406–412. [Google Scholar] [CrossRef]
- Lovat, P.E.; Di Sano, F.; Corazzari, M.; Fazi, B.; Donnorso, R.P.; Pearson, A.D.; Hall, A.G.; Redfern, C.P.; Piacentini, M. Gangliosides link the acidic sphingomyelinase mediated induction of ceramide to 12-lipoxygenase dependent apoptosis of neuroblastoma in response to fenretinide. J. Natl. Cancer Inst. 2004, 96, 1288–1299. [Google Scholar] [CrossRef]
- Melchiorri, D.; Martini, F.; Lococo, E.; Gradini, R.; Barletta, E.; de Maria, R.; Caricasole, A.; Nicoletti, F.; Lenti, L. An early increase in the disialoganglioside GD3 contributes to the development of neuronal apoptosis in culture. Cell Death Differ. 2002, 9, 609–615. [Google Scholar] [CrossRef]
- De Maria, R.; Lenti, L.; Malisan, F.; d’Agostino, F.; Tomassini, B.; Zeuner, A.; Rippo, M.R.; Testi, R. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science 1997, 277, 1652–1655. [Google Scholar] [CrossRef]
- Malorni, W.; Giammarioli, A.M.; Garofalo, T.; Sorice, M. Dynamics of lipid raft components during lymphocyte apoptosis: The paradigmatic role of GD3. Apoptosis 2007, 12, 941–949. [Google Scholar] [CrossRef]
- Sorice, M.; Garofalo, T.; Misasi, R.; Manganelli, V.; Vona, R.; Malorni, W. Ganglioside GD3 as a raft component in cell death regulation. Anticancer Agents Med. Chem. 2012, 12, 376–382. [Google Scholar] [CrossRef]
- Ando, S.; Toyoda, Y.; Nagai, Y.; Ikuta, F. Alterations in brain gangliosides and other lipids with Creutzfeldt-Jakob disease and subacute sclerosing panencephalitis (SSPE). Jpn. J. Exp. Med. 1984, 54, 229–234. [Google Scholar]
- Ohtani, Y.; Tamai, Y.; Ohnuki, S.; Miura, S. Ganglioside alterations in the central and peripheral nervous systems of patients with Creutzfeldt-Jakob disease. Neurodegeneration 1996, 5, 331–338. [Google Scholar] [CrossRef]
- Yu, R.K.; Ledeen, R.W.; Eng, L.F. Ganglioside abnormalities in multiple sclerosis. J. Neurochem. 1974, 23, 169–174. [Google Scholar] [CrossRef]
- Malisan, F.; Testi, R. GD3 ganglioside and apoptosis. Biochim. Biophys. Acta 2002, 1585, 179–187. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, S.H.; Cho, H.Y.; Shin, H.S.; Sung, H.R.; Jung, J.R.; Quan, M.L.; Jiang, D.H.; Bae, H.R. GD3 accumulation in cell surface lipid rafts prior to mitochondrial targeting contributes to amyloid-β-induced apoptosis. J. Korean Med. Sci. 2010, 25, 1492–1498. [Google Scholar] [CrossRef]
- Rippo, M.R.; Malisan, F.; Ravagnan, L.; Tomassini, B.; Condo, I.; Costantini, P.; Susin, S.A.; Rufini, A.; Todaro, M.; Kroemer, G.; Testi, R. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 2000, 14, 2047–2054. [Google Scholar]
- García-Ruiz, C.; Colell, A.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J. Biol. Chem. 2002, 277, 36443–36448. [Google Scholar]
- Tempera, I.; Buchetti, B.; Lococo, E.; Gradini, R.; Mastronardi, A.; Mascellino, M.T.; Sale, P.; Mosca, L.; d’Erme, M.; Lenti, L. GD3 nuclear localization after apoptosis induction in HUT-78 cells. Biochem. Biophys. Res. Commun. 2008, 368, 495–500. [Google Scholar]
- Nakatsuji, Y.; Miller, R.H. Selective cell-cycle arrest and induction of apoptosis in proliferating neural cells by ganglioside GM3. Exp. Neurol. 2001, 168, 290–299. [Google Scholar]
- Ferrari, G.; Anderson, B.L.; Stephens, R.M.; Kaplan, D.R.; Greene, L.A. Prevention of apoptotic neuronal death by GM1 ganglioside. Involvement of Trk neurotrophin receptors. J. Biol. Chem. 1995, 270, 3074–3080. [Google Scholar]
- Mutoh, T.; Tokuda, A.; Miyadai, T.; Hamaguchi, M.; Fujiki, N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc. Natl. Acad. Sci. USA 1995, 92, 5087–5091. [Google Scholar] [CrossRef]
- Duchemin, A.M.; Ren, Q.; Mo, L.; Neff, N.H.; Hadjiconstantinou, M. GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J. Neurochem. 2002, 81, 696–707. [Google Scholar] [CrossRef]
- Rabin, S.J.; Bachis, A.; Mocchetti, I. Gangliosides activate Trk receptors by inducing the release of neurotrophins. J. Biol. Chem. 2002, 277, 49466–49472. [Google Scholar] [CrossRef]
- Lim, S.T.; Esfahani, K.; Avdoshina, V.; Mocchetti, I. Exogenous gangliosides increase the release of brain-derived neurotrophic factor. Neuropharmacology 2011, 60, 1160–1167. [Google Scholar] [CrossRef]
- Duchemin, A.M.; Ren, Q.; Neff, N.H.; Hadjiconstantinou, M. GM1-induced activation of phosphatidylinositol 3-kinase: Involvement of Trk receptors. J. Neurochem. 2008, 104, 1466–1477. [Google Scholar] [CrossRef]
- Suzuki, S.; Numakawa, T.; Shimazu, K.; Koshimizu, H.; Hara, T.; Hatanaka, H.; Mei, L.; Lu, B.; Kojima, M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: Roles in synaptic modulation. J. Cell Biol. 2004, 167, 1205–1215. [Google Scholar] [CrossRef]
- Wu, C.; Butz, S.; Ying, Y.; Anderson, R.G. Tyrosine kinase receptors concentrated in caveolae-like domains from neuronal plasma membrane. J. Biol. Chem. 1997, 272, 3554–3559. [Google Scholar] [CrossRef]
- Inokuchi, J. Neurotrophic and neuroprotective actions of an enhancer of ganglioside biosynthesis. Int. Rev. Neurobiol. 2009, 85, 319–336. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Wang, J.; Wang, Y.; Xie, X.; Meyenhofer, M.F.; Ledeen, R.W. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: Protection with LIGA 20, a membrane-permeant analog of GM1. J. Neurosci. 2005, 25, 11014–11022. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice lacking major brain gangliosides develop Parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef]
- Maglione, V.; Marchi, P.; Di Pardo, A.; Lingrell, S.; Horkey, M.; Tidmarsh, E.; Sipione, S. Impaired ganglioside metabolism in Huntington’s disease and neuroprotective role of GM1. J. Neurosci. 2010, 30, 4072–4080. [Google Scholar] [CrossRef]
- Svennerholm, L.; Brane, G.; Karlsson, I.; Lekman, A.; Ramstrom, I.; Wikkelso, C. Alzheimer disease-effect of continuous intracerebroventricular treatment with GM1 ganglioside and a systematic activation programme. Dement. Geriatr. Cogn. Disord. 2002, 14, 128–136. [Google Scholar] [CrossRef]
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148. [Google Scholar] [CrossRef]
- Schneider, J.S.; Roeltgen, D.P.; Mancall, E.L.; Chapas-Crilly, J.; Rothblat, D.S.; Tatarian, G.T. Parkinson’s disease: Improved function with GM1 ganglioside treatment in a randomized placebo-controlled study. Neurology 1998, 50, 1630–1636. [Google Scholar] [CrossRef]
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 ganglioside in Parkinson’s disease: Results of a five year open study. J. Neurol. Sci. 2010, 292, 45–51. [Google Scholar] [CrossRef]
- Geisler, F.H.; Dorsey, F.C.; Coleman, W.P. Past and current clinical studies with GM-1 ganglioside in acute spinal cord injury. Ann. Emerg. Med. 1993, 22, 1041–1047. [Google Scholar] [CrossRef]
- Lenzi, G.L.; Grigoletto, F.; Gent, M.; Roberts, R.S.; Walker, M.D.; Easton, J.D.; Carolei, A.; Dorsey, F.C.; Rocca, W.A.; Bruno, R.; et al. Early treatment of stroke with monosialoganglioside GM1. Efficacy and safety results of the early stroke trial. Stroke 1994, 25, 1552–1558. [Google Scholar] [CrossRef]
- Omodeo-Salé, F.; Palestini, P. Chronic ethanol effects on glycoconjugates and glycosyltransferases of rat brain. Alcohol 1994, 11, 301–306. [Google Scholar] [CrossRef]
- Omodeo-Salé, F.; Gornati, R.; Palestini, P. Ganglioside long-chain base composition of rat brain subcellular fractions after chronic ethanol administration. Alcohol 1996, 13, 291–295. [Google Scholar] [CrossRef]
- Sonnino, S.; Chigorno, V. Ganglioside molecular species containing C18- and C20-sphingosine in mammalian nervous tissues and neuronal cell cultures. Biochim. Biophys. Acta 2000, 1469, 63–77. [Google Scholar] [CrossRef]
- Ghosh, P.; Ender, I.; Hale, E.A. Long-term ethanol consumption selectively impairs ganglioside pathway in rat brain. Alcohol. Clin. Exp. Res. 1998, 22, 1220–1226. [Google Scholar] [CrossRef]
- Azuine, M.A.; Patel, S.J.; Lakshman, M.R. Effects of chronic ethanol administration on the activities and relative synthetic rates of myelin and synaptosomal plasma membrane-associated sialidase in the rat brain. Neurochem. Int. 2006, 48, 67–74. [Google Scholar] [CrossRef]
- Garige, M.; Azuine, M.A.; Lakshman, M.R. Chronic ethanol consumption down-regulates CMP-NeuAc:GM3 alpha2,8-sialyltransferase (ST8Sia-1) gene in the rat brain. Neurochem. Int. 2006, 49, 312–318. [Google Scholar] [CrossRef]
- Hungund, B.L.; Ross, D.C.; Gokhale, V.S. Ganglioside GM1 reduces fetal alcohol effects in rat pups exposed to ethanol in utero. Alcohol. Clin. Exp. Res. 1994, 18, 1248–1251. [Google Scholar] [CrossRef]
- Prasad, V.V. Effect of prenatal and postnatal exposure to ethanol on rat central nervous system gangliosides and glycosidases. Lipids 1992, 27, 344–348. [Google Scholar]
- Noronha, A.B.; Druse, M.J.; Gnaedinger, J.M.; Kelly, G.M. Gangliosides in axolemmal and synaptic membrane fractions from developing rats: Effects of maternal ethanol consumption on offspring. Alcohol. Clin. Exp. Res. 1985, 9, 531–534. [Google Scholar]
- Chen, S.Y.; Periasamy, A.; Yang, B.; Herman, B.; Jacobson, K.; Sulik, K.K. Differential sensitivity of mouse neural crest cells to ethanol-induced toxicity. Alcohol 2000, 20, 75–81. [Google Scholar] [CrossRef]
- Chen, S.Y.; Yang, B.; Jacobson, K.; Sulik, K.K. The membrane disordering effect of ethanol on neural crest cells in vitro and the protective role of GM1 ganglioside. Alcohol 1996, 13, 589–595. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Swanson, D.J.; Walker, D.W. Ethanol neurotoxicity in vitro: Effects of GM1 ganglioside and protein synthesis inhibition. Brain Res. 1994, 654, 336–342. [Google Scholar] [CrossRef]
- Saito, M.; Berg, M.J.; Guidotti, A.; Marks, N. Gangliosides attenuate ethanol-induced apoptosis in rat cerebellar granule neurons. Neurochem. Res. 1999, 24, 1107–1115. [Google Scholar] [CrossRef]
- Hungund, B.L.; Mahadik, S.P. Role of gangliosides in behavioral and biochemical actions of alcohol: Cell membrane structure and function. Alcohol. Clin. Exp. Res. 1993, 17, 329–339. [Google Scholar]
- Hungund, B.L.; Gokhale, V.S. Reduction of fatty acid ethyl ester accumulation by ganglioside GM1 in rat fetus exposed to ethanol. Biochem. Pharmacol. 1994, 48, 2103–2108. [Google Scholar] [CrossRef]
- Hungund, B.L.; Zheng, Z.; Lin, L.; Barkai, A.I. Ganglioside GM1 reduces ethanol induced phospholipase A2 activity in synaptosomal preparations from mice. Neurochem. Int. 1994, 25, 321–325. [Google Scholar] [CrossRef]
- Saito, M.; Mao, R.F.; Wang, R.; Vadasz, C.; Saito, M. Effects of gangliosides on ethanol-induced neurodegeneration in the developing mouse brain. Alcohol. Clin. Exp. Res. 2007, 31, 665–674. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saito, M.; Saito, M. Involvement of Sphingolipids in Ethanol Neurotoxicity in the Developing Brain. Brain Sci. 2013, 3, 670-703. https://doi.org/10.3390/brainsci3020670
Saito M, Saito M. Involvement of Sphingolipids in Ethanol Neurotoxicity in the Developing Brain. Brain Sciences. 2013; 3(2):670-703. https://doi.org/10.3390/brainsci3020670
Chicago/Turabian StyleSaito, Mariko, and Mitsuo Saito. 2013. "Involvement of Sphingolipids in Ethanol Neurotoxicity in the Developing Brain" Brain Sciences 3, no. 2: 670-703. https://doi.org/10.3390/brainsci3020670