Melanocortins, Melanocortin Receptors and Multiple Sclerosis

Abstract
:1. Introduction
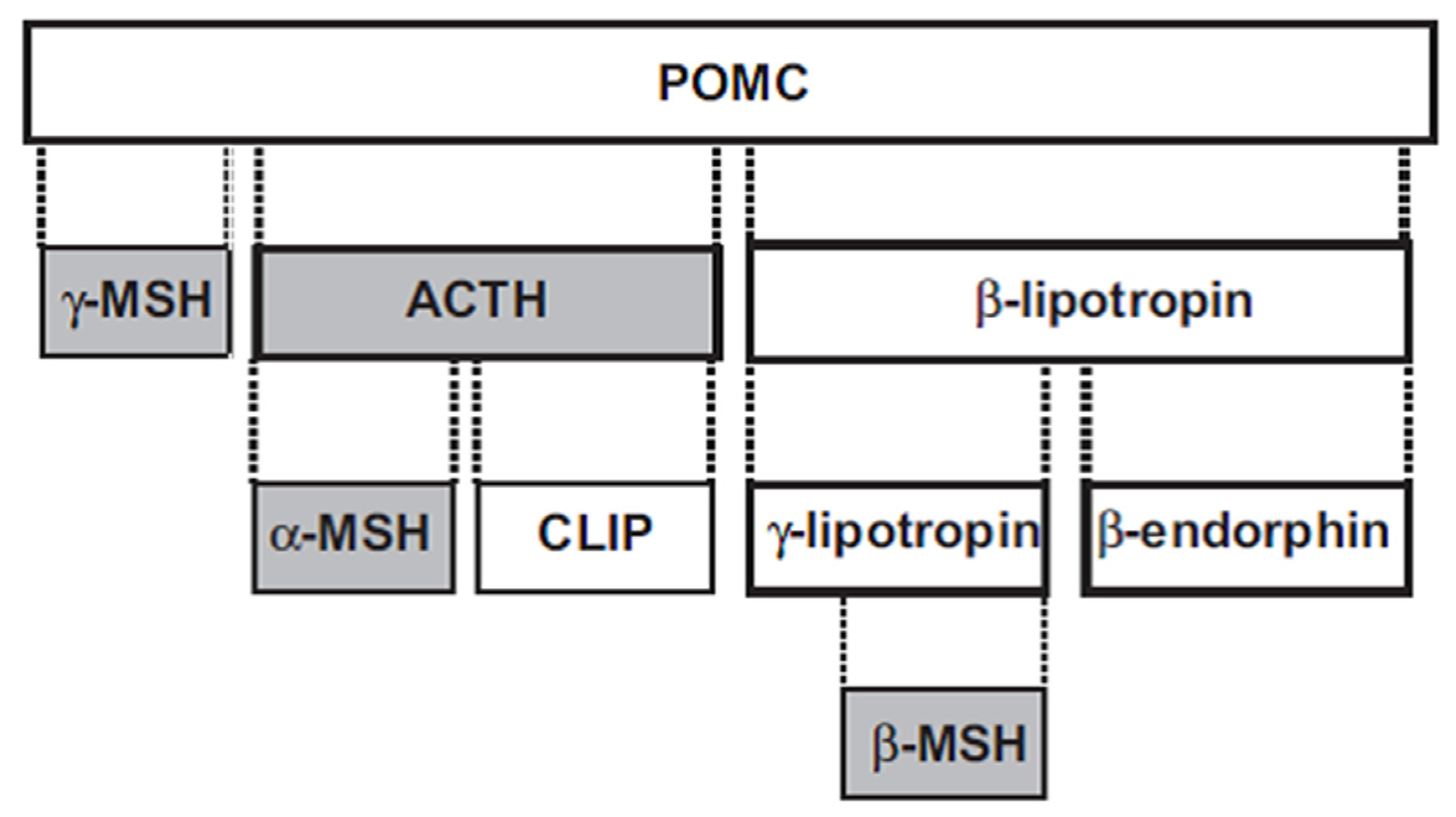
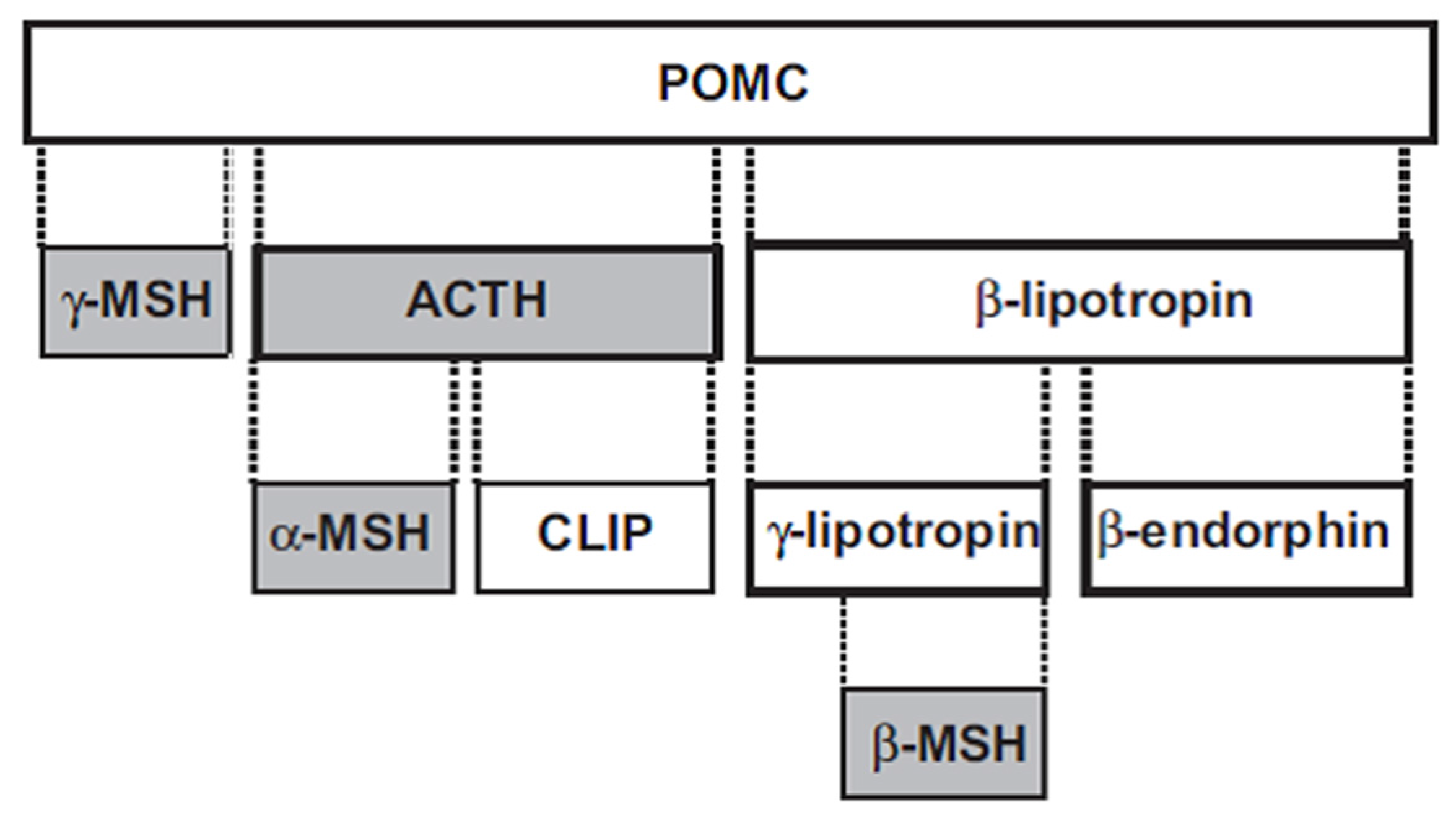
2. Melanocortins
3. Melanocortin Receptors
4. Melanocortin Receptor Signaling
5. Anti-Inflammatory Effects on the Peripheral Immune System
5.1. Corticosteroid Dependent Effects
5.2. Corticosteroid Independent Effects
6. Direct Effects in the CNS
6.1. Effects Mediated through Neuronal Regulation of Immune System Function
6.2. Effects on CNS Neurons
6.3. Effects on Glia
7. Effects on Endogenous Cells of the CNS with Potential Protective and Reparative Importance in MS and Other CNS Disorders
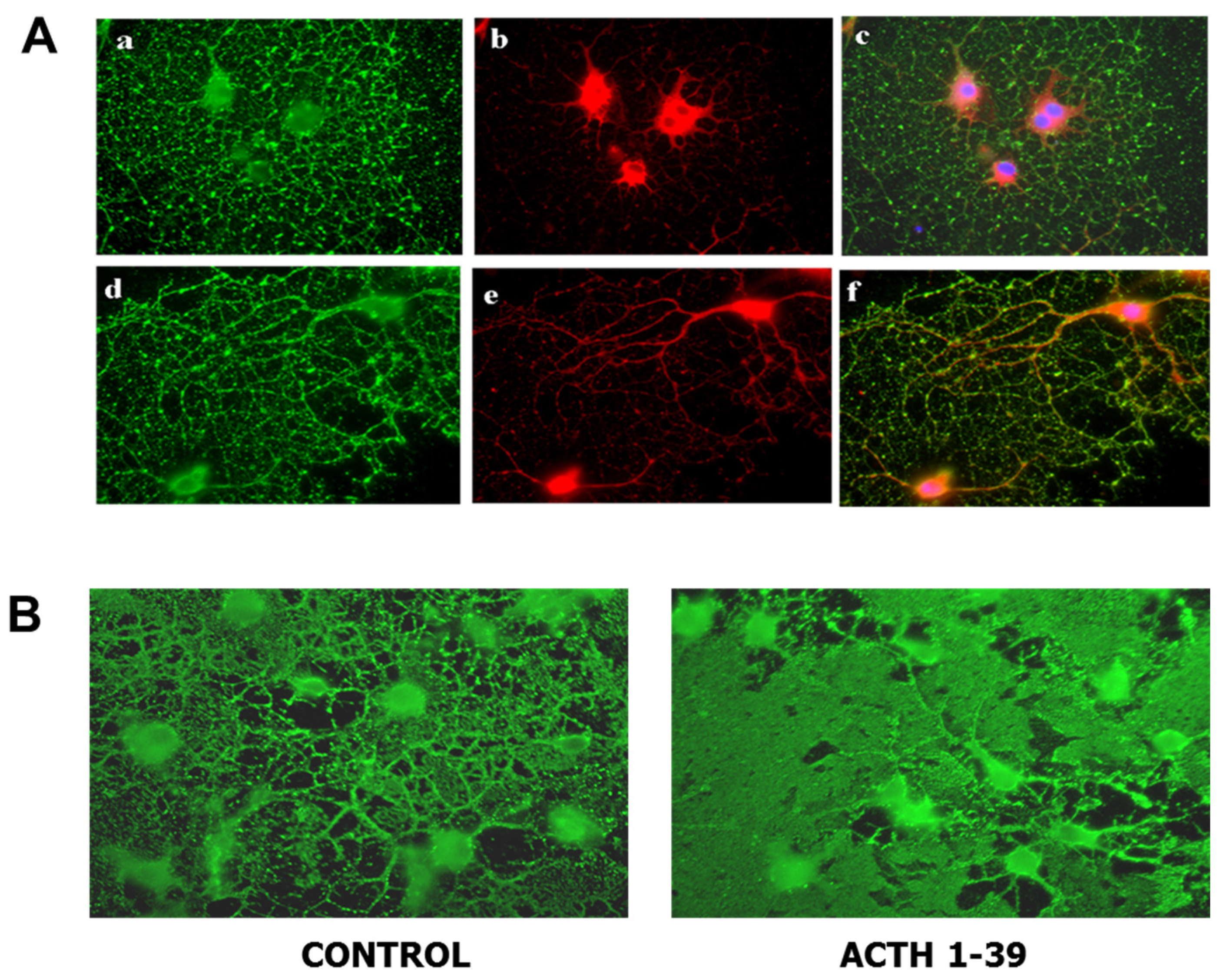
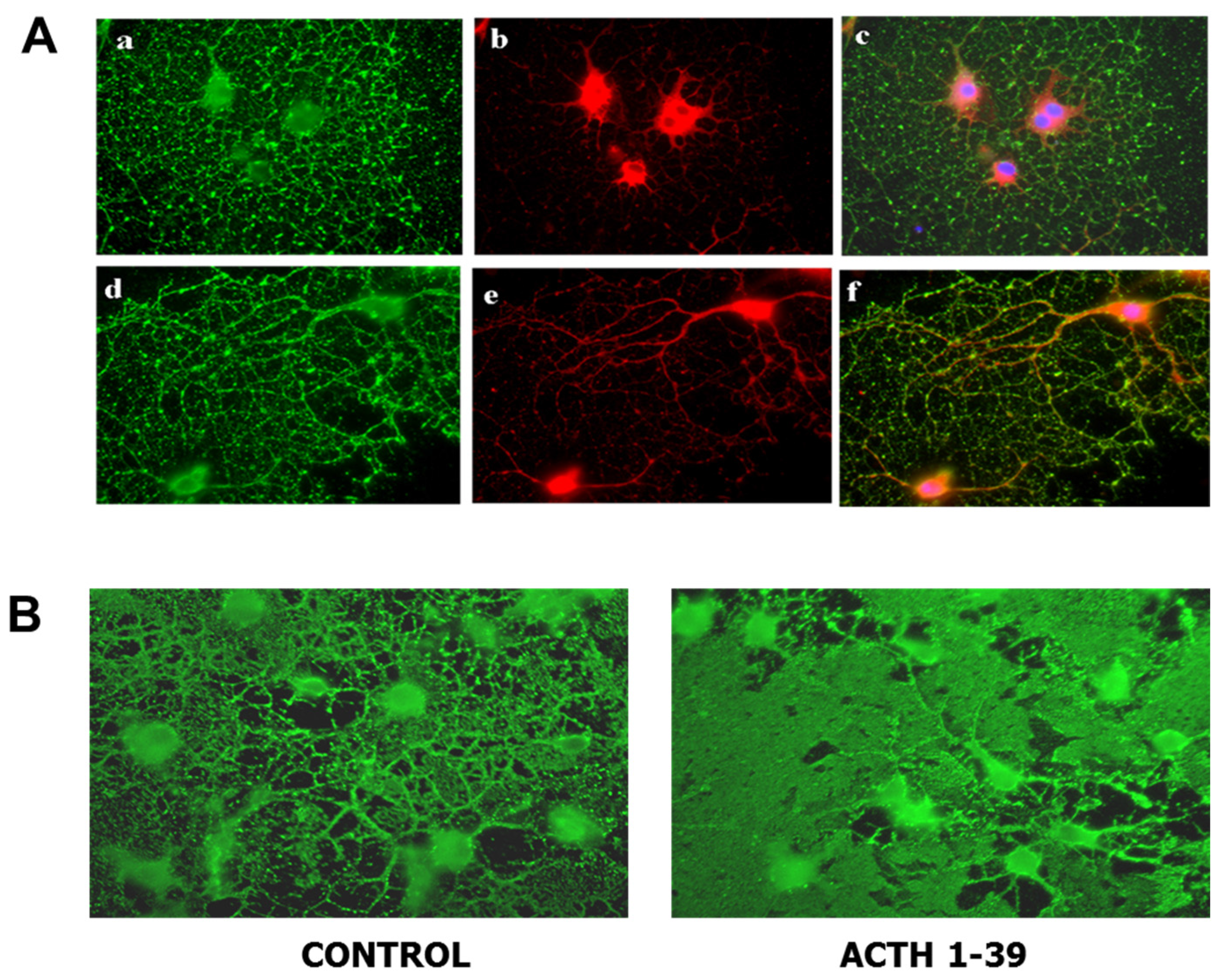
8. Melanocortin Receptor Signaling in Oligodendroglial Protection
9. Treatment of Human Neurologic Diseases with Melanocortins
10. Future Studies
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adan, R.A.; Gispen, W.H. Brain melanocortin receptors: From cloning to function. Peptides 1997, 18, 1279–1287. [Google Scholar] [CrossRef]
- Cone, R.D. Studies on the physiological functions of the melanocortin system. Endocr. Rev. 2006, 27, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Caruso, V.; Lagerstrom, M.C.; Olszewski, P.K.; Fredriksson, R.; Schioth, H.B. Synaptic changes induced by melanocortin signalling. Nat. Rev. Neurosci. 2014, 15, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Montero-Melendez, T. ACTH: The forgotten therapy. Semin. Immunol. 2015, 27, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Yao, T.; Kong, X.; Williams, K.W.; Liu, T. Melanocortin neurons: Multiple routes to regulation of metabolism. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Arnason, B.G.; Berkovich, R.; Catania, A.; Lisak, R.P.; Zaidi, M. Mechanisms of action of adrenocorticotropic hormone and other melanocortins relevant to the clinical management of patients with multiple sclerosis. Mult. Scler. 2013, 19, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Brzoska, T.; Luger, T.A.; Maaser, C.; Abels, C.; Bohm, M. Alpha-melanocyte-stimulating hormone and related tripeptides: Biochemistry, antiinflammatory and protective effects in vitro and in vivo, and future perspectives for the treatment of immune-mediated inflammatory diseases. Endocr. Rev. 2008, 29, 581–602. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Gatti, S.; Colombo, G.; Lipton, J.M. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol. Rev. 2004, 56, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Lonati, C.; Sordi, A.; Carlin, A.; Leonardi, P.; Gatti, S. The melanocortin system in control of inflammation. Sci. World J. 2010, 10, 1840–1853. [Google Scholar] [CrossRef] [PubMed]
- Muceniece, R.; Dambrova, M. Melanocortins in brain inflammation: The role of melanocortin receptor subtypes. Adv. Exp. Med. Biol. 2010, 681, 61–70. [Google Scholar] [PubMed]
- Bertolini, A.; Tacchi, R.; Vergoni, A.V. Brain effects of melanocortins. Pharmacol. Res. 2009, 59, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Catania, A. Neuroprotective actions of melanocortins: A therapeutic opportunity. Trends Neurosci. 2008, 31, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Dores, R.M.; Londraville, R.L.; Prokop, J.; Davis, P.; Dewey, N.; Lesinski, N. Molecular evolution of GPCRs: Melanocortin/melanocortin receptors. J. Mol. Endocrinol. 2014, 52, T29–T42. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G. Distribution and Function of Melanocortin Receptors within the Brain. Adv. Exp. Med. Biol. 2010, 681, 29–48. [Google Scholar] [PubMed]
- Schioth, H.B.; Haitina, T.; Ling, M.K.; Ringholm, A.; Fredriksson, R.; Cerda-Reverter, J.M.; Klovins, J. Evolutionary conservation of the structural, pharmacological, and genomic characteristics of the melanocortin receptor subtypes. Peptides 2005, 26, 1886–1900. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Structure, function and regulation of the melanocortin receptors. Eur. J. Pharmacol. 2011, 660, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Harmon, C.M. Molecular signatures of human melanocortin receptors for ligand binding and signaling. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Switonski, M.; Mankowska, M.; Salamon, S. Family of melanocortin receptor (MCR) genes in mammals-mutations, polymorphisms and phenotypic effects. J. Appl. Genet. 2013, 54, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Malek, Z.A.; Swope, V.B.; Starner, R.J.; Koikov, L.; Cassidy, P.; Leachman, S. Melanocortins and the melanocortin 1 receptor, moving translationally towards melanoma prevention. Arch. Biochem. Biophys. 2014, 563, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Demidowich, A.P.; Jun, J.Y.; Yanovski, J.A. Polymorphisms and mutations in the melanocortin-3 receptor and their relation to human obesity. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, C.; Garcia-Borron, J.C.; Jimenez-Cervantes, C.; Olivares, C. MC1R signaling.Intracellular partners and pathophysiological implications. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef]
- Friedman, A.P. Do hyporesponsive genetic variants of the melanocortin 1 receptor contribute to the etiology of multiple sclerosis? Med. Hypotheses 2004, 62, 49–52. [Google Scholar] [CrossRef]
- Partridge, J.M.; Weatherby, S.J.; Woolmore, J.A.; Highland, D.J.; Fryer, A.A.; Mann, C.L.; Boggild, M.D.; Ollier, W.E.; Strange, R.C.; Hawkins, C.P. Susceptibility and outcome in MS: Associations with polymorphisms in pigmentation-related genes. Neurology 2004, 62, 2323–2325. [Google Scholar] [CrossRef] [PubMed]
- Strange, R.C.; Ramachandran, S.; Zeegers, M.P.; Emes, R.D.; Abraham, R.; Raveendran, V.; Boggild, M.; Gilford, J.; Hawkins, C.P. The Multiple Sclerosis Severity Score: Associations with MC1R single nucleotide polymorphisms and host response to ultraviolet radiation. Mult. Scler. 2010, 16, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.X.; Zou, L.P.; He, B.; Yue, W.H.; Liu, Z.L.; Zhang, D. ACTH receptor (MC2R) promoter variants associated with infantile spasms modulate MC2R expression and responsiveness to ACTH. Pharmacogenet Genom. 2010, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; He, B.; Fang, F.; Tang, C.Y.; Zou, L.P. Genetic polymorphisms of MC2R gene associated with responsiveness to adrenocorticotropic hormone therapy in infantile spasms. Chin. Med. J. 2008, 121, 1627–1632. [Google Scholar] [PubMed]
- Farooqi, I.S.; Yeo, G.S.; Keogh, J.M.; Aminian, S.; Jebb, S.A.; Butler, G.; Cheetham, T.; O’Rahilly, S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Investig. 2000, 106, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Hinney, A.; Volckmar, A.L.; Knoll, N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog. Mol. Biol. Transl. Sci. 2013, 114, 147–191. [Google Scholar] [PubMed]
- Miller, C.L.; Murakami, P.; Ruczinski, I.; Ross, R.G.; Sinkus, M.; Sullivan, B.; Leonard, S. Two complex genotypes relevant to the kynurenine pathway and melanotropin function show association with schizophrenia and bipolar disorder. Schizophr. Res. 2009, 113, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Valli-Jaakola, K.; Suviolahti, E.; Schalin-Jantti, C.; Ripatti, S.; Silander, K.; Oksanen, L.; Salomaa, V.; Peltonen, L.; Kontula, K. Further evidence for the role of ENPP1 in obesity: Association with morbid obesity in Finns. Obesity 2008, 16, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Eves, P.C.; Haycock, J.W. Melanocortin signalling mechanisms. Adv. Exp. Med. Biol. 2010, 681, 19–28. [Google Scholar] [PubMed]
- Rodrigues, A.R.; Almeida, H.; Gouveia, A.M. Intracellular signaling mechanisms of the melanocortin receptors: Current state of the art. Cell. Mol. Life Sci. 2015, 72, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, W.; Tao, Y.X. Pharmacological chaperones for the misfolded melanocortin-4 receptor associated with human obesity. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Rouault, A.A.J.; Srinivasan, D.K.; Yin, T.C.; Lee, A.A.; Sebag, J.A. Melanocortin Receptor Accessory Proteins (MRAPs): Functions in the melanocortin system and beyond. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Ericson, M.D.; Lensing, C.J.; Fleming, K.A.; Schlasner, K.N.; Doering, S.R.; Haskell-Luevano, C. Bench-top to clinical therapies: A review of melanocortin ligands from 1954 to 2016. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Pantel, J.; Williams, S.Y.; Mi, D.; Sebag, J.; Corbin, J.D.; Weaver, C.D.; Cone, R.D. Development of a high throughput screen for allosteric modulators of melanocortin-4 receptor signaling using a real time cAMP assay. Eur. J. Pharmacol. 2011, 660, 139–147. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef] [PubMed]
- Breit, A.; Buch, T.R.; Boekhoff, I.; Solinski, H.J.; Damm, E.; Gudermann, T. Alternative G protein coupling and biased agonism: New insights into melanocortin-4 receptor signalling. Mol. Cell. Endocrinol. 2011, 331, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X. Constitutive activity in melanocortin-4 receptor: Biased signaling of inverse agonists. Adv. Pharmacol. 2014, 70, 135–154. [Google Scholar] [PubMed]
- Clark, A.J.; Forfar, R.; Hussain, M.; Jerman, J.; McIver, E.; Taylor, D.; Chan, L. ACTH Antagonists. Front. Endocrinol. 2016, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Ghaddhab, C.; Vuissoz, J.M.; Deladoey, J. From Bioinactive ACTH to ACTH Antagonist: The Clinical Perspective. Front. Endocrinol. 2017, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Catania, A. The melanocortin system in leukocyte biology. J. Leukoc. Biol. 2007, 81, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Baron, P.; Meda, L.; Prat, E.; Scarpini, E.; Delgado, R.; Catania, A.; Lipton, J.M.; Scarlato, G. Alpha-MSH peptides inhibit production of nitric oxide and tumor necrosis factor-alpha by microglial cells activated with beta-amyloid and interferon gamma. Biochem. Biophys. Res. Commun. 1999, 263, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.M.; Catania, A. Mechanisms of antiinflammatory action of the neuroimmunomodulatory peptide alpha-MSH. Ann. N. Y. Acad. Sci. 1998, 840, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Namba, K. In vitro induction of CD25+ CD4+ regulatory T cells by the neuropeptide alpha-melanocyte stimulating hormone (alpha-MSH). Immunol. Cell Biol. 2001, 79, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W.; Yee, D.G.; Nishida, T.; Namba, K. Neuropeptide regulation of immunity. The immunosuppressive activity of alpha-melanocyte-stimulating hormone (alpha-MSH). Ann. N. Y. Acad. Sci. 2000, 917, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Reder, A.; Birnbaum, G. B-cell differentiation in multiple sclerosis and the effect of intravenous ACTH. Neurology 1983, 33, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W.; Kitaichi, N.; Biros, D. Melanocortin 5 receptor and ocular immunity. Cell. Mol. Biol. 2006, 52, 53–59. [Google Scholar] [PubMed]
- Biros, D.J.; Namba, K.; Taylor, A.W. Alpha-MSH regulates protein ubiquitination in T cells. Cell. Mol. Biol. 2006, 52, 33–38. [Google Scholar] [PubMed]
- Getting, S.J.; Christian, H.C.; Flower, R.J.; Perretti, M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis Rheum. 2002, 46, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Lee, C.; Funahashi, H.; Friedman, J.; Lee, S.; Elmquist, J. Melanocortin-4 receptor expression in a vago-vagal circuitry involved in postprandial functions. J. Comp. Neurol. 2010, 518, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, D.; Ottani, A.; Neri, L.; Zaffe, D.; Grieco, P.; Jochem, J.; Cavallini, G.M.; Catania, A.; Guarini, S. Multiple beneficial effects of melanocortin MC4 receptor agonists in experimental neurodegenerative disorders: Therapeutic perspectives. Prog. Neurobiol. 2017, 148, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Gee, C.E.; Chen, C.L.; Roberts, J.L.; Thompson, R.; Watson, S.J. Identification of proopiomelanocortin neurones in rat hypothalamus by in situ cDNA-mRNA hybridization. Nature 1983, 306, 374–376. [Google Scholar] [CrossRef]
- Kishi, T.; Aschkenasi, C.J.; Lee, C.E.; Mountjoy, K.G.; Saper, C.B.; Elmquist, J.K. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J. Comp. Neurol. 2003, 457, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G.; Mortrud, M.T.; Low, M.J.; Simerly, R.B.; Cone, R.D. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol. Endocrinol. 1994, 8, 1298–1308. [Google Scholar] [PubMed]
- Spaccapelo, L.; Bitto, A.; Galantucci, M.; Ottani, A.; Irrera, N.; Minutoli, L.; Altavilla, D.; Novellino, E.; Grieco, P.; Zaffe, D.; et al. Melanocortin MC(4) receptor agonists counteract late inflammatory and apoptotic responses and improve neuronal functionality after cerebral ischemia. Eur. J. Pharmacol. 2011, 670, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Browning, K.N.; Coleman, F.H.; Sutton, G.; Zheng, H.; Butler, A.; Berthoud, H.R.; Travagli, R.A. Presynaptic melanocortin-4 receptors on vagal afferent fibers modulate the excitability of rat nucleus tractus solitarius neurons. J. Neurosci. 2008, 28, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.; Kaplan, J.M.; Grill, H.J. The role of the dorsal vagal complex and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology 2000, 141, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Yang, H.; Ulloa, L.; Al-Abed, Y.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Lipton, J.M. Peptide modulation of fever and inflammation within the brain. Ann. N. Y. Acad. Sci. 1998, 856, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Tanida, M.; Shintani, N.; Hashimoto, H. The melanocortin system is involved in regulating autonomic nerve activity through central pituitary adenylate cyclase-activating polypeptide. Neurosci. Res. 2011, 70, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.; Balthasar, N.; Olson, D.; Scott, M.; Berglund, E.; Lee, C.E.; Choi, M.J.; Lauzon, D.; Lowell, B.B.; Elmquist, J.K. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011, 13, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Agosti, F.; Cordisco Gonzalez, S.; Martinez Damonte, V.; Tolosa, M.J.; Di Siervi, N.; Schioth, H.B.; Davio, C.; Perello, M.; Raingo, J. Melanocortin 4 receptor constitutive activity inhibits L-type voltage-gated calcium channels in neurons. Neuroscience 2017, 346, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, D.; Neri, L.; Canalini, F.; Calevro, A.; Ottani, A.; Vandini, E.; Sena, P.; Zaffe, D.; Guarini, S. NDP-alpha-MSH induces intense neurogenesis and cognitive recovery in Alzheimer transgenic mice through activation of melanocortin MC4 receptors. Mol. Cell. Neurosci. 2015, 67, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lisak, R.P.; Nedelkoska, L.; Bealmear, B.; Benjamins, J.A. Melanocortin receptor agonist ACTH 1–39 protects rat forebrain neurons from apoptotic, excitotoxic and inflammation-related damage. Exp. Neurol. 2015, 273, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Carniglia, L.; Durand, D.; Scimonelli, T.N.; Lasaga, M. Astrocytes: New targets of melanocortin 4 receptor actions. J. Mol. Endocrinol. 2013, 51, R33–R50. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Durand, D.; Schioth, H.B.; Rey, R.; Seilicovich, A.; Lasaga, M. Activation of melanocortin 4 receptors reduces the inflammatory response and prevents apoptosis induced by lipopolysaccharide and interferon-gamma in astrocytes. Endocrinology 2007, 148, 4918–4926. [Google Scholar] [CrossRef] [PubMed]
- Zohar, M.; Salomon, Y. Melanocortins stimulate proliferation and induce morphological changes in cultured rat astrocytes by distinct transducing mechanisms. Brain Res. 1992, 576, 49–58. [Google Scholar] [CrossRef]
- Ramirez, D.; Saba, J.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Melanocortin 4 receptor activates ERK-cFos pathway to increase brain-derived neurotrophic factor expression in rat astrocytes and hypothalamus. Mol. Cell. Endocrinol. 2015, 411, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Carniglia, L.; Durand, D.; Caruso, C.; Lasaga, M. Effect of NDP-alpha-MSH on PPAR-gamma and -beta expression and anti-inflammatory cytokine release in rat astrocytes and microglia. PLoS ONE 2013, 8, e57313. [Google Scholar] [CrossRef] [PubMed]
- Delgado, R.; Carlin, A.; Airaghi, L.; Demitri, M.T.; Meda, L.; Galimberti, D.; Baron, P.; Lipton, J.M.; Catania, A. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J. Leukoc. Biol. 1998, 63, 740–745. [Google Scholar] [PubMed]
- Carniglia, L.; Ramirez, D.; Durand, D.; Saba, J.; Caruso, C.; Lasaga, M. [Nle4, D-Phe7]-alpha-MSH Inhibits Toll-Like Receptor (TLR)2- and TLR4-Induced Microglial Activation and Promotes a M2-Like Phenotype. PLoS ONE 2016, 11, e0158564. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, R.; Yadegarazadi, M.J.; Amini, K. Peripheral nerve regeneration following transection injury to rat sciatic nerve by local application of adrenocorticotropic hormone. J. Craniomaxillofac. Surg. 2014, 42, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Teare, K.A.; Pearson, R.G.; Shakesheff, K.M.; Haycock, J.W. Alpha-MSH inhibits inflammatory signalling in Schwann cells. Neuroreport 2004, 15, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Dyer, J.K.; Philipsen, H.L.; Tonnaer, J.A.; Hermkens, P.H.; Haynes, L.W. Melanocortin analogue Org2766 binds to rat Schwann cells, upregulates NGF low-affinity receptor p75, and releases neurotrophic activity. Peptides 1995, 16, 515–522. [Google Scholar] [CrossRef]
- Van der Zee, C.E.; Brakkee, J.H.; Gispen, W.H. alpha-MSH and Org.2766 in peripheral nerve regeneration: Different routes of delivery. Eur. J. Pharmacol. 1988, 147, 351–357. [Google Scholar] [CrossRef]
- Lisak, R.; Kies, M. Experimental allergic encephalomyelitis as a tool for evaluating immunsuppressant activity of drugs. In Immunopharmacology; Rosenthale, M., Mansmann, H., Jr., Eds.; Spectrum Publications: New York, NY, USA, 1976; pp. 173–185. [Google Scholar]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Moyer, A.W.; Jervis, G.A.; Black, J.; Koprowski, H.; Cox, H.R. Action of adrenocorticotropic hormone (ACTH) in experimental allergic encephalomyelitis of the guinea pig. Proc. Soc. Exp. Biol. Med. 1950, 75, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Cusick, M.F.; Libbey, J.E.; Oh, L.; Jordan, S.; Fujinami, R.S. Acthar gel treatment suppresses acute exacerbations in a murine model of relapsing-remitting multiple sclerosis. Autoimmunity 2015, 48, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W.; Kitaichi, N. The diminishment of experimental autoimmune encephalomyelitis (EAE) by neuropeptide alpha-melanocyte stimulating hormone (alpha-MSH) therapy. Brain Behav. Immun. 2008, 22, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Luby, T.M.; Chen, H.; Etemad-Moghadam, B.; Lee, D.; Aziz, N.; Ramstedt, U.; Hedley, M.L. Generation of expression constructs that secrete bioactive alphaMSH and their use in the treatment of experimental autoimmune encephalomyelitis. Gene Ther. 2003, 10, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Duckers, H.J.; Verhaagen, J.; de Bruijn, E.; Gispen, W.H. Effective use of a neurotrophic ACTH4-9 analogue in the treatment of a peripheral demyelinating syndrome (experimental allergic neuritis). An intervention study. Brain 1994, 117, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Han, D.; Hong, J.; Zhang, H.; Ying, Y.; Tian, Y.; Zhang, L.; Lin, J. SValpha-MSH, a novel alpha-melanocyte stimulating hormone analog, ameliorates autoimmune encephalomyelitis through inhibiting autoreactive CD4(+) T cells activation. J. Neuroimmunol. 2014, 269, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Mykicki, N.; Herrmann, A.M.; Schwab, N.; Deenen, R.; Sparwasser, T.; Limmer, A.; Wachsmuth, L.; Klotz, L.; Köhrer, K.; Faber, C.; et al. Melanocortin-1 receptor activation is neuroprotective in mouse models of neuroinflammatory disease. Sci. Transl. Med. 2016, 8, 362ra146. [Google Scholar] [CrossRef] [PubMed]
- Forslin Aronsson, A.; Spulber, S.; Oprica, M.; Winblad, B.; Post, C.; Schultzberg, M. Alpha-MSH rescues neurons from excitotoxic cell death. J. Mol. Neurosci. 2007, 33, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Gatti, S.; Lonati, C.; Acerbi, F.; Sordi, A.; Leonardi, P.; Carlin, A.; Gaini, S.M.; Catania, A. Protective action of NDP-MSH in experimental subarachnoid hemorrhage. Exp. Neurol. 2012, 234, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Schaible, E.V.; Steinstrasser, A.; Jahn-Eimermacher, A.; Luh, C.; Sebastiani, A.; Kornes, F.; Pieter, D.; Schafer, M.K.; Engelhard, K.; Thal, S.C. Single administration of tripeptide alpha-MSH(11–13) attenuates brain damage by reduced inflammation and apoptosis after experimental traumatic brain injury in mice. PLoS ONE 2013, 8, e71056. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, W.A.; Schotman, P.; Jennekens, F.G.; Gispen, W.H.; De Wied, D. The enhanced recovery of sensorimotor function in rats is related to the melanotropic moiety of ACTH/MSH neuropeptides. Eur. J. Pharmacol. 1983, 92, 231–236. [Google Scholar] [CrossRef]
- Gispen, W.H.; Verhaagen, J.; Bar, D. ACTH/MSH-derived peptides and peripheral nerve plasticity: Neuropathies, neuroprotection and repair. Prog. Brain Res. 1994, 100, 223–229. [Google Scholar] [PubMed]
- Bar, P.R.; Mandys, V.; Turecek, R.; Gispen, W.H. Alpha-melanocyte-stimulating hormone has protective properties against the toxic effect of cisplatin on cultured dorsal root ganglia. Ann. N. Y. Acad. Sci. 1993, 680, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Chai, B.; Li, J.Y.; Zhang, W.; Newman, E.; Ammori, J.; Mulholland, M.W. Melanocortin-4 receptor-mediated inhibition of apoptosis in immortalized hypothalamic neurons via mitogen-activated protein kinase. Peptides 2006, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Bar, P.R.; Hol, E.M.; Gispen, W.H. Trophic effects of melanocortins on neuronal cells in culture. Ann. N. Y. Acad. Sci. 1993, 692, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Joosten, E.A.; Verhaagh, S.; Martin, D.; Robe, P.; Franzen, R.; Hooiveld, M.; Doornbos, R.; Bar, P.R.; Moonen, G. Alpha-MSH stimulates neurite outgrowth of neonatal rat corticospinal neurons in vitro. Brain Res 1996, 736, 91–98. [Google Scholar] [CrossRef]
- Benjmains, J.; Nedelkoska, L.; Lisak, R. Melanocortin receptor subtypes are expressed on cells in the oligodendroglial lineage and signal ACTH protection. J. Neurosci. Res. 2017, in press. [Google Scholar]
- Benjamins, J.A.; Nedelkoska, L.; Bealmear, B.; Lisak, R.P. ACTH protects mature oligodendroglia from excitotoxic and inflammation-related damage in vitro. Glia 2013, 61, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Benjamins, J.A.; Nedelkoska, L.; Lisak, R.P. Adrenocorticotropin hormone 1–39 promotes proliferation and differentiation of oligodendroglial progenitor cells and protects from excitotoxic and inflammation-related damage. J. Neurosci. Res. 2014, 92, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Lisak, R.P.; Nedelkoska, L.; Benjamins, J.A. The melanocortin ACTH 1–39 promotes protection of oligodendrocytes by astroglia. J. Neurol. Sci. 2016, 362, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Lisak, R.; Nedellkoska, L.; Benjamins, J. Melanocortin receptor ACTH 1–39 may protect oigodendroglia by inhibiting prtein kinase C. In Proceedings of the Annual Meeting of the American Academy of Neurology, Vancouver, BC, Canada, 15–21 April 2016. [Google Scholar]
- Baram, T.Z.; Mitchell, W.G.; Tournay, A.; Snead, O.C.; Hanson, R.A.; Horton, E.J. High-dose corticotropin (ACTH) versus prednisone for infantile spasms: A prospective, randomized, blinded study. Pediatrics 1996, 97, 375–379. [Google Scholar] [PubMed]
- Stafstrom, C.E.; Arnason, B.G.; Baram, T.Z.; Catania, A.; Cortez, M.A.; Glauser, T.A.; Pranzatelli, M.R.; Riikonen, R.; Rogawski, M.A.; Shinnar, S.; et al. Treatment of infantile spasms: Emerging insights from clinical and basic science perspectives. J. Child Neurol. 2011, 26, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Shumiloff, N.A.; Lam, W.M.; Manasco, K.B. Adrenocorticotropic hormone for the treatment of West Syndrome in children. Ann. Pharmacother. 2013, 47, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Nalin, A.; Facchinetti, F.; Galli, V.; Petraglia, F.; Storchi, R.; Genazzani, A.R. Reduced ACTH content in cerebrospinal fluid of children affected by cryptogenic infantile spasms with hypsarrhythmia. Epilepsia 1985, 26, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.Z.; Mitchell, W.G.; Snead, O.C., 3rd; Horton, E.J.; Saito, M. Brain-adrenal axis hormones are altered in the CSF of infants with massive infantile spasms. Neurology 1992, 42, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Nagamitsu, S.; Matsuishi, T.; Yamashita, Y.; Shimizu, T.; Iwanaga, R.; Murakami, Y.; Miyazaki, M.; Hashimoto, T.; Kato, H. Decreased cerebrospinal fluid levels of beta-endorphin and ACTH in children with infantile spasms. J. Neural Transm. 2001, 108, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Rogawski, M.A. Stress-induced deoxycorticosterone-derived neurosteroids modulate GABA(A) receptor function and seizure susceptibility. J. Neurosci. 2002, 22, 3795–3805. [Google Scholar] [PubMed]
- Pranzatelli, M.R.; Chun, K.Y.; Moxness, M.; Tate, E.D.; Allison, T.J. Cerebrospinal fluid ACTH and cortisol in opsoclonus-myoclonus: Effect of therapy. Pediatr. Neurol. 2005, 33, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Glaser, G.H.; Merritt, H.H. Effects of ACTH and cortisone in multiple sclerosis. Trans. Am. Neurol. Assoc. 1951, 56, 130–133. [Google Scholar] [PubMed]
- Miller, H.; Newell, D.J.; Ridley, A. Multiple sclerosis. Treatment of acute exacerbations with corticotrophin (A.C.T.H.). Lancet 1961, 2, 1120–1122. [Google Scholar] [CrossRef]
- Filippini, G.; Brusaferri, F.; Sibley, W.A.; Citterio, A.; Ciucci, G.; Midgard, R.; Candelise, L. Corticosteroids or ACTH for acute exacerbations in multiple sclerosis. Cochrane Database Syst. Rev. 2000. [Google Scholar] [CrossRef]
- Rose, A.S.; Kuzma, J.W.; Kurtzke, J.F.; Namerow, N.S.; Sibley, W.A.; Tourtellotte, W.W. Cooperative study in the evaluation of therapy in multiple sclerosis. ACTH vs. placebo—Final report. Neurology 1970, 20, 1–59. [Google Scholar] [PubMed]
- Rose, A.S.; Kuzma, J.W.; Kurtzke, J.F.; Sibley, W.A.; Tourtellotte, W.W. Cooperative study in the evaluation of therapy in multiple sclerosis; ACTH vs. placebo in acute exacerbations. Preliminary report. Neurology 1968, 18, 1–10. [Google Scholar]
- Thompson, A.J.; Kennard, C.; Swash, M.; Summers, B.; Yuill, G.M.; Shepherd, D.I.; Roche, S.; Perkin, G.D.; Loizou, L.A.; Ferner, R.; et al. Relative efficacy of intravenous methylprednisolone and ACTH in the treatment of acute relapse in MS. Neurology 1989, 39, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Bell, S.; Challenger, R.; Hammock, V.; Nyberg, M.; Decker, D.; Becker, P.M.; Young, D. Pharmacodynamics and tolerability of repository corticotropin injection in healthy human subjects: A comparison with intravenous methylprednisolone. J. Clin. Pharmacol. 2016, 56, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Milanese, C.; La Mantia, L.; Salmaggi, A.; Campi, A.; Eoli, M.; Scaioli, V.; Nespolo, A.; Corridori, F. Double-blind randomized trial of ACTH versus dexamethasone versus methylprednisolone in multiple sclerosis bouts. Clinical, cerebrospinal fluid and neurophysiological results. Eur. Neurol. 1989, 29, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Berkovich, R.; Bakshi, R.; Amezcua, L.; Axtell, R.C.; Cen, S.Y.; Tauhid, S.; Neema, M.; Steinman, L. Adrenocorticotropic hormone versus methylprednisolone added to interferon beta in patients with multiple sclerosis experiencing breakthrough disease: A randomized, rater-blinded trial. Ther. Adv. Neurol. Disord. 2017, 10, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Tourtellotte, W.W.; Baumhefner, R.W.; Potvin, A.R.; Ma, B.I.; Potvin, J.H.; Mendez, M.; Syndulko, K. Multiple sclerosis de novo CNS IgG synthesis: Effect of ACTH and corticosteroids. Neurology 1980, 30, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, C.; Codella, M.; Rocca, M.A.; Gasperini, C.; Capra, R.; Bastianello, S.; Filippi, M. Disease activity in multiple sclerosis studied by weekly triple-dose magnetic resonance imaging. J. Neurol. 1999, 246, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Wolansky, L.J.; Skurnick, J.; Lincoln, J.; Cheriyan, J.; Szczepanowski, K.; Kamin, S.S.; Pachner, A.R.; Halper, J.; Cook, S.D. Efficacy of treatment of MS with IFNbeta-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology 2009, 72, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.D.; Dhib-Jalbut, S.; Dowling, P.; Durelli, L.; Ford, C.; Giovannoni, G.; Halper, J.; Harris, C.; Herbert, J.; Li, D.; et al. Use of Magnetic Resonance Imaging as Well as Clinical Disease Activity in the Clinical Classification of Multiple Sclerosis and Assessment of Its Course: A Report from an International CMSC Consensus Conference, March 5–7, 2010. Int. J. MS Care 2012, 14, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Campi, A.; Martinelli, V.; Colombo, B.; Yousry, T.; Canal, N.; Scotti, G.; Comi, G. Comparison of triple dose versus standard dose gadolinium-DTPA for detection of MRI enhancing lesions in patients with primary progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 1995, 59, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, J.S.; Narayana, P.A.; O’Connor, P.; Coyle, P.K.; Ford, C.; Johnson, K.; Miller, A.; Pardo, L.; Kadosh, S.; Ladkani, D. Glatiramer acetate in primary progressive multiple sclerosis: Results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann. Neurol. 2007, 61, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.P.; Montalban, X.; Uitdehaag, B.M.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, J.M.; Glanz, B.I.; Healy, B.C.; Arora, A.; Neema, M.; Benedict, R.H.; Guss, Z.D.; Tauhid, S.; Buckle, G.J.; Houtchens, M.K.; et al. Brain MRI lesion load at 1.5T and 3T versus clinical status in multiple sclerosis. J. Neuroimaging 2011, 21, e50–e56. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M.; Gerevini, S.; Filippi, M.; Falini, A. HIgh-field strength MRI (3.0T or more) in white matter diseases. In High Field Brain MRI. Use in Clincal Practice; Scarabino, T., Pollice, S., Popolizio, T., Eds.; Springer International PUblishing: Basel, Switzerland, 2017. [Google Scholar]
- Vorbrodt, A.W.; Lassmann, H.; Wisniewski, H.M.; Lossinsky, A.S. Ultracytochemical studies of the blood-meningeal barrier (BMB) in rat spinal cord. Acta Neuropathol. 1981, 55, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Angelov, D.N. Ultrastructural investigation of the meningeal compartment of the blood-cerebrospinal fluid-barrier in rats and cats. A horseradish peroxidase study. Z. Mikrosk. Anat. Forsch. 1990, 104, 1–16. [Google Scholar] [PubMed]
- Zheng, W.; Zhao, Q.; Graziano, J.H. Primary culture of choroidal epithelial cells: Characterization of an in vitro model of blood-CSF barrier. In Vitro Cell. Dev. Biol. Anim. 1998, 34, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Drewes, L.R. What is the blood-brain barrier? A molecular perspective. Cerebral vascular biology. Adv. Exp. Med. Biol. 1999, 474, 111–122. [Google Scholar] [PubMed]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P. Pericytes: Pluripotent cells of the blood brain barrier. Curr. Pharm. Des. 2008, 14, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Blood-brain barrier: Recent developments and clinical correlations. Neurology 2012, 78, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Monnot, A.D.; Zheng, W. Culture of choroid plexus epithelial cells and in vitro model of blood-CSF barrier. Methods Mol. Biol. 2013, 945, 13–29. [Google Scholar] [PubMed]
- Chaput, N.; Thery, C. Exosomes: Immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440. [Google Scholar] [CrossRef] [PubMed]
- Pusic, A.; Lusic, K.; Kraig, R. What are exosomes and how can they be used in multiple sclerosis therapy? Expert Rev. Neurother. 2014, 14, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, I.; Mycko, M.P.; Raine, C.S.; Selmaj, K.W. The role of exosomes in CNS inflammation and their involvement in multiple sclerosis. J. Neuroimmunol. 2017, 306, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target. 2002, 10, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gelperina, S.E.; Khalansky, A.S.; Skidan, I.N.; Smirnova, Z.S.; Bobruskin, A.I.; Severin, S.E.; Turowski, B.; Zanella, F.E.; Kreuter, J. Toxicological studies of doxorubicin bound to polysorbate 80-coated poly(butyl cyanoacrylate) nanoparticles in healthy rats and rats with intracranial glioblastoma. Toxicol. Lett. 2002, 126, 131–141. [Google Scholar] [CrossRef]
- LaVan, D.A.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, T.K.; Castrucci, A.M.; Staples, D.J.; Affholter, J.A.; De Vaux, A.; Hruby, V.J.; Hadley, M.E. Structure-activity relationships of [Nle4, D-Phe7]alpha-MSH. Discovery of a tripeptidyl agonist exhibiting sustained bioactivity. Ann. N. Y. Acad. Sci. 1993, 680, 597–599. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Toxic Agent | OL | OPC | Neurons |
|---|---|---|---|
| Glutamate | + | + | + |
| Staurosporine | + | + | + |
| Quinolinic acid | + | + | + |
| Kynurenic acid | none | none | none |
| H2O2 (reactive oxygen species | + | + | + |
| Nitric oxide (slow release) | none | slight | none |
| Nitric oxide (rapid release) | none | none | slight |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisak, R.P.; Benjamins, J.A. Melanocortins, Melanocortin Receptors and Multiple Sclerosis. Brain Sci. 2017, 7, 104. https://doi.org/10.3390/brainsci7080104
Lisak RP, Benjamins JA. Melanocortins, Melanocortin Receptors and Multiple Sclerosis. Brain Sciences. 2017; 7(8):104. https://doi.org/10.3390/brainsci7080104
Chicago/Turabian StyleLisak, Robert P., and Joyce A. Benjamins. 2017. "Melanocortins, Melanocortin Receptors and Multiple Sclerosis" Brain Sciences 7, no. 8: 104. https://doi.org/10.3390/brainsci7080104
APA StyleLisak, R. P., & Benjamins, J. A. (2017). Melanocortins, Melanocortin Receptors and Multiple Sclerosis. Brain Sciences, 7(8), 104. https://doi.org/10.3390/brainsci7080104