Platelet Endothelial Cell Adhesion Molecule-1 and Oligodendrogenesis: Significance in Alcohol Use Disorders

 ,
, 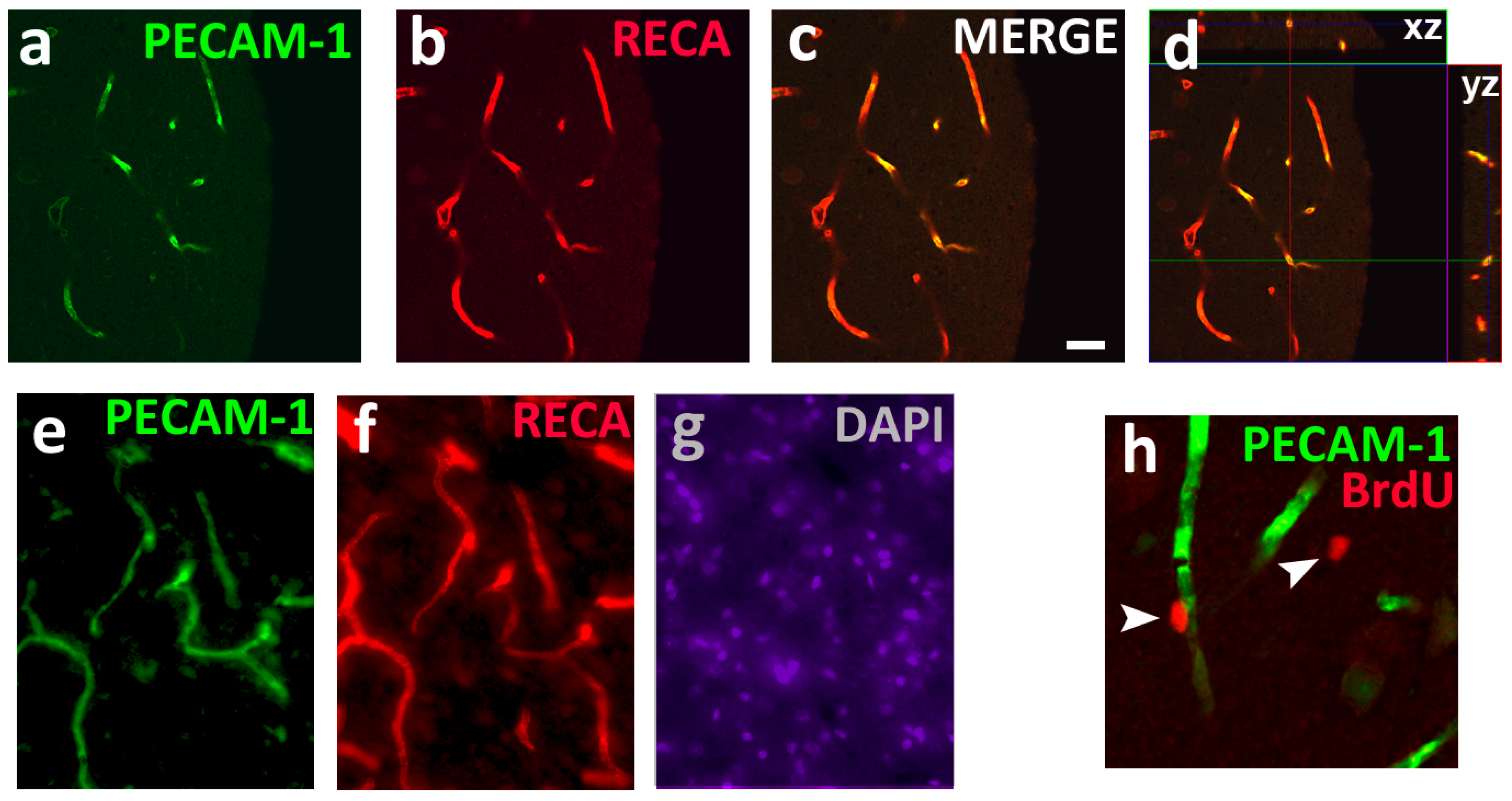{kind=link}
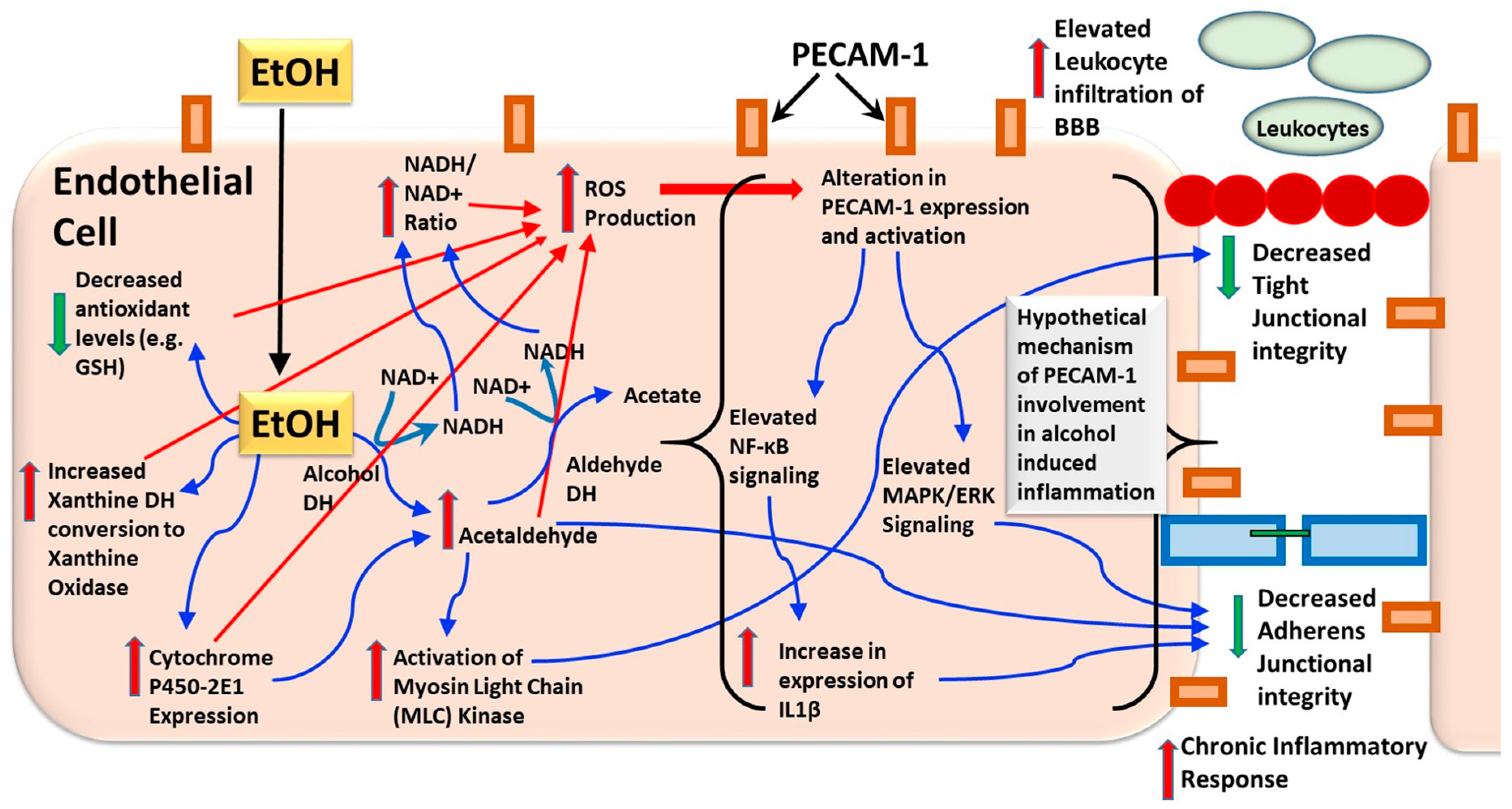{kind=link}
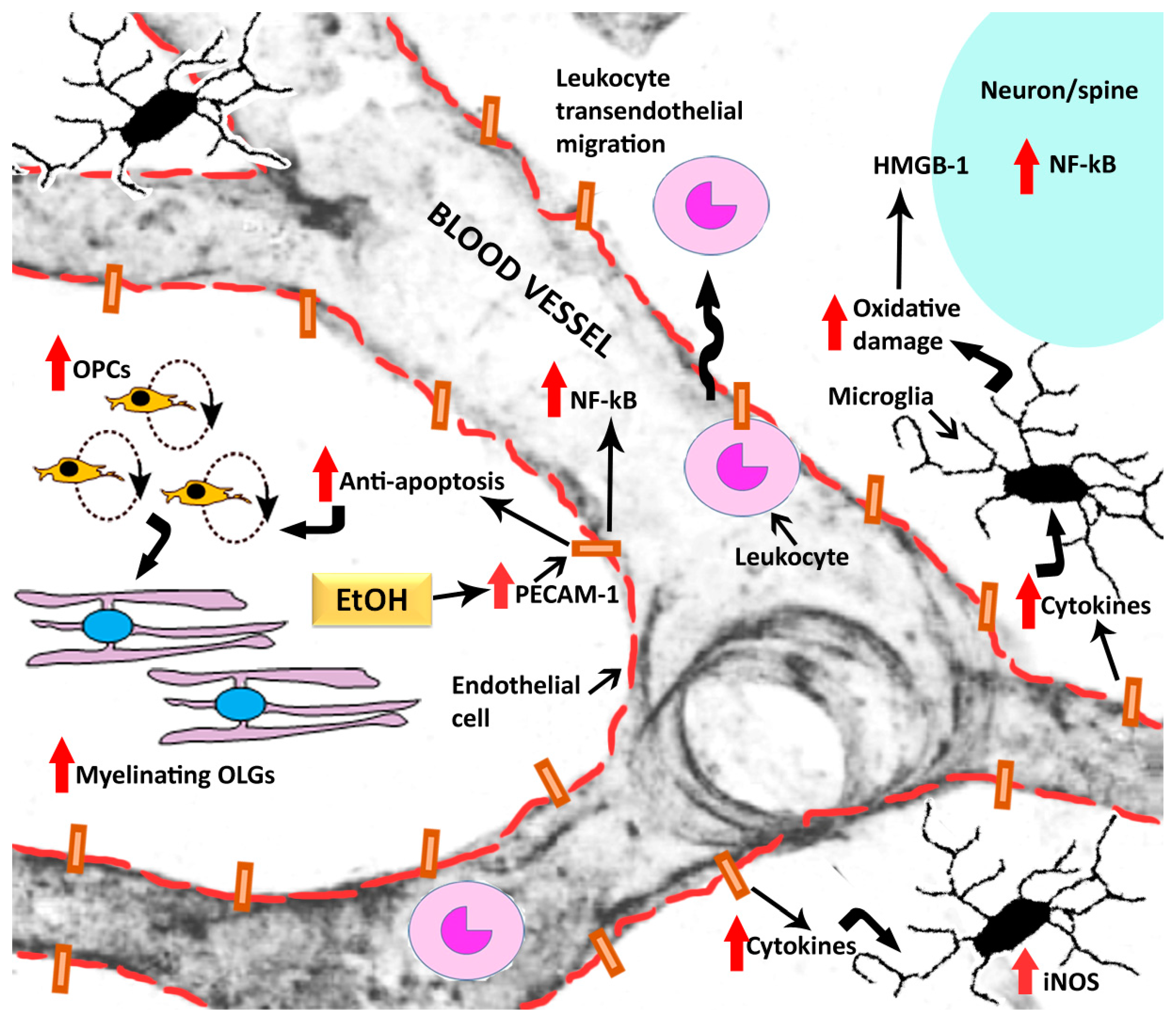{kind=link}
Abstract
:1. Alcohol Use Disorder: Focus on Pathology Associated with the Disorder
2. Platelet Endothelial Cell Adhesion Molecule-1: Source and Function
2.1. Source of PECAM-1
2.2. Function of PECAM-1 on the Endothelial Cells
2.3. Role in Inflammatory Responses
2.4. Role in Apoptosis
2.5. Role in AUD
3. Oligodendrocytes and Oligodendrogenesis
Role of OLGs and OPCs in AUD
4. Interaction of Oligodendrogenesis and PECAM-1 in AUD
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Traphagen, N.; Tian, Z.; Allen-Gipson, D. Chronic Ethanol Exposure: Pathogenesis of Pulmonary Disease and Dysfunction. Biomolecules 2015, 5, 2840–2853. [Google Scholar] [CrossRef] [PubMed]
- Mason, B.J. Emerging pharmacotherapies for alcohol use disorder. Neuropharmacology 2017, 122, 244–253. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, J.M.; Foege, W.H. Actual causes of death in the United States. JAMA 1993, 270, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Marks, J.S.; Stroup, D.F.; Gerberding, J.L. Actual causes of death in the United States, 2000. JAMA 2004, 291, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Munukutla, S.; Pan, G.; Deshpande, M.; Thandavarayan, R.A.; Krishnamurthy, P.; Palaniyandi, S.S. Alcohol Toxicity in Diabetes and Its Complications: A Double Trouble? Alcohol. Clin. Exp. Res. 2016, 40, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Koob, G.F. Addiction is a Reward Deficit and Stress Surfeit Disorder. Front. Psychiatry 2013, 4, 72. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Koob, G.F.; McLellan, A.T. Neurobiologic Advances from the Brain Disease Model of Addiction. N. Engl. J. Med. 2016, 374, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, P.; Cooksy, A.L. Role of diacetyl metabolite in alcohol toxicity and addiction via electron transfer and oxidative stress. Arch. Toxicol. 2005, 79, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reimers, E.; Santolaria-Fernandez, F.; Martin-Gonzalez, M.C.; Fernandez-Rodriguez, C.M.; Quintero-Platt, G. Alcoholism: A systemic proinflammatory condition. World J. Gastroenterol. 2014, 20, 14660–14671. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.K.; Thompson, J.; Chen, L.; Dagda, M.; Dertien, J.; Dossou, K.S.; Moaddel, R.; Bergeson, S.E.; Kruman, I.I. Differential sensitivity of prefrontal cortex and hippocampus to alcohol-induced toxicity. PLoS ONE 2014, 9, e106945. [Google Scholar] [CrossRef] [PubMed]
- Hovatta, I.; Juhila, J.; Donner, J. Oxidative stress in anxiety and comorbid disorders. Neurosci. Res. 2010, 68, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Vetreno, R.P. Neuroimmune basis of alcoholic brain damage. Int. Rev. Neurobiol. 2014, 118, 315–357. [Google Scholar] [PubMed]
- Vetreno, R.P.; Hall, J.M.; Savage, L.M. Alcohol-related amnesia and dementia: Animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiol. Learn. Mem. 2011, 96, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Abdul Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Haorah, J. Inhibitory effects of alcohol on glucose transport across the blood-brain barrier leads to neurodegeneration: Preventive role of acetyl-L: Carnitine. Psychopharmacology 2011, 214, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Keil, V.C.; Greschus, S.; Schneider, C.; Hadizadeh, D.R.; Schild, H.H. The Whole Spectrum of Alcohol-Related Changes in the CNS: Practical MR and CT Imaging Guidelines for Daily Clinical Use. Rofo 2015, 187, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Chastain, L.G.; Sarkar, D.K. Role of microglia in regulation of ethanol neurotoxic action. Int. Rev. Neurobiol. 2014, 118, 81–103. [Google Scholar] [PubMed]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leuk. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Alikunju, S.; Abdul Muneer, P.M.; Zhang, Y.; Szlachetka, A.M.; Haorah, J. The inflammatory footprints of alcohol-induced oxidative damage in neurovascular components. Brain Behav. Immun. 2011, 25 (Suppl. 1), S129–S136. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, S.; Zundorf, G.; Reiser, G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J. Neurosci. Res. 2005, 79, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, R. Alcohol and NMDA receptor: Current research and future direction. Front. Mol. Neurosci. 2013, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Basuroy, S.; Leffler, C.W.; Parfenova, H. CORM-A1 prevents blood-brain barrier dysfunction caused by ionotropic glutamate receptor-mediated endothelial oxidative stress and apoptosis. Am. J. Physiol. Cell Physiol. 2013, 304, C1105–C1115. [Google Scholar] [CrossRef] [PubMed]
- Betzen, C.; White, R.; Zehendner, C.M.; Pietrowski, E.; Bender, B.; Luhmann, H.J.; Kuhlmann, C.R. Oxidative stress upregulates the NMDA receptor on cerebrovascular endothelium. Free Radic. Biol. Med. 2009, 47, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J.; Liu, G.Q. Glutamate up-regulates P-glycoprotein expression in rat brain microvessel endothelial cells by an NMDA receptor-mediated mechanism. Life Sci. 2004, 75, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, S.S.; Fannon, M.J.; Nguyen, T.B.; Mandyam, C.D. Hyper-oligodendrogenesis at the vascular niche and reduced blood-brain barrier integrity in the prefrontal cortex during protracted abstinence. Neuroscience 2017, 362, 265–271. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, T.A.; Sheibani, N. PECAM-1 isoform-specific functions in PECAM-1-deficient brain microvascular endothelial cells. Microvasc. Res. 2008, 75, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Araiz, A.; Porcu, F.; Perez-Hernandez, M.; Garcia-Gutierrez, M.S.; Aracil-Fernandez, M.A.; Gutierrez-Lopez, M.D.; Guerri, C.; Manzanares, J.; O’Shea, E.; Colado, M.I. Disruption of blood-brain barrier integrity in postmortem alcoholic brain: Preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, A.; Losy, J. PECAM-1, a key player in neuroinflammation. Eur. J. Neurol. 2006, 13, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Baptista, S.; Goncalves, J.; Leal, E.; Milhazes, N.; Borges, F.; Ribeiro, C.F.; Quintela, O.; Lendoiro, E.; Lopez-Rivadulla, M.; et al. Methamphetamine transiently increases the blood-brain barrier permeability in the hippocampus: Role of tight junction proteins and matrix metalloproteinase-9. Brain Res. 2011, 1411, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Sambuy, Y. A sideways glance. Alcoholic breakdown of barriers: How ethanol can initiate a landslide towards disease. Genes Nutr. 2009, 4, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. Curr. Rev. 2015, 37, 331–341, 344–351. [Google Scholar]
- Ji, G.; O’Brien, C.D.; Feldman, M.; Manevich, Y.; Lim, P.; Sun, J.; Albelda, S.M.; Kotlikoff, M.I. PECAM-1 (CD31) regulates a hydrogen peroxide-activated nonselective cation channel in endothelial cells. J. Cell Biol. 2002, 157, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Maas, M.; Stapleton, M.; Bergom, C.; Mattson, D.L.; Newman, D.K.; Newman, P.J. Endothelial cell PECAM-1 confers protection against endotoxic shock. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H159–H164. [Google Scholar] [CrossRef] [PubMed]
- Woodfin, A.; Voisin, M.B.; Nourshargh, S. PECAM-1: A multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2514–2523. [Google Scholar] [CrossRef] [PubMed]
- Rattan, V.; Sultana, C.; Shen, Y.; Kalra, V.K. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. Am. J. Physiol. 1997, 273, E453–E461. [Google Scholar] [PubMed]
- Dianzani, U.; Malavasi, F. Lymphocyte adhesion to endothelium. Crit. Rev. Immunol. 1995, 15, 167–200. [Google Scholar] [CrossRef] [PubMed]
- Carlos, T.M.; Harlan, J.M. Leukocyte-endothelial adhesion molecules. Blood 1994, 84, 2068–2101. [Google Scholar] [PubMed]
- Kirschbaum, N.E.; Gumina, R.J.; Newman, P.J. Organization of the gene for human platelet/endothelial cell adhesion molecule-1 shows alternatively spliced isoforms and a functionally complex cytoplasmic domain. Blood 1994, 84, 4028–4037. [Google Scholar] [PubMed]
- Yu, Y.; Fuscoe, J.C.; Zhao, C.; Guo, C.; Jia, M.; Qing, T.; Bannon, D.I.; Lancashire, L.; Bao, W.; Du, T.; et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 2014, 5, 3230. [Google Scholar] [CrossRef] [PubMed]
- Gumina, R.J.; Kirschbaum, N.E.; Piotrowski, K.; Newman, P.J. Characterization of the human platelet/endothelial cell adhesion molecule-1 promoter: Identification of a GATA-2 binding element required for optimal transcriptional activity. Blood 1997, 89, 1260–1269. [Google Scholar] [PubMed]
- Almendro, N.; Bellon, T.; Rius, C.; Lastres, P.; Langa, C.; Corbi, A.; Bernabeu, C. Cloning of the human platelet endothelial cell adhesion molecule-1 promoter and its tissue-specific expression. Structural and functional characterization. J. Immunol. 1996, 157, 5411–5421. [Google Scholar] [PubMed]
- Park, S.; Sorenson, C.M.; Sheibani, N. PECAM-1 isoforms, eNOS and endoglin axis in regulation of angiogenesis. Clin. Sci. 2015, 129, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hemaol. 2016, 23, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, N.; Sorenson, C.M.; Frazier, W.A. Tissue specific expression of alternatively spliced murine PECAM-1 isoforms. Dev. Dyn. 1999, 214, 44–54. [Google Scholar] [CrossRef]
- Wang, Y.; Su, X.; Sorenson, C.M.; Sheibani, N. Tissue-specific distributions of alternatively spliced human PECAM-1 isoforms. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1008–H1017. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.Y.; Huber, M.; Beauchemin, N.; Famiglietti, J.; Albelda, S.M.; Veillette, A. Regulation of mouse PECAM-1 tyrosine phosphorylation by the Src and Csk families of protein-tyrosine kinases. J. Biol. Chem. 1998, 273, 15765–15772. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.M.; Chen, X.H.; Zhang, X. Roles of PECAM-1 in cell function and disease progression. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4082–4088. [Google Scholar] [PubMed]
- Kitazume, S.; Imamaki, R.; Kurimoto, A.; Ogawa, K.; Kato, M.; Yamaguchi, Y.; Tanaka, K.; Ishida, H.; Ando, H.; Kiso, M.; et al. Interaction of platelet endothelial cell adhesion molecule (PECAM) with alpha2,6-sialylated glycan regulates its cell surface residency and anti-apoptotic role. J. Biol. Chem. 2014, 289, 27604–27613. [Google Scholar] [CrossRef] [PubMed]
- Lertkiatmongkol, P.; Paddock, C.; Newman, D.K.; Zhu, J.; Thomas, M.J.; Newman, P.J. The Role of Sialylated Glycans in Human Platelet Endothelial Cell Adhesion Molecule 1 (PECAM-1)-mediated Trans Homophilic Interactions and Endothelial Cell Barrier Function. J. Biol. Chem. 2016, 291, 26216–26225. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Barreuther, M.; Davis, S.; Madri, J.A. Platelet endothelial cell adhesion molecule-1 is phosphorylatable by c-Src, binds Src-Src homology 2 domain, and exhibits immunoreceptor tyrosine-based activation motif-like properties. J. Biol. Chem. 1997, 272, 14442–14446. [Google Scholar] [CrossRef] [PubMed]
- Newman, P.J.; Newman, D.K. Signal transduction pathways mediated by PECAM-1: New roles for an old molecule in platelet and vascular cell biology. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Newman, P.J. PECAM-1: Regulator of endothelial junctional integrity. Cell Tissue Res. 2014, 355, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Labelle, M.; Regina, A.; Berthelet, F.; Beliveau, R. Isolation of endothelial cells from brain, lung, and kidney: Expression of the multidrug resistance P-glycoprotein isoforms. Biochem. Biophys. Res. Commun. 2011, 281, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Paddock, C.; Zhou, D.; Lertkiatmongkol, P.; Newman, P.J.; Zhu, J. Structural basis for PECAM-1 homophilic binding. Blood 2016, 127, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tzima, E. PECAM-1 is necessary for flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.K.; Zheng, Q.; Pan, S.; Jin, Z.G.; Berk, B.C. Flow activates ERK1/2 and endothelial nitric oxide synthase via a pathway involving PECAM1, SHP2, and Tie2. J. Biol. Chem. 2005, 280, 29620–29624. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Masuda, M.; Fujiwara, K. Fluid flow and osmotic stress induce tyrosine phosphorylation of an endothelial cell 128 kDa surface glycoprotein. Biochem. Biophys. Res. Commun. 1995, 214, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Akers, S.M.; O’Leary, H.A.; Minnear, F.L.; Craig, M.D.; Vos, J.A.; Coad, J.E.; Gibson, L.F. VE-cadherin and PECAM-1 enhance ALL migration across brain microvascular endothelial cell monolayers. Exp. Hematol. 2010, 38, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Couty, J.P.; Rampon, C.; Leveque, M.; Laran-Chich, M.P.; Bourdoulous, S.; Greenwood, J.; Couraud, P.O. PECAM-1 engagement counteracts ICAM-1-induced signaling in brain vascular endothelial cells. J. Neurochem. 2007, 103, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Urbani, S.; Bradfield, P.F.; Lee, B.P.; Imhof, B.A. Vascular and epithelial junctions: A barrier for leucocyte migration. Biochem. Soc. Trans. 2008, 36, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Newman, D.K.; Newman, P.J. PECAM-1: Conflicts of interest in inflammation. Life Sci. 2010, 87, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.S.; Andrew, D.P.; Takimoto, H.; Kaufman, S.A.; Yoshida, H.; Spellberg, J.; de la Pompa, J.L.; Elia, A.; Wakeham, A.; Karan-Tamir, B.; et al. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J. Immunol. 1999, 162, 3022–3030. [Google Scholar] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.S.; Goncalves, E.D.; Pedroso, G.S.; Farias, H.R.; Junqueira, S.C.; Marcon, R.; Tuon, T.; Cola, M.; Silveira, P.C.; Santos, A.R.; et al. Physical Exercise Attenuates Experimental Autoimmune Encephalomyelitis by Inhibiting Peripheral Immune Response and Blood-Brain Barrier Disruption. Mol. Neurobiol. 2016, 54, 4723–4737. [Google Scholar] [CrossRef] [PubMed]
- Graesser, D.; Solowiej, A.; Bruckner, M.; Osterweil, E.; Juedes, A.; Davis, S.; Ruddle, N.H.; Engelhardt, B.; Madri, J.A. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Investig. 2002, 109, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Zhao, R.W.; Ueno, K.; Hickey, W.F. PECAM-1 (CD31) expression in the central nervous system and its role in experimental allergic encephalomyelitis in the rat. J. Neurosci. Res. 1996, 45, 747–757. [Google Scholar] [CrossRef]
- Carrithers, M.; Tandon, S.; Canosa, S.; Michaud, M.; Graesser, D.; Madri, J.A. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am. J. Pathol. 2005, 166, 185–196. [Google Scholar] [CrossRef]
- Cheung, K.; Ma, L.; Wang, G.; Coe, D.; Ferro, R.; Falasca, M.; Buckley, C.D.; Mauro, C.; Marelli-Berg, F.M. CD31 signals confer immune privilege to the vascular endothelium. Proc. Natl. Acad. Sci. USA 2015, 112, E5815–E5824. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sheibani, N. Modulation of VE-cadherin and PECAM-1 mediated cell-cell adhesions by mitogen-activated protein kinases. J. Cell Biochem. 2003, 90, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Deddens, L.H.; van Tilborg, G.A.; van der Toorn, A.; de Vries, H.E.; Dijkhuizen, R.M. PECAM-1-targeted micron-sized particles of iron oxide as MRI contrast agent for detection of vascular remodeling after cerebral ischemia. Contrast Media Mol. Imaging 2013, 8, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Shao, G.; Yu, L.; Ren, C. Angiogenesis contributes to the neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Int. J. Neurosci. 2015, 125, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Liu, X.; Li, N.; Jiang, Y.; Chen, G.; Cao, X.; Wang, J. PECAM-1 ligation negatively regulates TLR4 signaling in macrophages. J. Immunol. 2007, 179, 7344–7351. [Google Scholar] [CrossRef] [PubMed]
- Cepinskas, G.; Savickiene, J.; Ionescu, C.V.; Kvietys, P.R. PMN transendothelial migration decreases nuclear NFkappaB in IL-1beta-activated endothelial cells: Role of PECAM-1. J. Cell Biol. 2003, 161, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Localized signals that regulate transendothelial migration. Curr. Opin. Immunol. 2016, 38, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Transendothelial migration: Unifying principles from the endothelial perspective. Immunol. Rev. 2016, 273, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Krombach, F.; Dejana, E. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J. Leuk. Biol. 2006, 80, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.T.; Larbi, K.Y.; Scheiermann, C.; Woodfin, A.; Gerwin, N.; Haskard, D.O.; Nourshargh, S. ICAM-2 mediates neutrophil transmigration in vivo: Evidence for stimulus specificity and a role in PECAM-1-independent transmigration. Blood 2006, 107, 4721–4727. [Google Scholar] [CrossRef] [PubMed]
- Woodfin, A.; Voisin, M.B.; Imhof, B.A.; Dejana, E.; Engelhardt, B.; Nourshargh, S. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood 2009, 113, 6246–6257. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Leskov, I.L.; Yurdagul, A., Jr.; Thiel, B.; Kevil, C.G.; Stokes, K.Y.; Orr, A.W. Recruitment of the adaptor protein Nck to PECAM-1 couples oxidative stress to canonical NF-kappaB signaling and inflammation. Sci. Signal. 2015, 8, ra20. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Tourdot, B.E.; Newman, D.K.; Newman, P.J. The anti-inflammatory actions of platelet endothelial cell adhesion molecule-1 do not involve regulation of endothelial cell NF-kappa B. J. Immunol. 2010, 184, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Botella, L.M.; Puig-Kroger, A.; Almendro, N.; Sanchez-Elsner, T.; Munoz, E.; Corbi, A.; Bernabeu, C. Identification of a functional NF-kappa B site in the platelet endothelial cell adhesion molecule-1 promoter. J. Immunol. 2000, 164, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Feaver, R.E.; Gelfand, B.D.; Blackman, B.R. Human haemodynamic frequency harmonics regulate the inflammatory phenotype of vascular endothelial cells. Nat. Commun. 2013, 4, 1525. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Lawrimore, C.J.; Walter, T.J.; Coleman, L.G., Jr. The role of neuroimmune signaling in alcoholism. Neuropharmacology 2017, 122, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, S.S.; Fannon-Pavlich, M.J.; Ghofranian, A.; Quigley, J.A.; Dutta, R.R.; Galinato, M.H.; Mandyam, C.D. Wheel running reduces ethanol seeking by increasing neuronal activation and reducing oligodendroglial/neuroinflammatory factors in the medial prefrontal cortex. Brain Behav. Immun. 2016, 58, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Challenges in neuronal apoptosis. Curr. Alzheimer Res. 2006, 3, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Velumian, A.A.; Fehlings, M.G. The role of excitotoxicity in secondary mechanisms of spinal cord injury: A review with an emphasis on the implications for white matter degeneration. J. Neurotrauma 2004, 21, 754–774. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, M.R.; Poggi, A. PECAM-1, apoptosis and CD34+ precursors. Leuk. Lymphoma 2004, 45, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Kang, J.Q.; Maiese, K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid. Redox Signal. 2004, 6, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Bergom, C.; Gao, C.; Newman, P.J. Mechanisms of PECAM-1-mediated cytoprotection and implications for cancer cell survival. Leuk. Lymphoma 2005, 46, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Sun, W.; Christofidou-Solomidou, M.; Sawada, M.; Newman, D.K.; Bergom, C.; Albelda, S.M.; Matsuyama, S.; Newman, P.J. PECAM-1 functions as a specific and potent inhibitor of mitochondrial-dependent apoptosis. Blood 2003, 102, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.X.; Harbour, S.N.; Wee, J.L.; Lau, L.M.; Andrews, R.K.; Jackson, D.E. Proteolytic cleavage of platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) is regulated by a calmodulin-binding motif. FEBS Lett. 2004, 568, 70–78. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.D.; Ji, G.; Wang, Y.X.; Sun, J.; Krymskaya, V.P.; Ruberg, F.L.; Kotlikoff, M.I.; Albelda, S.M. PECAM-1 (CD31) engagement activates a phosphoinositide-independent, nonspecific cation channel in endothelial cells. FASEB J. 2001, 15, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Sheth, P.; Seth, A.; Atkinson, K.J.; Gheyi, T.; Kale, G.; Giorgianni, F.; Desiderio, D.M.; Li, C.; Naren, A.; Rao, R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem. J. 2007, 402, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol. Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Zhao, H.; Mayhan, W.G.; Arrick, D.M.; Xiong, W.; Sun, H. Alcohol-induced exacerbation of ischemic brain injury: Role of NAD(P)H oxidase. Alcohol. Clin. Exp. Res. 2010, 34, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Gagliardi, S.; Minervini, G.M.; Ciotti, M.T.; Marra, E.; Calissano, P. Glutamate neurotoxicity in rat cerebellar granule cells: A major role for xanthine oxidase in oxygen radical formation. J. Neurochem. 1997, 68, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Radek, K.A.; Matthies, A.M.; Burns, A.L.; Heinrich, S.A.; Kovacs, E.J.; Dipietro, L.A. Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1084–H1090. [Google Scholar] [CrossRef] [PubMed]
- Young, C.; Klocke, B.J.; Tenkova, T.; Choi, J.; Labruyere, J.; Qin, Y.Q.; Holtzman, D.M.; Roth, K.A.; Olney, J.W. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003, 10, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, S.S.; Fannon, M.J.; Staples, M.C.; Zamora-Martinez, E.R.; Navarro, A.I.; Kim, A.; Quigley, J.A.; Edwards, S.; Mandyam, C.D. Alcohol dependence-induced regulation of the proliferation and survival of adult brain progenitors is associated with altered BDNF-TrkB signaling. Brain Struct. Funct. 2016, 221, 4319–4335. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Jiang, Y.; Gupta, S.; Benlhabib, E. Effects of chronic ethanol drinking on the blood brain barrier and ensuing neuronal toxicity in alcohol-preferring rats subjected to intraperitoneal LPS injection. Alcohol Alcohol. 2007, 42, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Most, D.; Ferguson, L.B.; Mayfield, J.; Harris, R.A.; Blednov, Y.A. Neuroimmune pathways in alcohol consumption: Evidence from behavioral and genetic studies in rodents and humans. Int. Rev. Neurobiol. 2014, 118, 13–39. [Google Scholar] [PubMed]
- Smith, M.L.; Lopez, M.F.; Archer, K.J.; Wolen, A.R.; Becker, H.C.; Miles, M.F. Time-Course Analysis of Brain Regional Expression Network Responses to Chronic Intermittent Ethanol and Withdrawal: Implications for Mechanisms Underlying Excessive Ethanol Consumption. PLoS ONE 2016, 11, e0146257. [Google Scholar] [CrossRef] [PubMed]
- Whitman, B.A.; Knapp, D.J.; Werner, D.F.; Crews, F.T.; Breese, G.R. The cytokine mRNA increase induced by withdrawal from chronic ethanol in the sterile environment of brain is mediated by CRF and HMGB1 release. Alcohol. Clin. Exp. Res. 2013, 37, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, F.; Hen, R. Young and excitable: The function of new neurons in the adult mammalian brain. Curr. Opin. Neurobiol. 2005, 15, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Horner, P.J.; Thallmair, M.; Gage, F.H. Defining the NG2-expressing cell of the adult CNS. J. Neurocytol. 2002, 31, 469–480. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Tripathi, R.B. The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 2008, 107, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhao, N.; Bai, X.; Karram, K.; Trotter, J.; Goebbels, S.; Scheller, A.; Kirchhoff, F. Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia 2014, 62, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, S.S.; Staples, M.C.; Galinato, M.H.; Fannon, M.J.; Mandyam, C.D. Role of NG2 expressing cells in addiction: A new approach for an old problem. Front. Pharmacol. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.R.; Levine, J.M.; Reynolds, R. NG2-expressing cells in the central nervous system: Are they oligodendroglial progenitors? J. Neurosci. Res. 2000, 61, 471–479. [Google Scholar] [CrossRef]
- De Chevigny, A.; Cooper, O.; Vinuela, A.; Reske-Nielsen, C.; Lagace, D.C.; Eisch, A.J.; Isacson, O. Fate mapping and lineage analyses demonstrate the production of a large number of striatal neuroblasts after transforming growth factor alpha and noggin striatal infusions into the dopamine-depleted striatum. Stem Cells 2008, 26, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.A.; Chittajallu, R.; Belachew, S.; Gallo, V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J. Cell Biol. 2004, 165, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Kuspert, M.; Hammer, A.; Bosl, M.R.; Wegner, M. Olig2 regulates Sox10 expression in oligodendrocyte precursors through an evolutionary conserved distal enhancer. Nucleic Acids Res. 2011, 39, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Meijer, D.H.; Kane, M.F.; Mehta, S.; Liu, H.; Harrington, E.; Taylor, C.M.; Stiles, C.D.; Rowitch, D.H. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat. Rev. Neurosci. 2012, 13, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, H.; Yoshida, S.; Sugimori, M.; Kosako, H.; Kominami, R.; Nakafuku, M.; Nabeshima, Y. Dynamic expression of basic helix-loop-helix Olig family members: Implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech. Dev. 2000, 99, 143–148. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.; Mao, M.; et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 2013, 152, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zuo, H.; Maher, B.J.; Serwanski, D.R.; LoTurco, J.J.; Lu, Q.R.; Nishiyama, A. Olig2-dependent developmental fate switch of NG2 cells. Development 2012, 139, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.L.; Kesari, S.; Kitada, M.; Sun, T.; Arnett, H.A.; Alberta, J.A.; Anderson, D.J.; Stiles, C.D.; Rowitch, D.H. Development of NG2 neural progenitor cells requires Olig gene function. Proc. Natl. Acad. Sci. USA 2006, 103, 7853–7858. [Google Scholar] [CrossRef] [PubMed]
- Gritti, A.; Bonfanti, L.; Doetsch, F.; Caille, I.; Alvarez-Buylla, A.; Lim, D.A.; Galli, R.; Verdugo, J.M.; Herrera, D.G.; Vescovi, A.L. Multipotent neural stem cells reside into the rostral extension and olfactory bulb of adult rodents. J. Neurosci. 2002, 22, 437–445. [Google Scholar] [PubMed]
- Dimou, L.; Simon, C.; Kirchhoff, F.; Takebayashi, H.; Gotz, M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 2008, 28, 10434–10442. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.; Verhoeff, J.J.; Immervoll, H.; Brogger, J.C.; Kmiecik, J.; Poli, A.; Netland, I.A.; Prestegarden, L.; Planaguma, J.; Torsvik, A.; et al. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 2011, 122, 495–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehninger, D.; Wang, L.P.; Klempin, F.; Romer, B.; Kettenmann, H.; Kempermann, G. Enriched environment and physical activity reduce microglia and influence the fate of NG2 cells in the amygdala of adult mice. Cell Tissue Res. 2011, 345, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Mandyam, C.D.; Wee, S.; Eisch, A.J.; Richardson, H.N.; Koob, G.F. Methamphetamine self-administration and voluntary exercise have opposing effects on medial prefrontal cortex gliogenesis. J. Neurosci. 2007, 27, 11442–11450. [Google Scholar] [CrossRef] [PubMed]
- El Waly, B.; Macchi, M.; Cayre, M.; Durbec, P. Oligodendrogenesis in the normal and pathological central nervous system. Front. Neurosci. 2014, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, A.; Mangin, J.M.; Aguirre, A.; Gallo, V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat. Neurosci. 2010, 13, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.; Gascon, S.; Masserdotti, G.; Deshpande, A.; Simon, C.; Fischer, J.; Dimou, L.; Chichung Lie, D.; Schroeder, T.; Berninger, B. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat. Cell Biol. 2013, 15, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Karadottir, R.; Hamilton, N.B.; Bakiri, Y.; Attwell, D. Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat. Neurosci. 2008, 11, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, E.R.; Howard, S.G.; Schonmann, V.; Campagnoni, A.T. Identification of the dopamine D3 receptor in oligodendrocyte precursors: Potential role in regulating differentiation and myelin formation. J. Neurosci. 1998, 18, 5344–5353. [Google Scholar] [PubMed]
- Butt, A.M. Neurotransmitter-mediated calcium signalling in oligodendrocyte physiology and pathology. Glia 2006, 54, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, D.J.; Perez-Cerda, F.; Matute, C. KA1-like kainate receptor subunit immunoreactivity in neurons and glia using a novel anti-peptide antibody. Brain Res. Mol. Brain Res. 2000, 81, 164–176. [Google Scholar] [CrossRef]
- Karadottir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Stegmuller, J.; Werner, H.; Nave, K.A.; Trotter, J. The proteoglycan NG2 is complexed with alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by the PDZ glutamate receptor interaction protein (GRIP) in glial progenitor cells. Implications for glial-neuronal signaling. J. Biol. Chem. 2003, 278, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- De Biase, L.M.; Kang, S.H.; Baxi, E.G.; Fukaya, M.; Pucak, M.L.; Mishina, M.; Calabresi, P.A.; Bergles, D.E. NMDA receptor signaling in oligodendrocyte progenitors is not required for oligodendrogenesis and myelination. J. Neurosci. 2011, 31, 12650–12662. [Google Scholar] [CrossRef] [PubMed]
- De Biase, L.M.; Nishiyama, A.; Bergles, D.E. Excitability and synaptic communication within the oligodendrocyte lineage. J. Neurosci. 2010, 30, 3600–3611. [Google Scholar] [CrossRef] [PubMed]
- Gudz, T.I.; Komuro, H.; Macklin, W.B. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J. Neurosci. 2006, 26, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Tomassy, G.S.; Dershowitz, L.B.; Arlotta, P. Diversity Matters: A Revised Guide to Myelination. Trends Cell Biol. 2016, 26, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Watkins, T.A.; Cosgaya, J.M.; Zhang, C.; Chen, L.; Reichardt, L.F.; Shooter, E.M.; Barres, B.A. NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron 2004, 43, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Eisen, A.M.; McBain, C.J.; Gallo, V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 1998, 125, 2901–2914. [Google Scholar] [PubMed]
- Bergles, D.E.; Jabs, R.; Steinhauser, C. Neuron-glia synapses in the brain. Brain Res. Rev. 2010, 63, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Pham, L.D.; Seo, J.H.; Kim, K.W.; Lo, E.H.; Arai, K. Crosstalk between cerebral endothelium and oligodendrocyte. Cell Mol. Life Sci. 2014, 71, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.B.; Noorbhai, I.; Poinsatte, K.; Kong, X.; Anderson, A.; Monson, N.L.; Stowe, A.M. Stroke induces a rapid adaptive autoimmune response to novel neuronal antigens. Discov. Med. 2015, 19, 381–392. [Google Scholar] [PubMed]
- Pham, L.D.; Hayakawa, K.; Seo, J.H.; Nguyen, M.N.; Som, A.T.; Lee, B.J.; Guo, S.; Kim, K.W.; Lo, E.H.; Arai, K. Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia 2012, 60, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E.H. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J. Neurosci. 2009, 29, 4351–4355. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.T.; Sheedy, D.; Kril, J.J. Neuropathology of alcoholism. Handb. Clin. Neurol. 2014, 125, 603–615. [Google Scholar] [PubMed]
- Mandyam, C.D.; Koob, G.F. The addicted brain craves new neurons: Putative role for adult-born progenitors in promoting recovery. Trends Neurosci. 2012, 35, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Richardson, H.N.; Chan, S.H.; Crawford, E.F.; Lee, Y.K.; Funk, C.K.; Koob, G.F.; Mandyam, C.D. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol. Dis. 2009, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Zamora-Martinez, E.R.; Edwards, S.; Mandyam, C.D. Structural reorganization of pyramidal neurons in the medial prefrontal cortex of alcohol dependent rats is associated with altered glial plasticity. Brain Struct. Funct. 2015, 220, 1705–1720. [Google Scholar] [CrossRef] [PubMed]
- Van Eijk, J.; Demirakca, T.; Frischknecht, U.; Hermann, D.; Mann, K.; Ende, G. Rapid partial regeneration of brain volume during the first 14 days of abstinence from alcohol. Alcohol. Clin. Exp. Res. 2013, 37, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Lacadie, C.M.; Tuit, K.; Hong, K.I.; Constable, R.T.; Sinha, R. Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry 2013, 70, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.I.; Mandyam, C.D. Protracted abstinence from chronic ethanol exposure alters the structure of neurons and expression of oligodendrocytes and myelin in the medial prefrontal cortex. Neuroscience 2015, 293, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Pla, A.; Minarro, J.; Guerri, C. Neuroimmune activation and myelin changes in adolescent rats exposed to high-dose alcohol and associated cognitive dysfunction: A review with reference to human adolescent drinking. Alcohol Alcohol. 2014, 49, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Kuhad, A.; Chopra, K. Suppression of neuro-inflammatory signaling cascade by tocotrienol can prevent chronic alcohol-induced cognitive dysfunction in rats. Behav. Brain Res. 2009, 203, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.A.; Lopez-Sanchez, R.C.; Rendon-Ramirez, A. Lipids and Oxidative Stress Associated with Ethanol-Induced Neurological Damage. Oxid. Med. Cell. Longev. 2016, 2016, 1543809. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Kim, D.W.; Yoo, K.Y.; Jung, B.K.; Song, J.H.; Jung, J.Y.; Choi, S.Y.; Kang, T.C.; Lee, J.Y.; Kwon, Y.G.; Won, M.H. Ischemia-induced changes of platelet endothelial cell adhesion molecule-1 in the hippocampal CA1 region in gerbils. Brain Res. 2005, 1048, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, W.I.; Murata, S.; Nelson, G.H.; Werner, P.K.; Ranken, R.; Harmon, R.C. Anti-CD31 delays platelet adhesion/aggregation at sites of endothelial injury in mouse cerebral arterioles. Am. J. Pathol. 1994, 145, 33–36. [Google Scholar] [PubMed]
- Harper, K.M.; Knapp, D.J.; Breese, G.R. Withdrawal from Chronic Alcohol Induces a Unique CCL2 mRNA Increase in Adolescent But Not Adult Brain—Relationship to Blood Alcohol Levels and Seizures. Alcohol. Clin. Exp. Res. 2015, 39, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.D.; Maki, T.; Ayata, C.; Kim, K.W.; Lo, E.H.; Arai, K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Investig. 2013, 123, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Gotz, M.; Dimou, L. Progenitors in the adult cerebral cortex: Cell cycle properties and regulation by physiological stimuli and injury. Glia 2011, 59, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Baktir, M.A.; Srivatsan, M.; Salehi, A. Neuroprotective effects of physical activity on the brain: A closer look at trophic factor signaling. Front. Cell Neurosci. 2014, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Lynch, W.J.; Peterson, A.B.; Sanchez, V.; Abel, J.; Smith, M.A. Exercise as a novel treatment for drug addiction: A neurobiological and stage-dependent hypothesis. Neurosci. Biobehav. Rev. 2013, 37, 1622–1644. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, S.S.; Staples, M.C.; Fannon, M.J.; Ghofranian, A.; Mandyam, C.D. Evaluating Exercise as a Therapeutic Intervention for Methamphetamine Addiction-Like Behavior. Brain Plasticity 2015, 63, 63–81. [Google Scholar] [CrossRef]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Ohab, J.J.; Carmichael, S.T. Poststroke neurogenesis: Emerging principles of migration and localization of immature neurons. Neuroscientist 2008, 14, 369–380. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandyam, C.D.; Villalpando, E.G.; Steiner, N.L.; Quach, L.W.; Fannon, M.J.; Somkuwar, S.S. Platelet Endothelial Cell Adhesion Molecule-1 and Oligodendrogenesis: Significance in Alcohol Use Disorders. Brain Sci. 2017, 7, 131. https://doi.org/10.3390/brainsci7100131
Mandyam CD, Villalpando EG, Steiner NL, Quach LW, Fannon MJ, Somkuwar SS. Platelet Endothelial Cell Adhesion Molecule-1 and Oligodendrogenesis: Significance in Alcohol Use Disorders. Brain Sciences. 2017; 7(10):131. https://doi.org/10.3390/brainsci7100131
Chicago/Turabian StyleMandyam, Chitra D., Emmanuel G. Villalpando, Noah L. Steiner, Leon W. Quach, McKenzie J. Fannon, and Sucharita S. Somkuwar. 2017. "Platelet Endothelial Cell Adhesion Molecule-1 and Oligodendrogenesis: Significance in Alcohol Use Disorders" Brain Sciences 7, no. 10: 131. https://doi.org/10.3390/brainsci7100131
APA StyleMandyam, C. D., Villalpando, E. G., Steiner, N. L., Quach, L. W., Fannon, M. J., & Somkuwar, S. S. (2017). Platelet Endothelial Cell Adhesion Molecule-1 and Oligodendrogenesis: Significance in Alcohol Use Disorders. Brain Sciences, 7(10), 131. https://doi.org/10.3390/brainsci7100131