Ethanol Neurotoxicity in the Developing Cerebellum: Underlying Mechanisms and Implications

{kind=link}
{kind=link}
{kind=link}
Abstract
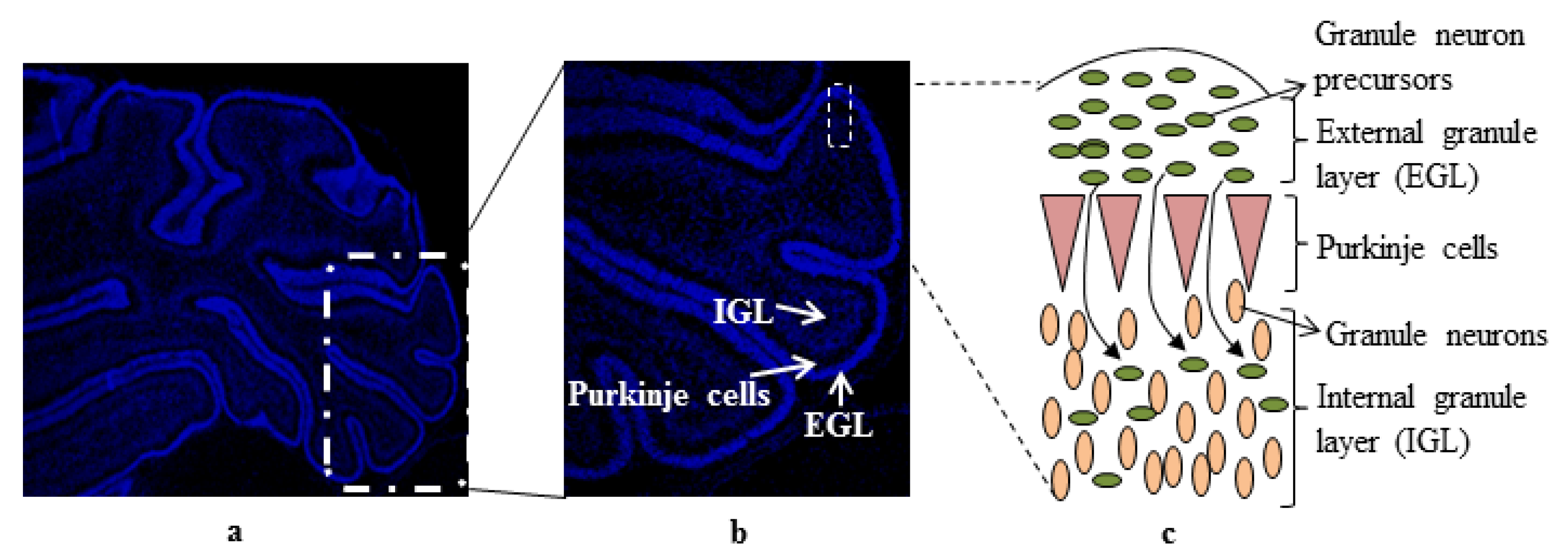
:1. Introduction

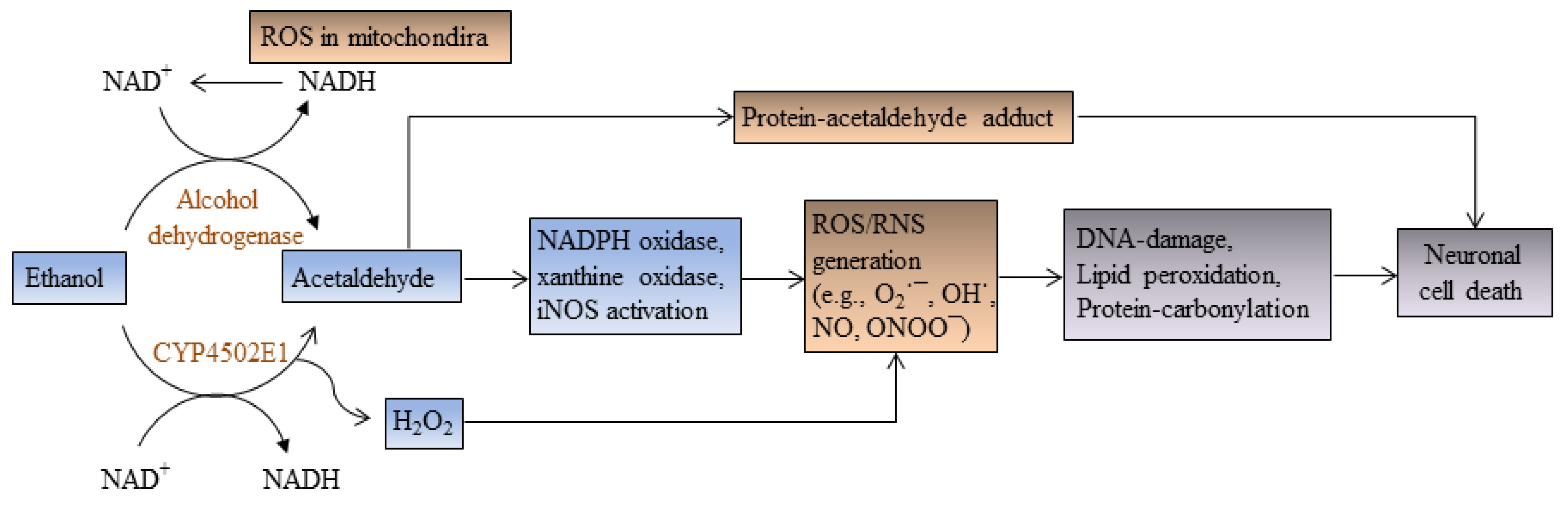
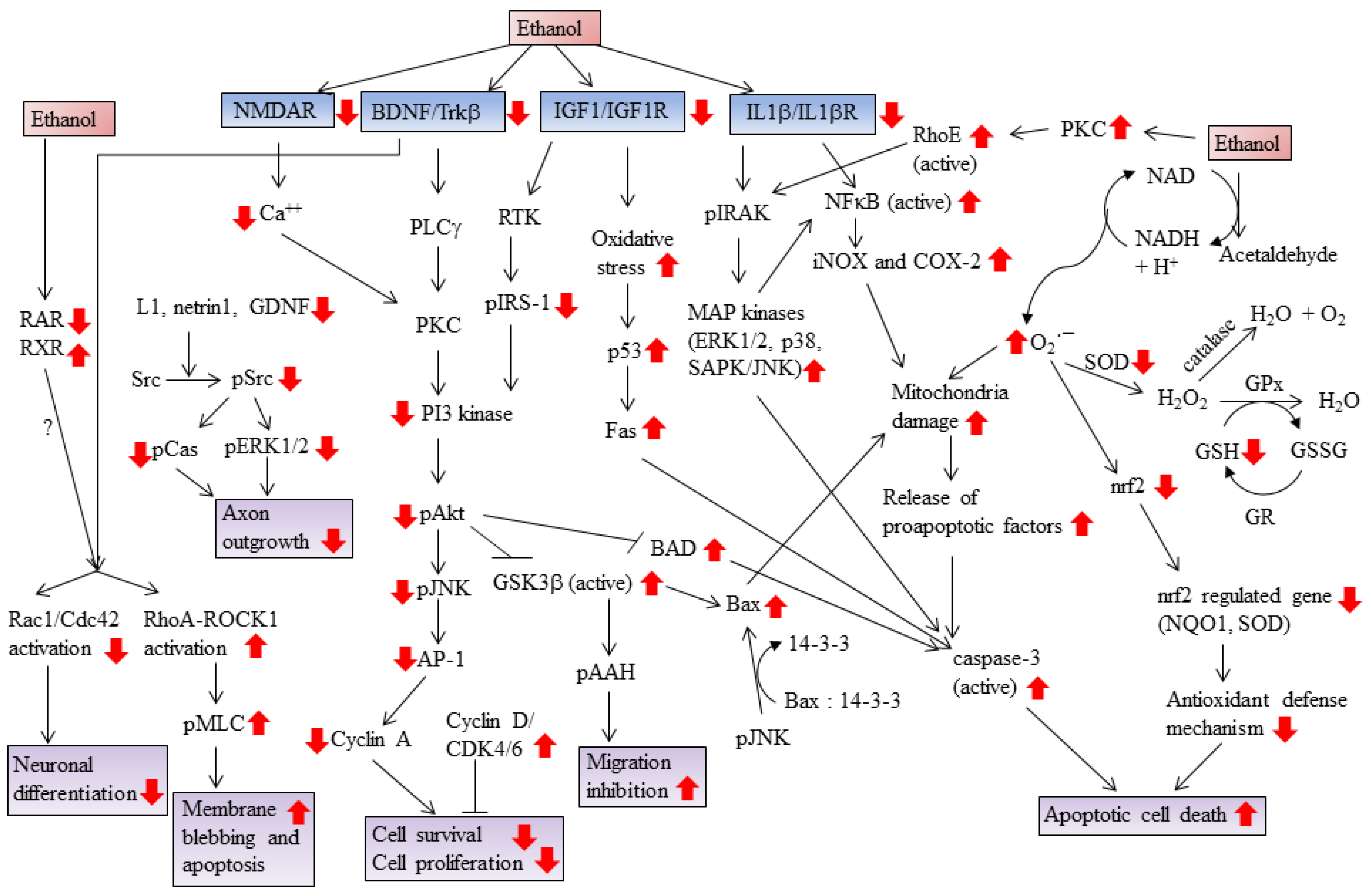
2. Ethanol Increases Oxidative Stress and Induces Apoptotic Cell Death

3. Ethanol Promotes Retinoic Acid Teratogenicity
4. Ethanol Impairs Brain-Derived Nerve Growth Factor (BDNF) Signaling
5. Ethanol Impairs Cytokine Signaling
6. Ethanol Impairs IGF-I Signaling
7. Ethanol Impairs N-Methyl-d-Aspartate (NMDA) Signaling
8. Ethanol Impairs RhoA GTPase Signaling
9. Ethanol Increases Prostaglandin Production
10. Ethanol Targets Glycogen Synthase Kinase 3
11. Ethanol Alters Cell Cycle Progression
12. Conclusion

Conflict of Interest
References
- Jiang, Y.; Kumada, T.; Cameron, D.B.; Komuro, H. Cerebellar granule cell migration and the effects of alcohol. Dev. Neurosci. 2008, 30, 7–23. [Google Scholar] [CrossRef]
- Guerri, C.; Bazinet, A.; Riley, E.P. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009, 44, 108–114. [Google Scholar] [CrossRef]
- Hamre, K.M.; West, J.R. The effects of the timing of ethanol exposure during the brain growth spurt on the number of cerebellar Purkinje and granule cell nuclear profiles. Alcohol. Clin. Exp. Res. 1993, 17, 610–622. [Google Scholar] [CrossRef]
- Pierce, D.R.; Goodlett, C.R.; West, J.R. Differential neuronal loss following early postnatal alcohol exposure. Teratology 1989, 40, 113–126. [Google Scholar] [CrossRef]
- Olney, J.W.; Ishimaru, M.J.; Bittigau, P.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing brain. Apoptosis 2000, 5, 515–521. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Bonthius, D.J.; West, J.R. Alcohol-induced neuronal loss in developing rats: Increased brain damage with binge exposure. Alcohol. Clin. Exp. Res. 1990, 14, 107–118. [Google Scholar] [CrossRef]
- Bauer-Moffett, C.; Altman, J. Ethanol-induced reductions in cerebellar growth of infant rats. Exp. Neurol. 1975, 48, 378–382. [Google Scholar] [CrossRef]
- LeBel, C.P.; Odunze, I.N.; Adams, J.D., Jr.; Bondy, S.C. Perturbations in cerebral oxygen radical formation and membrane order following vitamin E deficiency. Biochem. Biophys. Res. Commun. 1989, 163, 860–866. [Google Scholar] [CrossRef]
- Abel, E.L.; Hannigan, J.H. Maternal risk factors in fetal alcohol syndrome: Provocative and permissive influences. Neurotoxicol. Teratol. 1995, 17, 445–462. [Google Scholar] [CrossRef]
- Yacubova, E.; Komuro, H. Cellular and molecular mechanisms of cerebellar granule cell migration. Cell Biochem. Biophys. 2003, 37, 213–234. [Google Scholar] [CrossRef]
- Altman, J. Postnatal development of the cerebellar cortex in the rat. 3. Maturation of the components of the granular layer. J. Comp. Neurol. 1972, 145, 465–513. [Google Scholar] [CrossRef]
- Chedotal, A. Should I stay or should I go? Becoming a granule cell. Trends Neurosci. 2010, 33, 163–172. [Google Scholar] [CrossRef]
- Andersson, I.K.; Edwall, D.; Norstedt, G.; Rozell, B.; Skottner, A.; Hansson, H.A. Differing expression of insulin-like growth factor I in the developing and in the adult rat cerebellum. Acta Physiol. Scand. 1988, 132, 167–173. [Google Scholar] [CrossRef]
- Borghesani, P.R.; Peyrin, J.M.; Klein, R.; Rubin, J.; Carter, A.R.; Schwartz, P.M.; Luster, A.; Corfas, G.; Segal, R.A. BDNF stimulates migration of cerebellar granule cells. Development 2002, 129, 1435–1442. [Google Scholar]
- Hur, E.M.; Zhou, F.Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef]
- Lindholm, D.; Hamner, S.; Zirrgiebel, U. Neurotrophins and cerebellar development. Perspect. Dev. Neurobiol. 1997, 5, 83–94. [Google Scholar]
- Luo, J. Mechanisms of ethanol-induced death of cerebellar granule cells. Cerebellum 2012, 11, 145–154. [Google Scholar] [CrossRef]
- Light, K.E.; Belcher, S.M.; Pierce, D.R. Time course and manner of Purkinje neuron death following a single ethanol exposure on postnatal day 4 in the developing rat. Neuroscience 2002, 114, 327–337. [Google Scholar] [CrossRef]
- Idrus, N.M.; Napper, R.M. Acute and long-term Purkinje cell loss following a single ethanol binge during the early third trimester equivalent in the rat. Alcohol. Clin. Exp. Res. 2012, 36, 1365–1373. [Google Scholar] [CrossRef]
- Brocardo, P.S.; Gil-Mohapel, J.; Christie, B.R. The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res. Rev. 2011, 67, 209–225. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, C.K.; Lavoie, H.A.; Dipette, D.J.; Singh, U.S. Resveratrol restores Nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol. Pharmacol. 2011, 80, 446–457. [Google Scholar] [CrossRef]
- Henderson, G.I.; Chen, J.J.; Schenker, S. Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front. Biosci. 1999, 4, D541–D550. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Mayer, J.; Miller, R. Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci. Lett. 2002, 334, 83–86. [Google Scholar] [CrossRef]
- Henderson, G.I.; Devi, B.G.; Perez, A.; Schenker, S. In utero ethanol exposure elicits oxidative stress in the rat fetus. Alcohol. Clin. Exp. Res. 1995, 19, 714–720. [Google Scholar] [CrossRef]
- Ramachandran, V.; Watts, L.T.; Maffi, S.K.; Chen, J.; Schenker, S.; Henderson, G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003, 74, 577–588. [Google Scholar] [CrossRef]
- Lee, H.Y.; Li, S.P.; Park, M.S.; Bahk, Y.H.; Chung, B.C.; Kim, M.O. Ethanol’s effect on intracellular signal pathways in prenatal rat cortical neurons is GABAB1 dependent. Synapse 2007, 61, 622–628. [Google Scholar] [CrossRef]
- Smith, A.M.; Zeve, D.R.; Grisel, J.J.; Chen, W.J. Neonatal alcohol exposure increases malondialdehyde (MDA) and glutathione (GSH) levels in the developing cerebellum. Brain Res. Dev. Brain Res. 2005, 160, 231–238. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef]
- Antonio, A.M.; Druse, M.J. Antioxidants prevent ethanol-associated apoptosis in fetal rhombencephalic neurons. Brain Res. 2008, 1204, 16–23. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Kubovic, S.; Kotler, A.; Rogozinski, J.; Swanson, E.; Madorsky, V.; Posados, M. Differential effects of ethanol on c-jun N-terminal kinase, 14-3-3 proteins, and Bax in postnatal day 4 and postnatal day 7 rat cerebellum. Brain Res. 2012, 1432, 15–27. [Google Scholar] [CrossRef]
- Heaton, M.B.; Siler-Marsiglio, K.; Paiva, M.; Kotler, A.; Rogozinski, J.; Kubovec, S.; Coursen, M.; Madorsky, V. Ethanol influences on Bax associations with mitochondrial membrane proteins in neonatal rat cerebellum. Dev. Neurobiol. 2013, 73, 127–141. [Google Scholar] [CrossRef]
- Bremner, J.D.; McCaffery, P. The neurobiology of retinoic acid in affective disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 315–331. [Google Scholar] [CrossRef]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef]
- Duester, G. Involvement of alcohol dehydrogenase, short-chain dehydrogenase/reductase, aldehyde dehydrogenase, and cytochrome P450 in the control of retinoid signaling by activation of retinoic acid synthesis. Biochemistry 1996, 35, 12221–12227. [Google Scholar] [CrossRef]
- Boerman, M.H.; Napoli, J.L. Cellular retinol-binding protein-supported retinoic acid synthesis. Relative roles of microsomes and cytosol. J. Biol. Chem. 1996, 271, 5610–5616. [Google Scholar] [CrossRef]
- Grummer, M.A.; Zachman, R.D. Interaction of ethanol with retinol and retinoic acid in RAR beta and GAP-43 expression. Neurotoxicol. Teratol. 2000, 22, 829–836. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Wang, C.; Napoli, J.L. Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: A potential mechanism of ethanol toxicity. FASEB J. 2010, 24, 823–832. [Google Scholar] [CrossRef]
- Leo, M.A.; Lasker, J.M.; Raucy, J.L.; Kim, C.I.; Black, M.; Lieber, C.S. Metabolism of retinol and retinoic acid by human liver cytochrome P450IIC8. Arch. Biochem. Biophys. 1989, 269, 305–312. [Google Scholar] [CrossRef]
- Wolf, G. Tissue-specific increases in endogenous all-trans retinoic acid: Possible contributing factor in ethanol toxicity. Nutr. Rev. 2010, 68, 689–692. [Google Scholar] [CrossRef]
- Deltour, L.; Ang, H.L.; Duester, G. Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J. 1996, 10, 1050–1057. [Google Scholar]
- Connor, M.J.; Sidell, N. Retinoic acid synthesis in normal and Alzheimer diseased brain and human neural cells. Mol. Chem. Neuropathol. 1997, 30, 239–252. [Google Scholar] [CrossRef]
- McCaffery, P.; Koul, O.; Smith, D.; Napoli, J.L.; Chen, N.; Ullman, M.D. Ethanol increases retinoic acid production in cerebellar astrocytes and in cerebellum. Brain Res. Dev. Brain Res. 2004, 153, 233–241. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, C.K.; DiPette, D.D.; Singh, U.S. Ethanol impairs activation of retinoic acid receptors in cerebellar granule cells in a rodent model of fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2010, 34, 928–937. [Google Scholar] [CrossRef]
- Szondy, Z.; Reichert, U.; Fesus, L. Retinoic acids regulate apoptosis of T lymphocytes through an interplay between RAR and RXR receptors. Cell Death Differ. 1998, 5, 4–10. [Google Scholar]
- Papi, A.; Tatenhorst, L.; Terwel, D.; Hermes, M.; Kummer, M.P.; Orlandi, M.; Heneka, M.T. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J. Neurochem. 2009, 109, 1779–1790. [Google Scholar] [CrossRef]
- Zechel, C. Requirement of retinoic acid receptor isotypes alpha, beta, and gamma during the initial steps of neural differentiation of PCC7 cells. Mol. Endocrinol. 2005, 19, 1629–1645. [Google Scholar] [CrossRef]
- Joshi, S.; Guleria, R.; Pan, J.; DiPette, D.; Singh, U.S. Retinoic acid receptors and tissue-transglutaminase mediate short-term effect of retinoic acid on migration and invasion of neuroblastoma SH-SY5Y cells. Oncogene 2006, 25, 240–247. [Google Scholar]
- Joshi, S.; Guleria, R.S.; Pan, J.; Bayless, K.J.; Davis, G.E.; Dipette, D.; Singh, U.S. Ethanol impairs Rho GTPase signaling and differentiation of cerebellar granule neurons in a rodent model of fetal alcohol syndrome. Cell. Mol. Life Sci. 2006, 63, 2859–2870. [Google Scholar] [CrossRef]
- Cowansage, K.K.; LeDoux, J.E.; Monfils, M.H. Brain-derived neurotrophic factor: A dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmacol. 2010, 3, 12–29. [Google Scholar]
- Lipsky, R.H.; Marini, A.M. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann. N. Y. Acad. Sci. 2007, 1122, 130–143. [Google Scholar] [CrossRef]
- Lindholm, D.; Dechant, G.; Heisenberg, C.P.; Thoenen, H. Brain-derived neurotrophic factor is a survival factor for cultured rat cerebellar granule neurons and protects them against glutamate-induced neurotoxicity. Eur. J. Neurosci. 1993, 5, 1455–1464. [Google Scholar] [CrossRef]
- Rocamora, N.; Garcia-Ladona, F.J.; Palacios, J.M.; Mengod, G. Differential expression of brain-derived neurotrophic factor, neurotrophin-3, and low-affinity nerve growth factor receptor during the postnatal development of the rat cerebellar system. Brain Res. Mol. Brain Res. 1993, 17, 1–8. [Google Scholar] [CrossRef]
- Raivio, N.; Tiraboschi, E.; Saarikoski, S.T.; Castren, E.; Kiianmaa, K. Brain-derived neurotrophic factor expression after acute administration of ethanol. Eur. J. Pharmacol. 2012, 687, 9–13. [Google Scholar] [CrossRef]
- Logrip, M.L.; Janak, P.H.; Ron, D. Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J. Neurochem. 2009, 109, 1459–1468. [Google Scholar] [CrossRef]
- McGough, N.N.; He, D.Y.; Logrip, M.L.; Jeanblanc, J.; Phamluong, K.; Luong, K.; Kharazia, V.; Janak, P.H.; Ron, D. RACK1 and brain-derived neurotrophic factor: A homeostatic pathway that regulates alcohol addiction. J. Neurosci. 2004, 24, 10542–10552. [Google Scholar] [CrossRef]
- Kulkarny, V.V.; Wiest, N.E.; Marquez, C.P.; Nixon, S.C.; Valenzuela, C.F.; Perrone-Bizzozero, N.I. Opposite effects of acute ethanol exposure on GAP-43 and BDNF expression in the hippocampus versus the cerebellum of juvenile rats. Alcohol 2011, 45, 461–471. [Google Scholar] [CrossRef]
- Miki, T.; Kuma, H.; Yokoyama, T.; Sumitani, K.; Matsumoto, Y.; Kusaka, T.; Warita, K.; Wang, Z.Y.; Hosomi, N.; Imagawa, T.; et al. Early postnatal ethanol exposure induces fluctuation in the expression of BDNF mRNA in the developing rat hippocampus. Acta Neurobiol. Exp. 2008, 68, 484–493. [Google Scholar]
- Miki, T.; Harris, S.J.; Wilce, P.; Takeuchi, Y.; Bedi, K.S. Neurons in the hilus region of the rat hippocampus are depleted in number by exposure to alcohol during early postnatal life. Hippocampus 2000, 10, 284–295. [Google Scholar] [CrossRef]
- Miki, T.; Harris, S.J.; Wilce, P.A.; Takeuchi, Y.; Bedi, K.S. Effects of alcohol exposure during early life on neuron numbers in the rat hippocampus. I. Hilus neurons and granule cells. Hippocampus 2003, 13, 388–398. [Google Scholar] [CrossRef]
- Miki, T.; Harris, S.J.; Wilce, P.A.; Takeuchi, Y.; Bedi, K.S. Effects of age and alcohol exposure during early life on pyramidal cell numbers in the CA1-CA3 region of the rat hippocampus. Hippocampus 2004, 14, 124–134. [Google Scholar] [CrossRef]
- Heaton, M.B.; Mitchell, J.J.; Paiva, M.; Walker, D.W. Ethanol-induced alterations in the expression of neurotrophic factors in the developing rat central nervous system. Brain Res. Dev. Brain Res. 2000, 121, 97–107. [Google Scholar] [CrossRef]
- Light, K.E.; Ge, Y.; Belcher, S.M. Early postnatal ethanol exposure selectively decreases BDNF and truncated TrkB-T2 receptor mRNA expression in the rat cerebellum. Brain Res. Mol. Brain Res. 2001, 93, 46–55. [Google Scholar] [CrossRef]
- Ge, Y.; Belcher, S.M.; Light, K.E. Alterations of cerebellar mRNA specific for BDNF, p75NTR, and TrkB receptor isoforms occur within hours of ethanol administration to 4-day-old rat pups. Brain Res. Dev. Brain Res. 2004, 151, 99–109. [Google Scholar] [CrossRef]
- Climent, E.; Pascual, M.; Renau-Piqueras, J.; Guerri, C. Ethanol exposure enhances cell death in the developing cerebral cortex: Role of brain-derived neurotrophic factor and its signaling pathways. J. Neurosci. Res. 2002, 68, 213–225. [Google Scholar] [CrossRef]
- Li, Z.; Ding, M.; Thiele, C.J.; Luo, J. Ethanol inhibits brain-derived neurotrophic factor-mediated intracellular signaling and activator protein-1 activation in cerebellar granule neurons. Neuroscience 2004, 126, 149–162. [Google Scholar] [CrossRef]
- Lindsley, T.A.; Shah, S.N.; Ruggiero, E.A. Ethanol alters BDNF-induced Rho GTPase activation in axonal growth cones. Alcohol. Clin. Exp. Res. 2011, 35, 1321–1330. [Google Scholar] [CrossRef]
- Yeaney, N.K.; He, M.; Tang, N.; Malouf, A.T.; O’Riordan, M.A.; Lemmon, V.; Bearer, C.F. Ethanol inhibits L1 cell adhesion molecule tyrosine phosphorylation and dephosphorylation and activation of pp60(src). J. Neurochem. 2009, 110, 779–790. [Google Scholar] [CrossRef]
- Chen, S.; Charness, M.E. Ethanol disrupts axon outgrowth stimulated by netrin-1, GDNF, and L1 by blocking their convergent activation of Src family kinase signaling. J. Neurochem. 2012, 123, 602–612. [Google Scholar] [CrossRef]
- Valles, S.L.; Blanco, A.M.; Pascual, M.; Guerri, C. Chronic ethanol treatment enhances inflammatory mediators and cell death in the brain and in astrocytes. Brain Pathol. 2004, 14, 365–371. [Google Scholar]
- John, G.R.; Lee, S.C.; Song, X.; Rivieccio, M.; Brosnan, C.F. IL-1-regulated responses in astrocytes: Relevance to injury and recovery. Glia 2005, 49, 161–176. [Google Scholar] [CrossRef]
- Tetsuka, T.; Baier, L.D.; Morrison, A.R. Antioxidants inhibit interleukin-1-induced cyclooxygenase and nitric-oxide synthase expression in rat mesangial cells. Evidence for post-transcriptional regulation. J. Biol. Chem. 1996, 271, 11689–11693. [Google Scholar] [CrossRef]
- Militante, J.D.; Feinstein, D.L.; Syapin, P.J. Suppression by ethanol of inducible nitric oxide synthase expression in C6 glioma cells. J. Pharmacol. Exp. Ther. 1997, 281, 558–565. [Google Scholar]
- Syapin, P.J.; Militante, J.D.; Garrett, D.K.; Ren, L. Cytokine-induced iNOS expression in C6 glial cells: Transcriptional inhibition by ethanol. J. Pharmacol. Exp. Ther. 2001, 298, 744–752. [Google Scholar]
- Wang, J.Y.; Wang, J.Y.; Wang, J.Y.; Shum, A.Y.; Hwang, C.P. Ethanol modulates induction of nitric oxide synthase in glial cells by endotoxin. Life Sci. 1998, 63, 1571–1583. [Google Scholar] [CrossRef]
- Lee, H.; Jeong, J.; Son, E.; Mosa, A.; Cho, G.J.; Choi, W.S.; Ha, J.H.; Kim, I.K.; Lee, M.G.; Kim, C.Y.; Suk, K. Ethanol selectively modulates inflammatory activation signaling of brain microglia. J. Neuroimmunol. 2004, 156, 88–95. [Google Scholar] [CrossRef]
- Pruett, S.B.; Zheng, Q.; Fan, R.; Matthews, K.; Schwab, C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol 2004, 33, 147–155. [Google Scholar]
- Goral, J.; Kovacs, E.J. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J. Immunol. 2005, 174, 456–463. [Google Scholar]
- Blanco, A.M.; Valles, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar]
- Kouzoukas, D.E.; Li, G.; Takapoo, M.; Moninger, T.; Bhalla, R.C.; Pantazis, N.J. Intracellular calcium plays a critical role in the alcohol-mediated death of cerebellar granule neurons. J. Neurochem. 2013, 124, 323–335. [Google Scholar] [CrossRef]
- Garic, A.; Flentke, G.R.; Amberger, E.; Hernandez, M.; Smith, S.M. CaMKII activation is a novel effector of alcohol’s neurotoxicity in neural crest stem/progenitor cells. J. Neurochem. 2011, 118, 646–657. [Google Scholar] [CrossRef]
- Baron-Van Evercooren, A.; Olichon-Berthe, C.; Kowalski, A.; Visciano, G.; van Obberghen, E. Expression of IGF-I and insulin receptor genes in the rat central nervous system: A developmental, regional, and cellular analysis. J. Neurosci. Res. 1991, 28, 244–253. [Google Scholar] [CrossRef]
- Bondy, C.A. Transient IGF-I gene expression during the maturation of functionally related central projection neurons. J. Neurosci. 1991, 11, 3442–3455. [Google Scholar]
- De La Monte, S.M.; Wands, J.R. Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell. Mol. Life Sci. 2002, 59, 882–893. [Google Scholar] [CrossRef]
- Zhang, F.X.; Rubin, R.; Rooney, T.A. Ethanol induces apoptosis in cerebellar granule neurons by inhibiting insulin-like growth factor 1 signaling. J. Neurochem. 1998, 71, 196–204. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Neely, T.R.; Cannon, J.; Wands, J.R. Ethanol impairs insulin-stimulated mitochondrial function in cerebellar granule neurons. Cell. Mol. Life Sci. 2001, 58, 1950–1960. [Google Scholar] [CrossRef]
- Soscia, S.J.; Tong, M.; Xu, X.J.; Cohen, A.C.; Chu, J.; Wands, J.R.; de la Monte, S.M. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell. Mol. Life Sci. 2006, 63, 2039–2056. [Google Scholar] [CrossRef]
- Balazs, R.; Gallo, V.; Kingsbury, A. Effect of depolarization on the maturation of cerebellar granule cells in culture. Brain Res. 1988, 468, 269–276. [Google Scholar]
- Balazs, R.; Jorgensen, O.S.; Hack, N. N-methyl-d-aspartate promotes the survival of cerebellar granule cells in culture. Neuroscience 1988, 27, 437–451. [Google Scholar] [CrossRef]
- Gallo, V.; Kingsbury, A.; Balazs, R.; Jorgensen, O.S. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J. Neurosci. 1987, 7, 2203–2213. [Google Scholar]
- Xifro, X.; Malagelada, C.; Minano, A.; Rodriguez-Alvarez, J. Brief exposure to NMDA produces long-term protection of cerebellar granule cells from apoptosis. Eur. J. Neurosci. 2005, 21, 827–840. [Google Scholar] [CrossRef]
- Bhave, S.V.; Ghoda, L.; Hoffman, P.L. Brain-derived neurotrophic factor mediates the anti-apoptotic effect of NMDA in cerebellar granule neurons: Signal transduction cascades and site of ethanol action. J. Neurosci. 1999, 19, 3277–3286. [Google Scholar]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef]
- D'Angelo, E.; Rossi, P.; Garthwaite, J. Dual-component NMDA receptor currents at a single central synapse. Nature 1990, 346, 467–470. [Google Scholar]
- Hoffman, P.L.; Rabe, C.S.; Grant, K.A.; Valverius, P.; Hudspith, M.; Tabakoff, B. Ethanol and the NMDA receptor. Alcohol 1990, 7, 229–231. [Google Scholar] [CrossRef]
- Bhave, S.V.; Snell, L.D.; Tabakoff, B.; Hoffman, P.L. Mechanism of ethanol inhibition of NMDA receptor function in primary cultures of cerebral cortical cells. Alcohol. Clin. Exp. Res. 1996, 20, 934–941. [Google Scholar] [CrossRef]
- Grover, C.A.; Frye, G.D.; Griffith, W.H. Acute tolerance to ethanol inhibition of NMDA-mediated EPSPs in the CA1 region of the rat hippocampus. Brain Res. 1994, 642, 70–76. [Google Scholar] [CrossRef]
- Lima-Landman, M.T.; Albuquerque, E.X. Ethanol potentiates and blocks NMDA-activated single-channel currents in rat hippocampal pyramidal cells. FEBS Lett. 1989, 247, 61–67. [Google Scholar] [CrossRef]
- Lovinger, D.M.; White, G.; Weight, F.F. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 1989, 243, 1721–1724. [Google Scholar]
- Yang, X.; Criswell, H.E.; Simson, P.; Moy, S.; Breese, G.R. Evidence for a selective effect of ethanol on N-methyl-d-aspartate responses: Ethanol affects a subtype of the ifenprodil-sensitive N-methyl-d-aspartate receptors. J. Pharmacol. Exp. Ther. 1996, 278, 114–124. [Google Scholar]
- Randoll, L.A.; Wilson, W.R.; Weaver, M.S.; Spuhler-Phillips, K.; Leslie, S.W. N-methyl-d-aspartate-stimulated increases in intracellular calcium exhibit brain regional differences in sensitivity to inhibition by ethanol. Alcohol. Clin. Exp. Res. 1996, 20, 197–200. [Google Scholar] [CrossRef]
- Hoffman, P.L.; Rabe, C.S.; Moses, F.; Tabakoff, B. N-methyl-d-aspartate receptors and ethanol: Inhibition of calcium flux and cyclic GMP production. J. Neurochem. 1989, 52, 1937–1940. [Google Scholar] [CrossRef]
- Hardy, P.A.; Chen, W.; Wilce, P.A. Chronic ethanol exposure and withdrawal influence NMDA receptor subunit and splice variant mRNA expression in the rat cerebral cortex. Brain Res. 1999, 819, 33–39. [Google Scholar] [CrossRef]
- Follesa, P.; Ticku, M.K. Chronic ethanol-mediated up-regulation of the N-methyl-d-aspartate receptor polypeptide subunits in mouse cortical neurons in culture. J. Biol. Chem. 1996, 271, 13297–13299. [Google Scholar] [CrossRef]
- Trevisan, L.; Fitzgerald, L.W.; Brose, N.; Gasic, G.P.; Heinemann, S.F.; Duman, R.S.; Nestler, E.J. Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J. Neurochem. 1994, 62, 1635–1638. [Google Scholar]
- Follesa, P.; Ticku, M.K. Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Brain Res. Mol. Brain Res. 1995, 29, 99–106. [Google Scholar] [CrossRef]
- Chandler, L.J.; Norwood, D.; Sutton, G. Chronic ethanol upregulates NMDA and AMPA, but not kainate receptor subunit proteins in rat primary cortical cultures. Alcohol. Clin. Exp. Res. 1999, 23, 363–370. [Google Scholar] [CrossRef]
- Bao, X.; Hui, D.; Naassila, M.; Michaelis, E.K. Chronic ethanol exposure increases gene transcription of subunits of an N-methyl-d-aspartate receptor-like complex in cortical neurons in culture. Neurosci. Lett. 2001, 315, 5–8. [Google Scholar] [CrossRef]
- Bhave, S.V.; Hoffman, P.L. Ethanol promotes apoptosis in cerebellar granule cells by inhibiting the trophic effect of NMDA. J. Neurochem. 1997, 68, 578–586. [Google Scholar] [CrossRef]
- Hughes, P.D.; Kim, Y.N.; Randall, P.K.; Leslie, S.W. Effect of prenatal ethanol exposure on the developmental profile of the NMDA receptor subunits in rat forebrain and hippocampus. Alcohol. Clin. Exp. Res. 1998, 22, 1255–1261. [Google Scholar] [CrossRef]
- Nixon, K.; Hughes, P.D.; Amsel, A.; Leslie, S.W. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res. Dev. Brain Res. 2002, 139, 295–299. [Google Scholar] [CrossRef]
- Masood, K.; Wu, C.; Brauneis, U.; Weight, F.F. Differential ethanol sensitivity of recombinant N-methyl-d-aspartate receptor subunits. Mol. Pharmacol. 1994, 45, 324–329. [Google Scholar]
- Kuner, T.; Schoepfer, R.; Korpi, E.R. Ethanol inhibits glutamate-induced currents in heteromeric NMDA receptor subtypes. Neuroreport 1993, 5, 297–300. [Google Scholar] [CrossRef]
- Chu, B.; Anantharam, V.; Treistman, S.N. Ethanol inhibition of recombinant heteromeric NMDA channels in the presence and absence of modulators. J. Neurochem. 1995, 65, 140–148. [Google Scholar]
- Bhave, S.V.; Snell, L.D.; Tabakoff, B.; Hoffman, P.L. Ethanol sensitivity of NMDA receptor function in developing cerebellar granule neurons. Eur. J. Pharmacol. 1999, 369, 247–259. [Google Scholar] [CrossRef]
- Alvestad, R.M.; Grosshans, D.R.; Coultrap, S.J.; Nakazawa, T.; Yamamoto, T.; Browning, M.D. Tyrosine dephosphorylation and ethanol inhibition of N-Methyl-d-aspartate receptor function. J. Biol. Chem. 2003, 278, 11020–11025. [Google Scholar]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Govek, E.E.; Newey, S.E.; van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef]
- Guasch, R.M.; Tomas, M.; Minambres, R.; Valles, S.; Renau-Piqueras, J.; Guerri, C. RhoA and lysophosphatidic acid are involved in the actin cytoskeleton reorganization of astrocytes exposed to ethanol. J. Neurosci. Res. 2003, 72, 487–502. [Google Scholar] [CrossRef]
- Schaffert, C.S.; Todero, S.L.; Casey, C.A.; Thiele, G.M.; Sorrell, M.F.; Tuma, D.J. Chronic ethanol treatment impairs Rac and Cdc42 activation in rat hepatocytes. Alcohol. Clin. Exp. Res. 2006, 30, 1208–1213. [Google Scholar] [CrossRef]
- Higa, R.; Gonzalez, E.; Pustovrh, M.C.; White, V.; Capobianco, E.; Martinez, N.; Jawerbaum, A. PPARdelta and its activator PGI2 are reduced in diabetic embryopathy: Involvement of PPARdelta activation in lipid metabolic and signalling pathways in rat embryo early organogenesis. Mol. Hum. Reprod. 2007, 13, 103–110. [Google Scholar]
- Tomas, M.; Marin, P.; Megias, L.; Egea, G.; Renau-Piqueras, J. Ethanol perturbs the secretory pathway in astrocytes. Neurobiol. Dis. 2005, 20, 773–784. [Google Scholar] [CrossRef]
- Marin, M.P.; Esteban-Pretel, G.; Ponsoda, X.; Romero, A.M.; Ballestin, R.; Lopez, C.; Megias, L.; Timoneda, J.; Molowny, A.; Canales, J.J.; Renau-Piqueras, J. Endocytosis in cultured neurons is altered by chronic alcohol exposure. Toxicol. Sci. 2010, 115, 202–213. [Google Scholar] [CrossRef]
- Romero, A.M.; Esteban-Pretel, G.; Marin, M.P.; Ponsoda, X.; Ballestin, R.; Canales, J.J.; Renau-Piqueras, J. Chronic ethanol exposure alters the levels, assembly, and cellular organization of the actin cytoskeleton and microtubules in hippocampal neurons in primary culture. Toxicol. Sci. 2010, 118, 602–612. [Google Scholar] [CrossRef]
- Yanni, P.A.; Lindsley, T.A. Ethanol inhibits development of dendrites and synapses in rat hippocampal pyramidal neuron cultures. Brain Res. Dev. Brain Res. 2000, 120, 233–243. [Google Scholar] [CrossRef]
- Lindsley, T.A.; Kerlin, A.M.; Rising, L.J. Time-lapse analysis of ethanol’s effects on axon growth in vitro. Brain Res. Dev. Brain Res. 2003, 147, 191–199. [Google Scholar] [CrossRef]
- Chang, L.; Goldman, R.D. Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 2004, 5, 601–613. [Google Scholar] [CrossRef]
- Valles, S.; Pitarch, J.; Renau-Piqueras, J.; Guerri, C. Ethanol exposure affects glial fibrillary acidic protein gene expression and transcription during rat brain development. J. Neurochem. 1997, 69, 2484–2493. [Google Scholar]
- Minambres, R.; Guasch, R.M.; Perez-Arago, A.; Guerri, C. The RhoA/ROCK-I/MLC pathway is involved in the ethanol-induced apoptosis by anoikis in astrocytes. J. Cell Sci. 2006, 119, 271–282. [Google Scholar] [CrossRef]
- Guasch, R.M.; Blanco, A.M.; Perez-Arago, A.; Minambres, R.; Talens-Visconti, R.; Peris, B.; Guerri, C. RhoE participates in the stimulation of the inflammatory response induced by ethanol in astrocytes. Exp. Cell Res. 2007, 313, 3779–3788. [Google Scholar] [CrossRef]
- Sinervo, K.R.; Smith, G.N.; Bocking, A.D.; Patrick, J.; Brien, J.F. Effect of ethanol on the release of prostaglandins from ovine fetal brain stem during gestation. Alcohol. Clin. Exp. Res. 1992, 16, 443–448. [Google Scholar] [CrossRef]
- Anton, R.F.; Randall, C.L.; Becker, H.C. PGE measurement in mouse embryos and uterine/embryo tissue. Prostaglandins 1988, 36, 835–846. [Google Scholar]
- Luo, J.; Lindstrom, C.L.; Donahue, A.; Miller, M.W. Differential effects of ethanol on the expression of cyclo-oxygenase in cultured cortical astrocytes and neurons. J. Neurochem. 2001, 76, 1354–1363. [Google Scholar] [CrossRef]
- Knapp, D.J.; Crews, F.T. Induction of cyclooxygenase-2 in brain during acute and chronic ethanol treatment and ethanol withdrawal. Alcohol. Clin. Exp. Res. 1999, 23, 633–643. [Google Scholar] [CrossRef]
- Kumada, T.; Lakshmana, M.K.; Komuro, H. Reversal of neuronal migration in a mouse model of fetal alcohol syndrome by controlling second-messenger signalings. J. Neurosci. 2006, 26, 742–756. [Google Scholar] [CrossRef]
- Jing, H.; Li, Y. Effects of ethanol on mouse embryonic brain development and heat shock protein 73 expression. Toxicol. Vitro 2004, 18, 601–607. [Google Scholar] [CrossRef]
- Shimizu, T.; Kagawa, T.; Inoue, T.; Nonaka, A.; Takada, S.; Aburatani, H.; Taga, T. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol. Cell. Biol. 2008, 28, 7427–7441. [Google Scholar] [CrossRef]
- Cui, H.; Meng, Y.; Bulleit, R.F. Inhibition of glycogen synthase kinase 3beta activity regulates proliferation of cultured cerebellar granule cells. Brain Res. Dev. Brain Res. 1998, 111, 177–188. [Google Scholar] [CrossRef]
- Jin, L.; Hu, X.; Feng, L. NT3 inhibits FGF2-induced neural progenitor cell proliferation via the PI3K/GSK3 pathway. J. Neurochem. 2005, 93, 1251–1261. [Google Scholar] [CrossRef]
- Xu, J.; Yeon, J.E.; Chang, H.; Tison, G.; Chen, G.J.; Wands, J.; de la Monte, S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: Role of PTEN phosphatase. J. Biol. Chem. 2003, 278, 26929–26937. [Google Scholar]
- Luo, J. GSK3beta in ethanol neurotoxicity. Mol. Neurobiol. 2009, 40, 108–121. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.; Ma, C.; Bower, K.A.; Xu, M.; Fan, Z.; Shi, X.; Ke, Z.J.; Luo, J. Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J. Neurosci. Res. 2009, 87, 2793–2802. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Yao, W.; Lee, W. Lithium protects ethanol-induced neuronal apoptosis. Biochem. Biophys. Res. Commun. 2006, 350, 905–910. [Google Scholar] [CrossRef]
- Chakraborty, G.; Saito, M.; Mao, R.F.; Wang, R.; Vadasz, C.; Saito, M. Lithium blocks ethanol-induced modulation of protein kinases in the developing brain. Biochem. Biophys. Res. Commun. 2008, 367, 597–602. [Google Scholar] [CrossRef]
- Carter, J.J.; Tong, M.; Silbermann, E.; Lahousse, S.A.; Ding, F.F.; Longato, L.; Roper, N.; Wands, J.R.; de la Monte, S.M. Ethanol impaired neuronal migration is associated with reduced aspartyl-asparaginyl-beta-hydroxylase expression. Acta Neuropathol. 2008, 116, 303–315. [Google Scholar] [CrossRef]
- Copani, A.; Uberti, D.; Sortino, M.A.; Bruno, V.; Nicoletti, F.; Memo, M. Activation of cell-cycle-associated proteins in neuronal death: A mandatory or dispensable path? Trends Neurosci. 2001, 24, 25–31. [Google Scholar] [CrossRef]
- Arellano, M.; Moreno, S. Regulation of CDK/cyclin complexes during the cell cycle. Int. J. Biochem. Cell Biol. 1997, 29, 559–573. [Google Scholar] [CrossRef]
- Li, Z.; Miller, M.W.; Luo, J. Effects of prenatal exposure to ethanol on the cyclin-dependent kinase system in the developing rat cerebellum. Brain Res. Dev. Brain Res. 2002, 139, 237–245. [Google Scholar] [CrossRef]
- Santillano, D.R.; Kumar, L.S.; Prock, T.L.; Camarillo, C.; Tingling, J.D.; Miranda, R.C. Ethanol induces cell-cycle activity and reduces stem cell diversity to alter both regenerative capacity and differentiation potential of cerebral cortical neuroepithelial precursors. BMC Neurosci. 2005, 6, 59. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kumar, A.; LaVoie, H.A.; DiPette, D.J.; Singh, U.S. Ethanol Neurotoxicity in the Developing Cerebellum: Underlying Mechanisms and Implications. Brain Sci. 2013, 3, 941-963. https://doi.org/10.3390/brainsci3020941
Kumar A, LaVoie HA, DiPette DJ, Singh US. Ethanol Neurotoxicity in the Developing Cerebellum: Underlying Mechanisms and Implications. Brain Sciences. 2013; 3(2):941-963. https://doi.org/10.3390/brainsci3020941
Chicago/Turabian StyleKumar, Ambrish, Holly A. LaVoie, Donald J. DiPette, and Ugra S. Singh. 2013. "Ethanol Neurotoxicity in the Developing Cerebellum: Underlying Mechanisms and Implications" Brain Sciences 3, no. 2: 941-963. https://doi.org/10.3390/brainsci3020941