The Role of Peripheral CNS‐Directed Antibodies in Promoting Inflammatory CNS Demyelination

{kind=link}
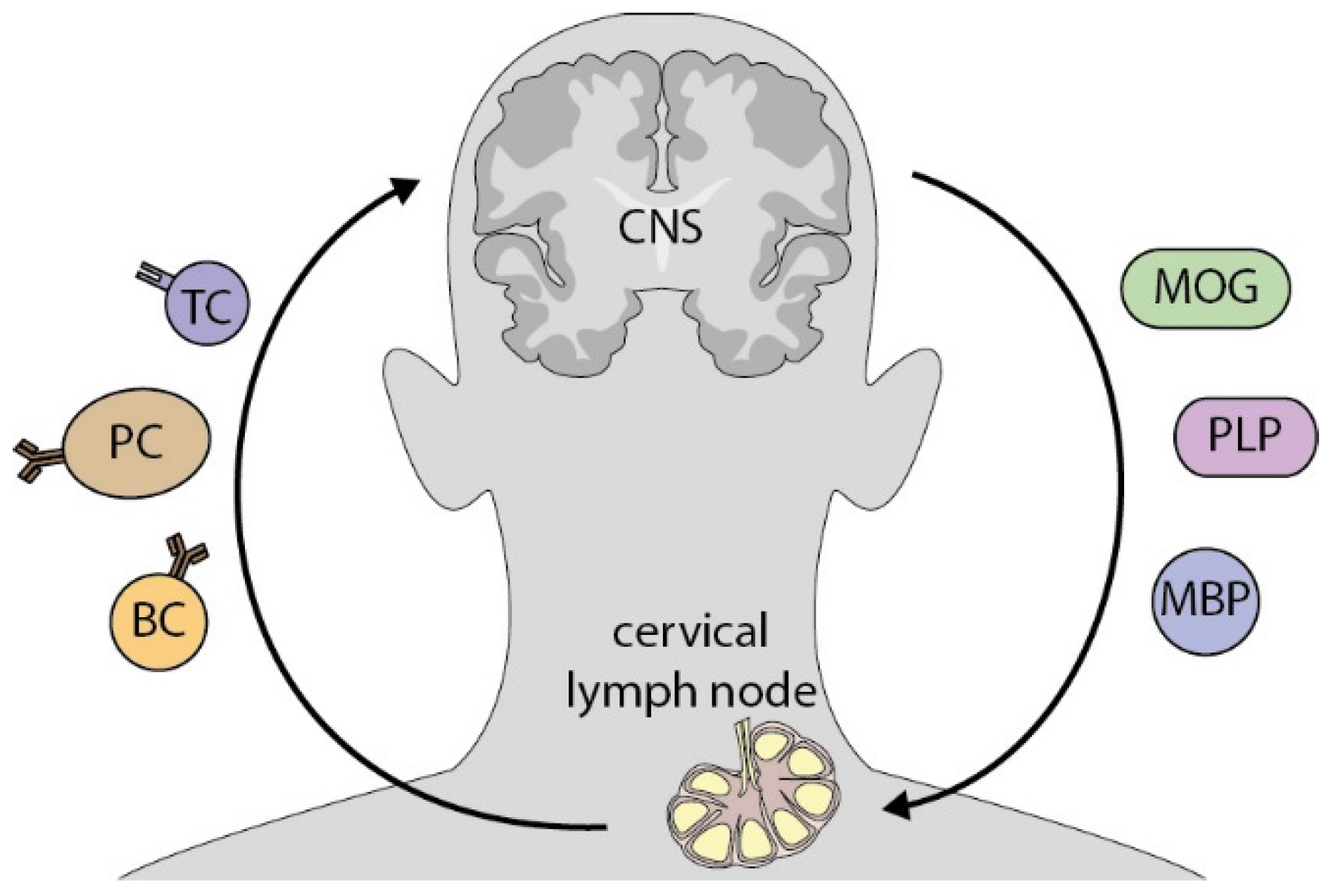{kind=link}
Abstract
:1. Introduction
2. Towards a Mechanistic Understating on the Role of Autoantibodies in CNS-Demyelinating Disorders
3. Stratifying Human Demyelinating Disorders by the Involvement of Distinct Autoantibodies
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Kinzel, S.; Weber, M.S.B. Cell-Directed Therapeutics in Multiple Sclerosis: Rationale and Clinical Evidence. CNS Drug 2016, 30, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.L.; O’Connor, K.C.; Bar-Or, A.; Zamvil, S.S.; Hemmer, B.; Tedder, T.F.; von Büdingen, H.C.; Stuve, O.; Yeaman, M.R.; Smith, T.J.; et al. B lymphocytes in neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflam. 2015, 2, e104. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.; Lamb, S.; Morgan, K.; Chen, A.; Waubant, E.; Genain, C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005, 64, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Kronsbein, H.C.; Weber, M.S. Targeting B cells in the treatment of multiple sclerosis: Recent advances and remaining challenges. Ther. Adv. Neurol. Disord. 2013, 6, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.; Schweitzer, N.; West, J.; Ranney, P.; Bottomly, K.B. lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J. Immunol. 1995, 155, 3734–3741. [Google Scholar] [PubMed]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002, 125, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Hinson, S.R.; Pittock, S.J.; Lucchinetti, C.F.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007, 69, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Wildemann, B.; Jarius, S.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Immunopathogenesis of neuromyelitis optica. Adv. Immunol. 2014, 121, 213–242. [Google Scholar] [PubMed]
- Waters, P.J.; McKeon, A.; Leite, M.I.; Rajasekharan, S.; Lennon, V.A.; Villalobos, A.; Palace, J.; Mandrekar, J.N.; Vincent, A.; Bar-Or, A.; et al. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology 2012, 78, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Majed, M.; Fryer, J.P.; McKeon, A.; Lennon, V.A.; Pittock, S.J. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol. Neuroimmunol. Neuroinflam. 2016, 3, e231. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A.; Freedman, D.A. A study of the crystalline albumin, gamma globulin and total protein in the cerebrospinal fluid of 100 cases of multiple sclerosis and in other diseases. Am. J. Med. Sci. 1950, I, 55–64. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kümpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Von Budingen, H.C.; Gulati, M.; Kuenzle, S.; Fischer, K.; Rupprecht, T.A.; Goebels, N. Clonally expanded plasma cells in the cerebrospinal fluid of patients with central nervous system autoimmune demyelination produce “oligoclonal bands”. J. Neuroimmunol. 2010, 218, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Storch, M.K.; Piddlesden, S.; Haltia, M.; Iivanainen, M.; Morgan, P.; Lassmann, H. Multiple sclerosis: In situ evidence for antibody- and complement-mediated demyelination. Ann. Neurol. 1998, 43, 465–471. [Google Scholar]
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Van der Goes, A.; Kortekaas, M.; Hoekstra, K.; Dijkstra, C.D.; Amor, S. The role of anti-myelin (auto)-antibodies in the phagocytosis of myelin by macrophages. J. Neuroimmunol. 1999, 101, 61–67. [Google Scholar] [CrossRef]
- Kinzel, S.; Lehmann-Horn, K.; Torke, S.; Häusler, D.; Winkler, A.; Stadelmann, C.; Payne, N.; Feldmann, L.; Saiz, A.; Reindl, M.; et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016, 132, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.; Sant’Angelo, D.; Pasqualini, T.; Taylor, T.; Levin, D.; Flavell, R.; Bottomly, K. Peptide and protein antigens require distinct antigen-presenting cell subsets for the priming of CD4+ T cells. J. Immunol. 1995, 154, 4915–4923. [Google Scholar] [PubMed]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Hemmer, B. Cooperation of B cells and T cells in the pathogenesis of multiple sclerosis. Results Probl. Cell Differ. 2010, 51, 115–126. [Google Scholar] [PubMed]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bösl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Prod’homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.; Slavin, A.J.; Linington, C.; et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010, 68, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Lalive, P.H.; Molnarfi, N.; Benkhoucha, M.; Weber, M.S.; Santiago-Raber, M.L. Antibody response in MOG35–55 induced EAE. J. Neuroimmunol. 2011, 240–241, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Schluesener, H.J.; Sobel, R.A.; Linington, C.; Weiner, H.L. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J. Immunol. 1987, 139, 4016–4021. [Google Scholar] [PubMed]
- Flach, A.C.; Litke, T.; Strauss, J.; Haberl, M.; Gómez, C.C.; Reindl, M.; Saiz, A.; Fehling, H.J.; Wienands, J.; Odoardi, F.; et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc. Natl. Acad. Sci. USA 2016, 113, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.R.; Lyon, G.M.; Ruddle, N.H. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J. Immunol. 2003, 171, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Storch, M.K.; Stefferl, A.; Brehm, U.; Weissert, R.; Wallström, E.; Kerschensteiner, M.; Olsson, T.; Linington, C.; Lassmann, H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998, 8, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Urich, E.; Gutcher, I.; Prinz, M.; Becher, B. Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 18697–18702. [Google Scholar] [CrossRef] [PubMed]
- Trotter, J.; DeJong, L.J.; Smith, M.E. Opsonization with antimyelin antibody increases the uptake and intracellular metabolism of myelin in inflammatory macrophages. J. Neurochem. 1986, 47, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.A.; Ramsbottom, M.J.; Cross, A.H. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 2002, 32, 1905–1913. [Google Scholar] [CrossRef]
- Seifert, C.L.; Wegner, C.; Sprenger, T.; Weber, M.S.; Brück, W.; Hemmer, B.; Sellner, J. Favourable response to plasma exchange in tumefactive CNS demyelination with delayed B-cell response. Mult. Scler. 2012, 18, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Mathey, E.K.; Derfuss, T.; Storch, M.K.; Williams, K.R.; Hales, K.; Woolley, D.R.; Al-Hayani, A.; Davies, S.N.; Rasband, M.N.; Olsson, T.; et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007, 204, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kümpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.G.; Catz, I. Relative frequency of autoantibodies to myelin basic protein and proteolipid protein in optic neuritis and multiple sclerosis cerebrospinal fluid. J. Neurol. Sci. 1994, 121, 66–73. [Google Scholar] [CrossRef]
- Lalive, P.H.; Menge, T.; Delarasse, C.; Williams, K.R.; Hales, K.; Woolley, D.R.; Al-Hayani, A.; Davies, S.N.; Rasband, M.N.; Olsson, T.; et al. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc. Natl Acad. Sci. USA 2006, 103, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti–aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Franciotta, D.; Waters, P.; Zipp, F.; Hohlfeld, R.; Vincent, A.; Wildemann, B. Mechanisms of Disease, aquaporin-4 antibodies in neuromyelitis optica. Nat. Clin. Pract. Neurol. 2008, 4, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, A.; Bergamaschi, R.; Martinelli, V.; Trojano, M.; Tola, M.R.; Merelli, E.; Mancardi, L.; Gallo, P.; Filippi, M.; Zaffaroni, M.; et al. Clinical characteristics, course and prognosis of relapsing Devic’s Neuromyelitis Optica. J. Neurol. 2004, 251, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Ruprecht, K.; Wildemann, B.; Kuempfel, T.; Ringelstein, M.; Geis, C.; Kleiter, I.; Kleinschnitz, C.; Berthele, A.; Brettschneider, J.; et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica, A multicentre study of 175 patients. J. Neuroinflam. 2012, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Franciotta, D.; Paul, F.; Bergamaschi, R.; Rommer, P.S.; Ruprecht, K.; Ringelstein, M.; Aktas, O.; Kristoferitsch, W.; Wildemann, B. Testing for antibodies to human aquaporin-4 by ELISA, Sensitivity, specificity, and direct comparison with immunohistochemistry. J. Neurol. Sci. 2012, 320, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, S.; Ito, T.; Misu, T.; Takahashi, T.; Kikuchi, A.; Suzuki, N.; Jin, K.; Aoki, M.; Fujihara, K.; Itoyama, Y. A case of NMO seropositive for aquaporin-4 antibody more than 10 years before onset. Neurology 2009, 72, 1960–1961. [Google Scholar] [CrossRef] [PubMed]
- Kleiter, I.; Gahlen, A.; Borisow, N.; Fischer, K.; Wernecke, K.D.; Wegner, B.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Neuromyelitis optica, Evaluation of 871 attacks and 1,153 treatment courses. Ann. Neurol. 2016, 79, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Obermeier, B.; Cotleur, A.C.; Spampinato, S.F.; Shimizu, F.; Yamamoto, E.; Sano, Y.; Kryzer, T.J.; Lennon, V.A.; Kanda, T.; et al. Effects of neuromyelitis optica–IgG at the blood–brain barrier in vitro. Neurol. Neuroimmunol. Neuroinflam. 2017, 4, e311. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Probstel, A.K.; Rudolf, G.; Dornmair, K.; Collongues, N.; Chanson, J.-B.; Sanderson, N.S.R.; Lindberg, R.L.P.; Kappos, L.; de Seze, J.; Derfuss, T. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J. Neuroinflam. 2015, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Varrin-Doyer, M.; Shetty, A.; Spencer, C.M.; Schulze-Topphoff, U.; Weber, M.S.; Bernard, C.C.; Forsthuber, T.; Cree, B.A.; Slavin, A.J.; Zamvil, S.S. MOG transmembrane and cytoplasmic domains contain highly stimulatory T-cell epitopes in MS. Neurol. Neuroimmunol. Neuroinflam. 2014, 1, e20. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.S.; Reid, H.H.; Beddoe, T.; Tynan, F.E.; Perugini, M.A.; Johns, T.G.; Bernard, C.C.; Rossjohn, J. The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 11059–11064. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Sabater, L.; Sospedra, M.; Ferrer-Francesch, X.; Escudero, D.; Martínez-Cáceres, E.; Pujol-Borrell, R. Multiple sclerosis candidate autoantigens except myelin oligodendrocyte glycoprotein are transcribed in human thymus. Eur. J. Immunol. 2002, 32, 2737–2747. [Google Scholar] [CrossRef]
- Shetty, A.; Gupta, S.G.; Varrin-Doyer, M.; Weber, M.S.; Prod’homme, T.; Molnarfi, N.; Ji, N.; Nelson, P.A.; Patarroyo, J.C.; Schulze-Topphoff, U.; et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol. Neuroimmunol. Neuroinflam. 2014, 1, e22. [Google Scholar] [CrossRef] [PubMed]
- Kitley, J.; Waters, P.; Woodhall, M.; Leite, M.I.; Murchison, A.; George, J.; Küker, W.; Chandratre, S.; Vincent, A.; Palace, J. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: A comparative study. JAMA Neurol. 2014, 71, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kiyota, N.; Kuroda, H.; Sato, D.K.; Nishiyama, S.; Takahashi, T.; Misu, T.; Nakashima, I.; Fujihara, K.; Aoki, M. Severe demyelination but no astrocytopathy in clinically definite neuromyelitis optica with anti-myelin-oligodendrocyte glycoprotein antibody. Mult. Scler. 2015, 21, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Sato, D.K.; Nakashima, I.; Nishiyama, S.; Tanaka, S.; Marignier, R.; Hyun, J.W.; Oliveira, L.M.; Reindl, M.; Seifert-Held, T.; et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, M.; Gerdes, L.A.; Krumbholz, M.; Ertl-Wagner, B.; Thaler, F.S.; Schuh, E.; Metz, I.; Blaschek, A.; Dick, A.; Brück, W.; et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol. Neuroimmunol. Neuroinflam. 2016, 3, e257. [Google Scholar] [CrossRef] [PubMed]
- Zamvil, S.S.; Slavin, A.J. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol. Neuroimmunol. Neuroinflam. 2015, 2, e62. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Fabriek, B.O.; Zwemmer, J.N.; Teunissen, C.E.; Dijkstra, C.D.; Polman, C.H.; Laman, J.D.; Castelijns, J.A. In vivo detection of myelin proteins in cervical lymph nodes of MS patients using ultrasound-guided fine-needle aspiration cytology. J. Neuroimmunol. 2005, 161, 190–194. [Google Scholar] [CrossRef] [PubMed]
- De Vos, A.F.; van Meurs, M.; Brok, H.P.; Boven, L.A.; Hintzen, R.Q.; van der Valk, P.; Ravid, R.; Rensing, S.; Boon, L.; Laman, J.D.; et al. Transfer of Central Nervous System Autoantigens and Presentation in Secondary Lymphoid Organs. J. Immunol. 2002, 169, 5415–5423. [Google Scholar] [CrossRef] [PubMed]
- Van Zwam, M.; Huizinga, R.; Heijmans, N.; van Meurs, M.; Wierenga-Wolf, A.F.; Melief, M.J.; Hintzen, R.Q.; ’t Hart, B.A.; Amor, S.; Boven, L.A.; et al. Surgical excision of CNS-draining lymph nodes reduces relapse severity in chronic-relapsing experimental autoimmune encephalomyelitis. J. Pathol. 2009, 217, 543–551. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinzel, S.; Weber, M.S. The Role of Peripheral CNS‐Directed Antibodies in Promoting Inflammatory CNS Demyelination. Brain Sci. 2017, 7, 70. https://doi.org/10.3390/brainsci7070070
Kinzel S, Weber MS. The Role of Peripheral CNS‐Directed Antibodies in Promoting Inflammatory CNS Demyelination. Brain Sciences. 2017; 7(7):70. https://doi.org/10.3390/brainsci7070070
Chicago/Turabian StyleKinzel, Silke, and Martin S. Weber. 2017. "The Role of Peripheral CNS‐Directed Antibodies in Promoting Inflammatory CNS Demyelination" Brain Sciences 7, no. 7: 70. https://doi.org/10.3390/brainsci7070070
APA StyleKinzel, S., & Weber, M. S. (2017). The Role of Peripheral CNS‐Directed Antibodies in Promoting Inflammatory CNS Demyelination. Brain Sciences, 7(7), 70. https://doi.org/10.3390/brainsci7070070