Adolescent Alcohol Drinking Renders Adult Drinking BLA-Dependent: BLA Hyper-Activity as Contributor to Comorbid Alcohol Use Disorder and Anxiety Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Apparatus
2.3. Adolescent Voluntary Intermittent Access to Alcohol
2.4. Dependent Measures
2.5. Surgical Procedures
2.6. Adult Voluntary Intermittent Access to Alcohol
2.7. Histological Procedures
2.8. Statistics
3. Results
3.1. Histology
3.2. Alcohol and QuAD Drinking in Adolescence
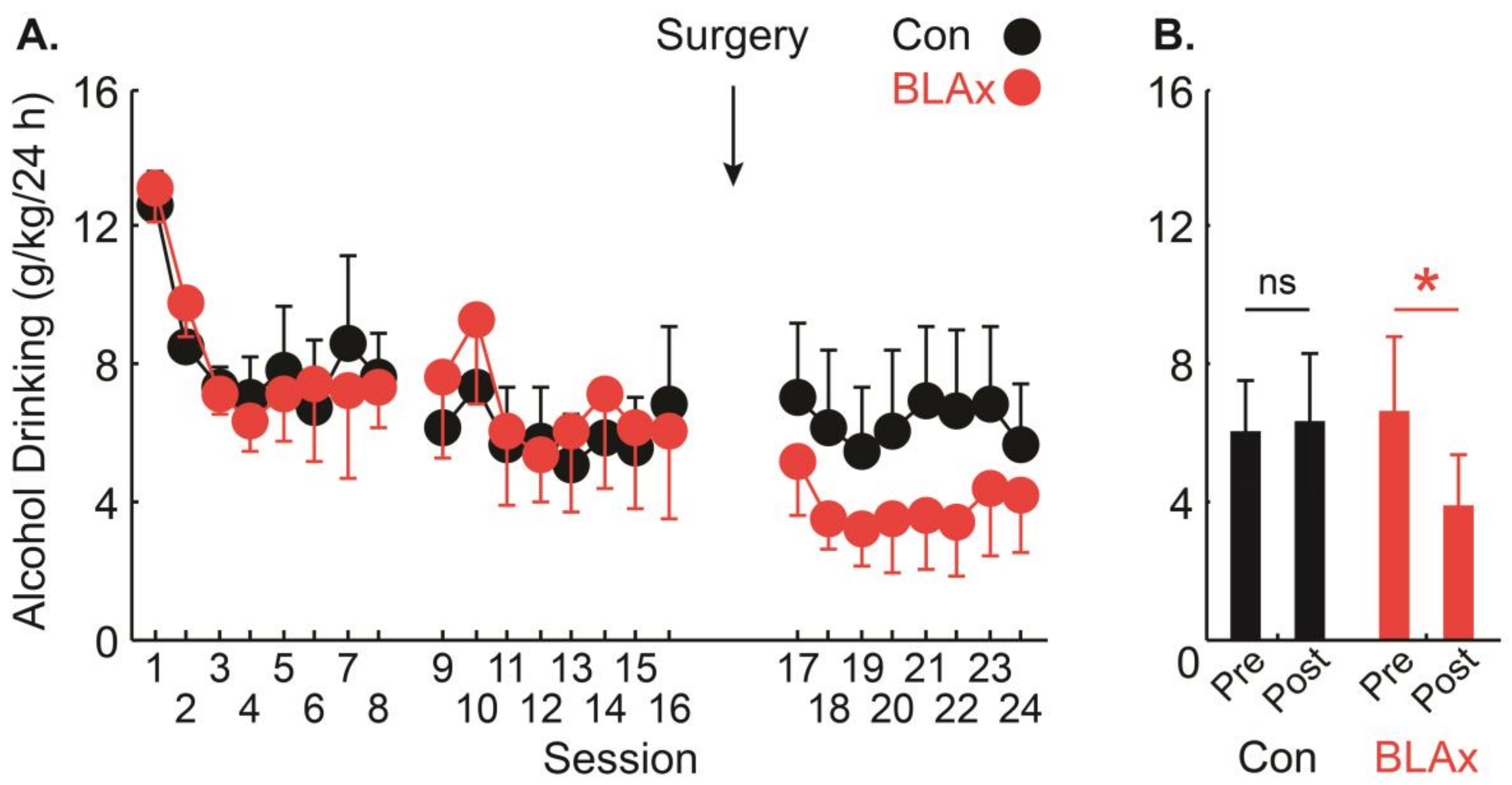
3.3. Adult Alcohol Drinking Established in Adolescence Depends on the BLA
3.4. Adult Alcohol Drinking Established in Adulthood Does not Depend on the BLA
4. Discussion
4.1. BLA Contribution to Fear
4.2. BLA Contribution to Alcohol-Related Behavior
4.3. Impact of Alcohol Drinking on BLA Function
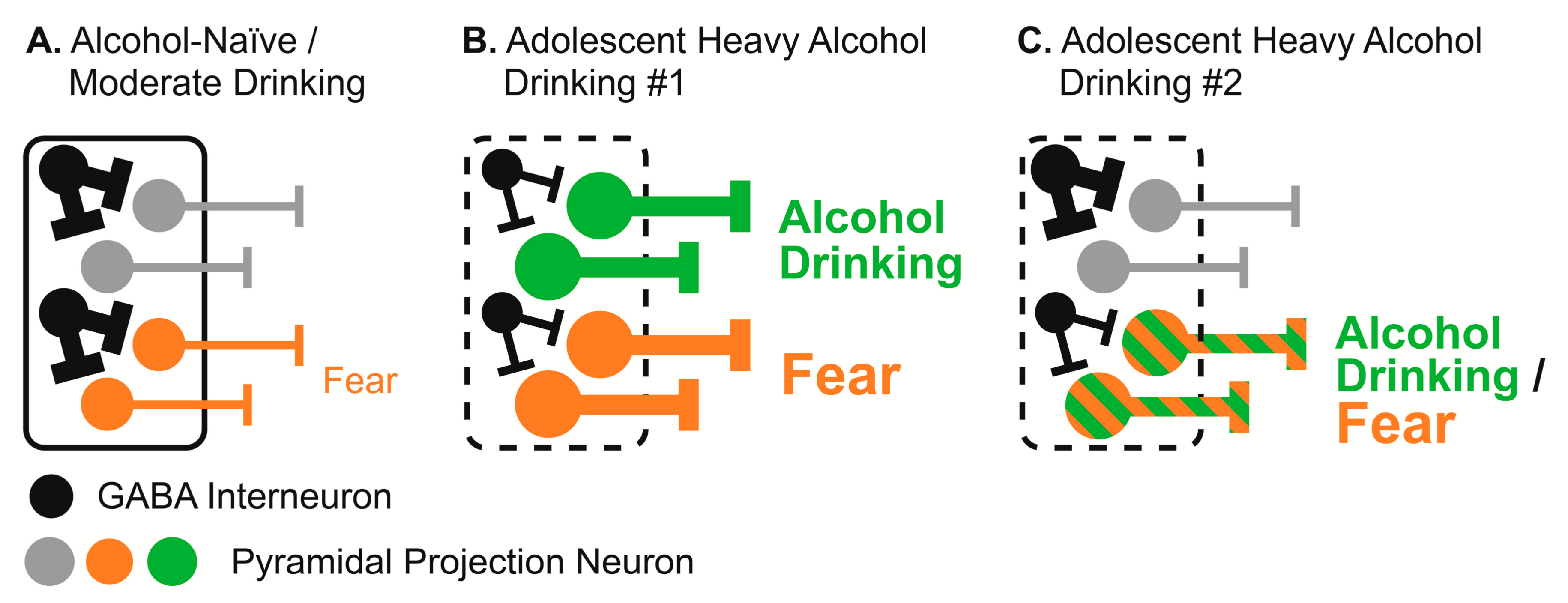
4.4. Proposed Alteration of BLA Function by Adolescent Alcohol Drinking
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Substance Abuse and Mental Health Services Administration. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings; Rockville, M.D., Ed.; Department of Health and Human Services: Washington, DC, USA, 2014; Volume NSDUH Series H-48. [Google Scholar]
- Hingson, R.W.; Heeren, T.; Winter, M.R. Age at drinking onset and alcohol dependence-age at onset, duration, and severity. Arch. Pediatr. Adolesc. Med. 2006, 160, 739–746. [Google Scholar] [CrossRef] [PubMed]
- DeWit, D.J.; Adlaf, E.M.; Offord, D.R.; Ogborne, A.C. Age at first alcohol use: A risk factor for the development of alcohol disorders. Am. J. Psychiatry 2000, 157, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, L.R.; Kneiber, D.; Wills, D.N.; Ehlers, C.L. Alcohol drinking during adolescence increases consumptive responses to alcohol in adulthood in wistar rats. Alcohol 2017, 59, 43–51. [Google Scholar] [CrossRef] [PubMed]
- DiLeo, A.; Wright, K.M.; Mangone, E.; McDannald, M.A. Alcohol gains access to appetitive learning through adolescent heavy drinking. Behav. Neurosci. 2015, 129, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Spear, L.P. The adolescent brain and the college drinker: Biological basis of propensity to use and misuse alcohol. J. Stud. Alcohol Suppl. 2002, 63, 71–81. [Google Scholar] [CrossRef]
- DiLeo, A.; Wright, K.M.; McDannald, M.A. Sub-second fear discrimination in rats: Adult impairment in adolescent heavy alcohol drinkers. Learn. Mem. 2016, 23, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Sananes, C.B.; Davis, M. N-methyl-d-aspartate lesions of the lateral and basolateral nuclei of the amygdala block fear-potentiated startle and shock sensitization of startle. Behav. Neurosci. 1992, 106, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Maren, S.; Aharonov, G.; Stote, D.L.; Fanselow, M.S. N-methyl-d-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav. Neurosci. 1996, 110, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Fanselow, M.S.; LeDoux, J.E. Why we think plasticity underlying pavlovian fear conditioning occurs in the basolateral amygdala. Neuron 1999, 23, 229–232. [Google Scholar] [CrossRef]
- Gale, G.D.; Anagnostaras, S.G.; Godsil, B.P.; Mitchell, S.; Nozawa, T.; Sage, J.R.; Wiltgen, B.; Fanselow, M.S. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J. Neurosci. 2004, 24, 3810–3815. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.W.; Han, J.S.; Kim, J.J. Selective neurotoxic lesions of basolateral and central nuclei of the amygdala produce differential effects on fear conditioning. J. Neurosci. 2004, 24, 7654–7662. [Google Scholar] [CrossRef] [PubMed]
- McDannald, M.A.; Galarce, E.M. Measuring pavlovian fear with conditioned freezing and conditioned suppression reveals different roles for the basolateral amygdala. Brain Res. 2011, 1374, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Doremus, T.L.; Brunell, S.C.; Rajendran, P.; Spear, L.P. Factors influencing elevated ethanol consumption in adolescent relative to adult rats. Alcohol. Clin. Exp. Res. 2005, 29, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Simms, J.A.; Steensland, P.; Medina, B.; Abernathy, K.E.; Chandler, L.J.; Wise, R.; Bartlett, S.E. Intermittent access to 20% ethanol induces high ethanol consumption in long-evans and wistar rats. Alcohol. Clin. Exp. Res. 2008, 32, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Spear, L.P. The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 2000, 24, 417–463. [Google Scholar] [CrossRef]
- Pickens, C.L.; Fisher, H.; Bright, N.; Gallo, M.; Ray, M.H.; Anji, A.; Kumari, M. Prior alcohol consumption does not impair go/no-go discrimination learning, but causes over-responding on go trials, in rats. Behav. Brain Res. 2016, 312, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Fisher, H.; Bright, N.; Gallo, M.; Pajser, A.; Pickens, C.L. Relationship of low doses of alcohol voluntarily consumed during adolescence and early adulthood with subsequent behavioral flexibility. Behav. Pharmacol. 2017, 28, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Moller, C.; Wiklund, L.; Sommer, W.; Thorsell, A.; Heilig, M. Decreased experimental anxiety and voluntary ethanol consumption in rats following central but not basolateral amygdala lesions. Brain Res. 1997, 760, 94–101. [Google Scholar] [CrossRef]
- McClintick, J.N.; McBride, W.J.; Bell, R.L.; Ding, Z.-M.; Liu, Y.; Xuei, X.; Edenberg, H.J. Gene expression changes in the ventral hippocampus and medial prefrontal cortex of adolescent alcohol-preferring (p) rats following binge-like-alcohol drinking. Alcohol 2017. [Google Scholar] [CrossRef]
- Liu, W.; Crews, F.T. Adolescent intermittent ethanol exposure enhances ethanol activation of the nucleus accumbens while blunting the prefrontal cortex responses in adult rat. Neuroscience 2015, 293, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Karanikas, C.A.; Lu, Y.L.; Richardson, H.N. Adolescent drinking targets corticotropin-releasing factor peptide-labeled cells in the central amygdala of male and female rats. Neuroscience 2013, 249, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Boix, J.; Felipo, V.; Guerri, C. Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J. Neurochem. 2009, 108, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, N.A.; Clark, J.J.; Collins, A.L.; Akers, C.A.; Phillips, P.E.; Bernstein, I.L. Risk preference following adolescent alcohol use is associated with corrupted encoding of costs but not rewards by mesolimbic dopamine. Proc. Natl. Acad. Sci. USA 2011, 108, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Alaux-Cantin, S.; Warnault, V.; Legastelois, R.; Botia, B.; Pierrefiche, O.; Vilpoux, C.; Naassila, M. Alcohol intoxications during adolescence increase motivation for alcohol in adult rats and induce neuroadaptations in the nucleus accumbens. Neuropharmacology 2013, 67, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Vargas, W.M.; Bengston, L.; Gilpin, N.W.; Whitcomb, B.W.; Richardson, H.N. Alcohol binge drinking during adolescence or dependence during adulthood reduces prefrontal myelin in male rats. J. Neurosci. 2014, 34, 14777–14782. [Google Scholar] [CrossRef] [PubMed]
- McMurray, M.S.; Amodeo, L.R.; Roitman, J.D. Consequences of adolescent ethanol consumption on risk preference and orbitofrontal cortex encoding of reward. Neuropsychopharmacology 2016, 41, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Kitai, S.T. Amygdaloid projections to the frontal-cortex and the striatum in the rat. J. Comp. Neurol. 1990, 298, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Pare, D.; Smith, Y.; Pare, J.F. Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat-phaseolus-vulgaris-leukoagglutinin anterograde tracing at the light and electron-microscopic level. Neuroscience 1995, 69, 567–583. [Google Scholar] [CrossRef]
- Bell, R.L.; Rodd, Z.A.; Lumeng, L.; Murphy, J.M.; McBride, W.J. The alcohol-preferring p rat and animal models of excessive alcohol drinking. Addict. Biol. 2006, 11, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Marchant, N.J.; Khuc, T.N.; Pickens, C.L.; Bonci, A.; Shaham, Y. Context-induced relapse to alcohol seeking after punishment in a rat model. Biol. Psychiatry 2013, 73, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Rizley, R.C.; Rescorla, R.A. Associations in second-order conditioning and sensory preconditioning. J. Comp. Physiol. Psychol. 1972, 81, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Killcross, S.; Robbins, T.W.; Everitt, B.J. Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature 1997, 388, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Petrovich, G.D.; Ross, C.A.; Mody, P.; Holland, P.C.; Gallagher, M. Central, but not basolateral, amygdala is critical for control of feeding by aversive learned cues. J. Neurosci. 2009, 29, 15205–15212. [Google Scholar] [CrossRef] [PubMed]
- LeDoux, J.E.; Cicchetti, P.; Xagoraris, A.; Romanski, L.M. The lateral amygdaloid nucleus: Sensory interface of the amygdala in fear conditioning. J. Neurosci. 1990, 10, 1062–1069. [Google Scholar] [PubMed]
- Bouton, M.E.; Bolles, R.C. Conditioned fear assessed by freezing and by the suppression of three different baselines. Anim. Learn. Behav. 1980, 8, 429–434. [Google Scholar] [CrossRef]
- Maren, S.; Aharonov, G.; Fanselow, M.S. Retrograde abolition of conditional fear after excitotoxic lesions in the basolateral amygdala of rats: Absence of a temporal gradient. Behav. Neurosci. 1996, 110, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Galarce, E.M.; McDannald, M.A.; Holland, P.C. The basolateral amygdala mediates the effects of cues associated with meal interruption on feeding behavior. Brain Res. 2010, 1350, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Walker, D.; Davis, M. Lack of a temporal gradient of retrograde amnesia following nmda-induced lesions of the basolateral amygdala assessed with the fear-potentiated startle paradigm. Behav. Neurosci. 1996, 110, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Campeau, S.; Davis, M. Involvement of the central nucleus and basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J. Neurosci. 1995, 15, 2301–2311. [Google Scholar] [PubMed]
- Muller, J.; Corodimas, K.P.; Fridel, Z.; LeDoux, J.E. Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav. Neurosci. 1997, 111, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Helmstetter, F.J.; Bellgowan, P.S. Effects of muscimol applied to the basolateral amygdala on acquisition and expression of contextual fear conditioning in rats. Behav. Neurosci. 1994, 108, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Mercado, D.; Padilla-Coreano, N.; Quirk, G.J. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 2011, 36, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.H.; Fibiger, H.C. Conditioned fear-induced changes in behavior and in the expression of the immediate early gene c-fos: With and without diazepam pretreatment. J. Neurosci. 1995, 15, 709–720. [Google Scholar] [PubMed]
- Rosen, J.B.; Fanselow, M.S.; Young, S.L.; Sitcoske, M.; Maren, S. Immediate-early gene expression in the amygdala following footshock stress and contextual fear conditioning. Brain Res. 1998, 796, 132–142. [Google Scholar] [CrossRef]
- Thomas, K.L.; Hall, J.; Everitt, B.J. Cellular imaging with zif268 expression in the rat nucleus accumbens and frontal cortex further dissociates the neural pathways activated following the retrieval of contextual and cued fear memory. Eur. J. Neurosci. 2002, 16, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Campeau, S.; Hayward, M.D.; Hope, B.T.; Rosen, J.B.; Nestler, E.J.; Davis, M. Induction of the c-fos proto-oncogene in rat amygdala during unconditioned and conditioned fear. Brain Res. 1991, 565, 349–352. [Google Scholar] [CrossRef]
- Scicli, A.P.; Petrovich, G.D.; Swanson, L.W.; Thompson, R.F. Contextual fear conditioning is associated with lateralized expression of the immediate early gene c-fos in the central and basolateral amygdalar nuclei. Behav. Neurosci. 2004, 118, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Quirk, G.J.; Repa, C.; LeDoux, J.E. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: Parallel recordings in the freely behaving rat. Neuron 1995, 15, 1029–1039. [Google Scholar] [CrossRef]
- Wolff, S.B.; Grundemann, J.; Tovote, P.; Krabbe, S.; Jacobson, G.A.; Muller, C.; Herry, C.; Ehrlich, I.; Friedrich, R.W.; Letzkus, J.J.; et al. Amygdala interneuron subtypes control fear learning through disinhibition. Nature 2014, 509, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Sangha, S.; Chadick, J.Z.; Janak, P.H. Safety encoding in the basal amygdala. J. Neurosci. 2013, 33, 3744–3751. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.; Fox, R.; Proulx, C.D.; Lin, J.Y.; Tsien, R.Y.; Malinow, R. Engineering a memory with ltd and ltp. Nature 2014, 511, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.P.; LeDoux, J.E.; Nader, K. Fear conditioning and ltp in the lateral amygdala are sensitive to the same stimulus contingencies. Nat. Neurosci. 2001, 4, 687–688. [Google Scholar] [CrossRef] [PubMed]
- Rainnie, D.G.; Asprodini, E.K.; Shinnick-Gallagher, P. Inhibitory transmission in the basolateral amygdala. J. Neurophysiol. 1991, 66, 999–1009. [Google Scholar] [PubMed]
- Davis, P.; Zaki, Y.; Maguire, J.; Reijmers, L.G. Cellular and oscillatory substrates of fear extinction learning. Nat. Neurosci. 2017, 20, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Lucas, E.K.; Jegarl, A.M.; Morishita, H.; Clem, R.L. Multimodal and site-specific plasticity of amygdala parvalbumin interneurons after fear learning. Neuron 2016, 91, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, I.; Humeau, Y.; Grenier, F.; Ciocchi, S.; Herry, C.; Luthi, A. Amygdala inhibitory circuits and the control of fear memory. Neuron 2009, 62, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Whalen, P.J.; Rauch, S.L.; Etcoff, N.L.; McInerney, S.C.; Lee, M.B.; Jenike, M.A. Masked presentations of emotional facial expressions modulate amygdala activity without explicit knowledge. J. Neurosci. 1998, 18, 411–418. [Google Scholar] [PubMed]
- Vuilleumier, P.; Armony, J.L.; Driver, J.; Dolan, R.J. Effects of attention and emotion on face processing in the human brain: An event-related fmri study. Neuron 2001, 30, 829–841. [Google Scholar] [CrossRef]
- Hariri, A.R.; Tessitore, A.; Mattay, V.S.; Fera, F.; Weinberger, D.R. The amygdala response to emotional stimuli: A comparison of faces and scenes. Neuroimage 2002, 17, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, I.; Phan, K.L.; Wood, A.; Angstadt, M.; Chua, P.; Heinrichs, M.; Stout, J.C.; Nathan, P.J. Oxytocin attenuates amygdala reactivity to fear in generalized social anxiety disorder. Neuropsychopharmacology 2010, 35, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.C.; Wright, C.I.; Wedig, M.M.; Gold, A.L.; Pollack, M.H.; Rauch, S.L. A functional mri study of amygdala responses to angry schematic faces in social anxiety disorder. Depression Anxiety 2008, 25, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Straube, T.; Mentzel, H.J.; Miltner, W.H.R. Neural mechanisms of automatic and direct processing of phobogenic stimuli in specific phobia. Biol. Psychiatry 2006, 59, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.L.; Whalen, P.J.; Shin, L.M.; McInerney, S.C.; Macklin, M.L.; Lasko, N.B.; Orr, S.P.; Pitman, R.K. Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: A functional mri study. Biol. Psychiatry 2000, 47, 769–776. [Google Scholar] [CrossRef]
- Etkin, A.; Wager, T.D. Functional neuroimaging of anxiety: A meta-analysis of emotional processing in ptsd, social anxiety disorder, and specific phobia. Am. J. Psychiatry 2007, 164, 1476–1488. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, T.; Kazama, A.; Bachevalier, J.; Davis, M. Impaired safety signal learning may be a biomarker of ptsd. Neuropharmacology 2012, 62, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Genud-Gabai, R.; Klavir, O.; Paz, R. Safety signals in the primate amygdala. J. Neurosci. 2013, 33, 17986–17994. [Google Scholar] [CrossRef] [PubMed]
- Likhtik, E.; Stujenske, J.M.; Topiwala, M.A.; Harris, A.Z.; Gordon, J.A. Prefrontal entrainment of amygdala activity signals safety in learned fear and innate anxiety. Nat. Neurosci. 2014, 17, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.P.; Jennings, J.H.; Ragole, T.; Flyer, J.G.; Benison, A.M.; Barth, D.S.; Watkins, L.R.; Maier, S.F. Safety signals mitigate the consequences of uncontrollable stress via a circuit involving the sensory insular cortex and bed nucleus of the stria terminalis. Biol. Psychiatry 2011, 70, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Millan, E.Z.; Reese, R.M.; Grossman, C.D.; Chaudhri, N.; Janak, P.H. Nucleus accumbens and posterior amygdala mediate cue-triggered alcohol seeking and suppress behavior during the omission of alcohol-predictive cues. Neuropsychopharmacology 2015, 40, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, J.M.; Reese, R.M.; Janak, P.H.; Chaudhri, N. Alcohol-seeking triggered by discrete pavlovian cues is invigorated by alcohol contexts and mediated by glutamate signaling in the basolateral amygdala. Neuropsychopharmacology 2015, 40, 2801–2812. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, C.M.; Cleva, R.M.; Hood, L.E.; Olive, M.F.; Gass, J.T. Mglur5 receptors in the basolateral amygdala and nucleus accumbens regulate cue-induced reinstatement of ethanol-seeking behavior. Pharmacol. Biochem. Behav. 2012, 101, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, N.; Woods, C.A.; Sahuque, L.L.; Gill, T.M.; Janak, P.H. Unilateral inactivation of the basolateral amygdala attenuates context-induced renewal of pavlovian-conditioned alcohol-seeking. Eur. J. Neurosci. 2013, 38, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, P.W.; Funk, D.; Juzytsch, W.; Le, A.D. Opioid receptors in the basolateral amygdala but not dorsal hippocampus mediate context-induced alcohol seeking. Behav. Brain Res. 2010, 211, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.T.; Sinclair, C.M.; Cleva, R.M.; Widholm, J.J.; Olive, M.F. Alcohol-seeking behavior is associated with increased glutamate transmission in basolateral amygdala and nucleus accumbens as measured by glutamate-oxidase-coated biosensors. Addict. Biol. 2011, 16, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, K.; Wrobel, E.; Korkosz, A.; Rogowski, A.; Kostowski, W.; Bienkowski, P.; Kaczmarek, L. Alcohol relapse induced by discrete cues activates components of ap-1 transcription factor and erk pathway in the rat basolateral and central amygdala. Neuropsychopharmacology 2008, 33, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- McCool, B.A.; Christian, D.T.; Fetzer, J.A.; Chappell, A.M. Lateral/basolateral amygdala serotonin type-2 receptors modulate operant self-administration of a sweetened ethanol solution via inhibition of principal neuron activity. Front. Integr. Neurosci. 2014, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.R.; Chappell, A.M.; Weiner, J.L. Effect of beta 3 adrenoceptor activation in the basolateral amygdala on ethanol seeking behaviors. Psychopharmacology 2014, 231, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Millan, E.Z.; Kim, H.A.; Janak, P.H. Optogenetic activation of amygdala projections to nucleus accumbens can arrest conditioned and unconditioned alcohol consummatory behavior. Neuroscience 2017, 360, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Floyd, D.W.; Jung, K.Y.; McCool, B.A. Chronic ethanol ingestion facilitates n-methyl-d-aspartate receptor function and expression in rat lateral/basolateral amygdala neurons. J. Pharmacol. Exp. Ther. 2003, 307, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Lack, A.K.; Diaz, M.R.; Chappell, A.; DuBois, D.W.; McCool, B.A. Chronic ethanol and withdrawal differentially modulate pre- and postsynaptic function at glutamatergic synapses in rat basolateral amygdala. J. Neurophysiol. 2007, 98, 3185–3196. [Google Scholar] [CrossRef] [PubMed]
- Lack, A.K.; Christian, D.T.; Diaz, M.R.; McCool, B.A. Chronic ethanol and withdrawal effects on kainate receptor-mediated excitatory neurotransmission in the rat basolateral amygdala. Alcohol 2009, 43, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Christian, D.T.; Alexander, N.J.; Diaz, M.R.; McCool, B.A. Thalamic glutamatergic afferents into the rat basolateral amygdala exhibit increased presynaptic glutamate function following withdrawal from chronic intermittent ethanol. Neuropharmacology 2013, 65, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Varodayan, F.P.; Bajo, M.; Soni, N.; Luu, G.; Madamba, S.G.; Schweitzer, P.; Roberto, M. Chronic alcohol exposure disrupts cb1 regulation of gabaergic transmission in the rat basolateral amygdala. Addict. Biol. 2017, 22, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Lindemeyer, A.K.; Liang, J.; Marty, V.N.; Meyer, E.M.; Suryanarayanan, A.; Olsen, R.W.; Spigelman, I. Ethanol-induced plasticity of gabaa receptors in the basolateral amygdala. Neurochem. Res. 2014, 39, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- McCool, B.A.; Frye, G.D.; Pulido, M.D.; Botting, S.K. Effects of chronic ethanol consumption on rat gaba(a) and strychnine-sensitive glycine receptors expressed by lateral/basolateral amygdala neurons. Brain Res. 2003, 963, 165–177. [Google Scholar] [CrossRef]
- Diaz, M.R.; Christian, D.T.; Anderson, N.J.; McCool, B.A. Chronic ethanol and withdrawal differentially modulate lateral/basolateral amygdala paracapsular and local gabaergic synapses. J. Pharmacol. Exp. Ther. 2011, 337, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Wille-Bille, A.; de Olmos, S.; Marengo, L.; Chiner, F.; Pautassi, R.M. Long-term ethanol self-administration induces delta fosb in male and female adolescent, but not in adult, wistar rats. Progress Neuro-Psychopharmacol. Biol. Psychiatry 2017, 74, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Ozburn, A.R.; Mayfield, R.D.; Ponomarev, I.; Jones, T.A.; Blednov, Y.A.; Harris, R.A. Chronic self-administration of alcohol results in elevated delta fosb: Comparison of hybrid mice with distinct drinking patterns. BMC Neurosci. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Spanagel, R.; Krystal, J.H. Glutamatergic targets for new alcohol medications. Psychopharmacology (Berl.) 2013, 229, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Kryger, R.; Wilce, P.A. The effects of alcoholism on the human basolateral amygdala. Neuroscience 2010, 167, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, T.; Han, J.S.; Conley, M.; Gallagher, M.; Holland, P. Neurotoxic lesions of basolateral, but not central, amygdala interfere with pavlovian second-order conditioning and reinforcer devaluation effects. J. Neurosci. 1996, 16, 5256–5265. [Google Scholar] [PubMed]
- Schoenbaum, G.; Chiba, A.A.; Gallagher, M. Changes in functional connectivity in orbitofrontal cortex and basolateral amygdala during learning and reversal training. J. Neurosci. 2000, 20, 5179–5189. [Google Scholar] [PubMed]
- Christianson, J.P.; Ragole, T.; Amat, J.; Greenwood, B.N.; Strong, P.V.; Paul, E.D.; Fleshner, M.; Watkins, L.R.; Maier, S.F. 5-hydroxytryptamine 2c receptors in the basolateral amygdala are involved in the expression of anxiety after uncontrollable traumatic stress. Biol. Psychiatry 2010, 67, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Di Ciano, P.; Everitt, B.J. Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. J. Neurosci. 2004, 24, 7167–7173. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.D.; Ewing, S.W.F.; Filbey, F.M.; Sabbineni, A.; Hutchison, K.E. Identifying neurobiological phenotypes associated with alcohol use disorder severity. Neuropsychopharmacology 2011, 36, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Tapert, S.F.; Brown, G.G.; Baratta, M.V.; Brown, S.A. Fmri bold response to alcohol stimuli in alcohol dependent young women. Addict. Behav. 2004, 29, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Schiller, D.; Levy, I.; Niv, Y.; LeDoux, J.E.; Phelps, E.A. From fear to safety and back: Reversal of fear in the human brain. J. Neurosci. 2008, 28, 11517–11525. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, N.W.; Herman, M.A.; Roberto, M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol. Psychiatry 2015, 77, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, N.W.; Karanikas, C.A.; Richardson, H.N. Adolescent binge drinking leads to changes in alcohol drinking, anxiety, and amygdalar corticotropin releasing factor cells in adulthood in male rats. PLoS ONE 2012, 7, e31466. [Google Scholar] [CrossRef] [PubMed]
- Kushner, M.G.; Abrams, K.; Thuras, P.; Hanson, K.L.; Brekke, M.; Sletten, S. Follow-up study of anxiety disorder and alcohol dependence in comorbid alcoholism treatment patients. Alcohol. Clin. Exp. Res. 2005, 29, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Driessen, M.; Meier, S.; Hill, A.; Wetterling, T.; Lange, W.; Junghanns, K. The course of anxiety, depression and drinking behaviours after completed detoxification in alcoholics with and without comorbid anxiety and depressive disorders. Alcohol Alcohol. 2001, 36, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Back, S.E.; Brady, K.T.; Sonne, S.C.; Verduin, M.L. Symptom improvement in co-occurring ptsd and alcohol dependence. J. Nerv. Ment. Dis. 2006, 194, 690–696. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moaddab, M.; Mangone, E.; Ray, M.H.; McDannald, M.A. Adolescent Alcohol Drinking Renders Adult Drinking BLA-Dependent: BLA Hyper-Activity as Contributor to Comorbid Alcohol Use Disorder and Anxiety Disorders. Brain Sci. 2017, 7, 151. https://doi.org/10.3390/brainsci7110151
Moaddab M, Mangone E, Ray MH, McDannald MA. Adolescent Alcohol Drinking Renders Adult Drinking BLA-Dependent: BLA Hyper-Activity as Contributor to Comorbid Alcohol Use Disorder and Anxiety Disorders. Brain Sciences. 2017; 7(11):151. https://doi.org/10.3390/brainsci7110151
Chicago/Turabian StyleMoaddab, Mahsa, Elizabeth Mangone, Madelyn H. Ray, and Michael A. McDannald. 2017. "Adolescent Alcohol Drinking Renders Adult Drinking BLA-Dependent: BLA Hyper-Activity as Contributor to Comorbid Alcohol Use Disorder and Anxiety Disorders" Brain Sciences 7, no. 11: 151. https://doi.org/10.3390/brainsci7110151
APA StyleMoaddab, M., Mangone, E., Ray, M. H., & McDannald, M. A. (2017). Adolescent Alcohol Drinking Renders Adult Drinking BLA-Dependent: BLA Hyper-Activity as Contributor to Comorbid Alcohol Use Disorder and Anxiety Disorders. Brain Sciences, 7(11), 151. https://doi.org/10.3390/brainsci7110151