Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
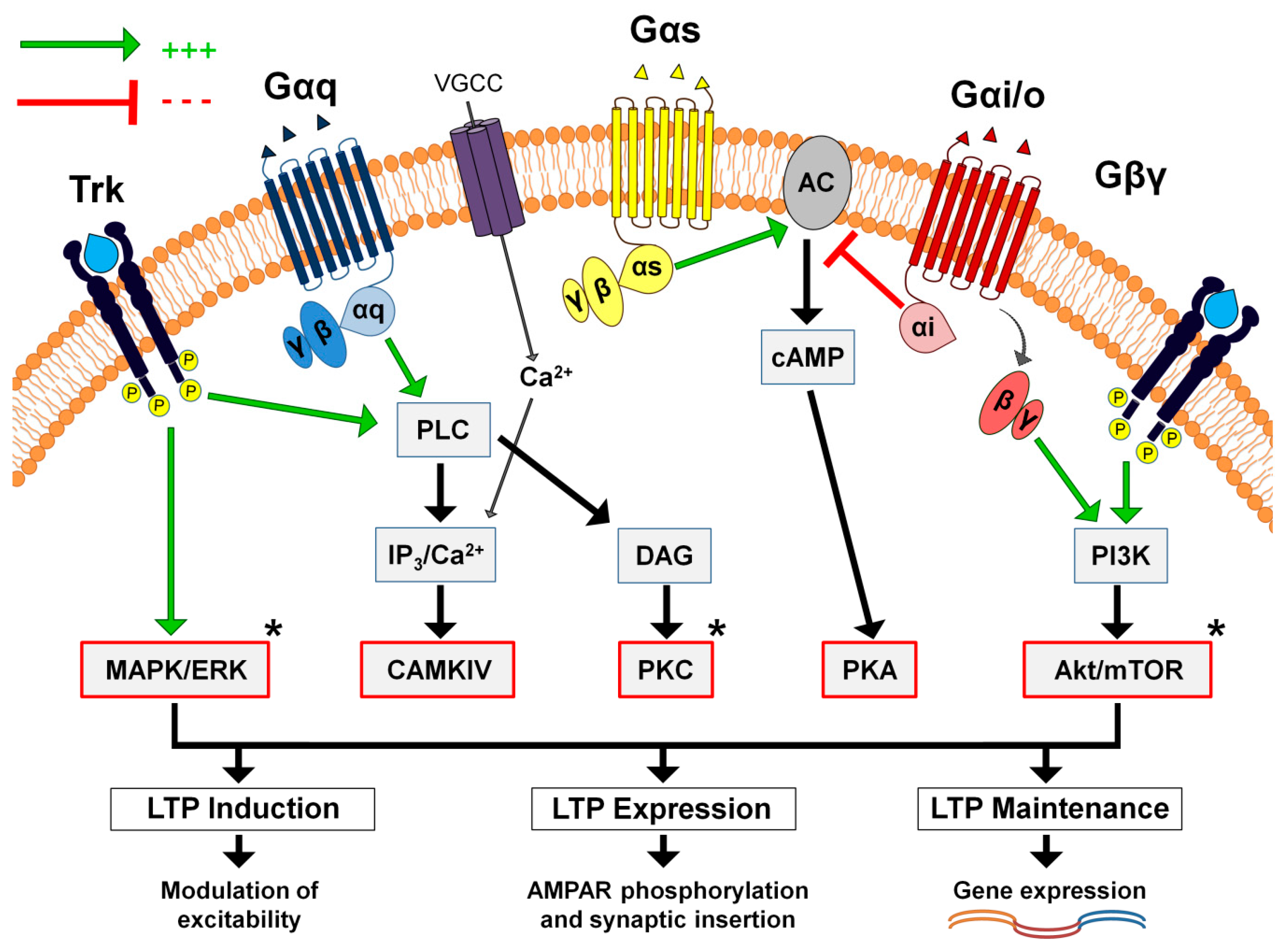
2. Metabotropic Pathways Are Necessary for Long-Term Synaptic Plasticity
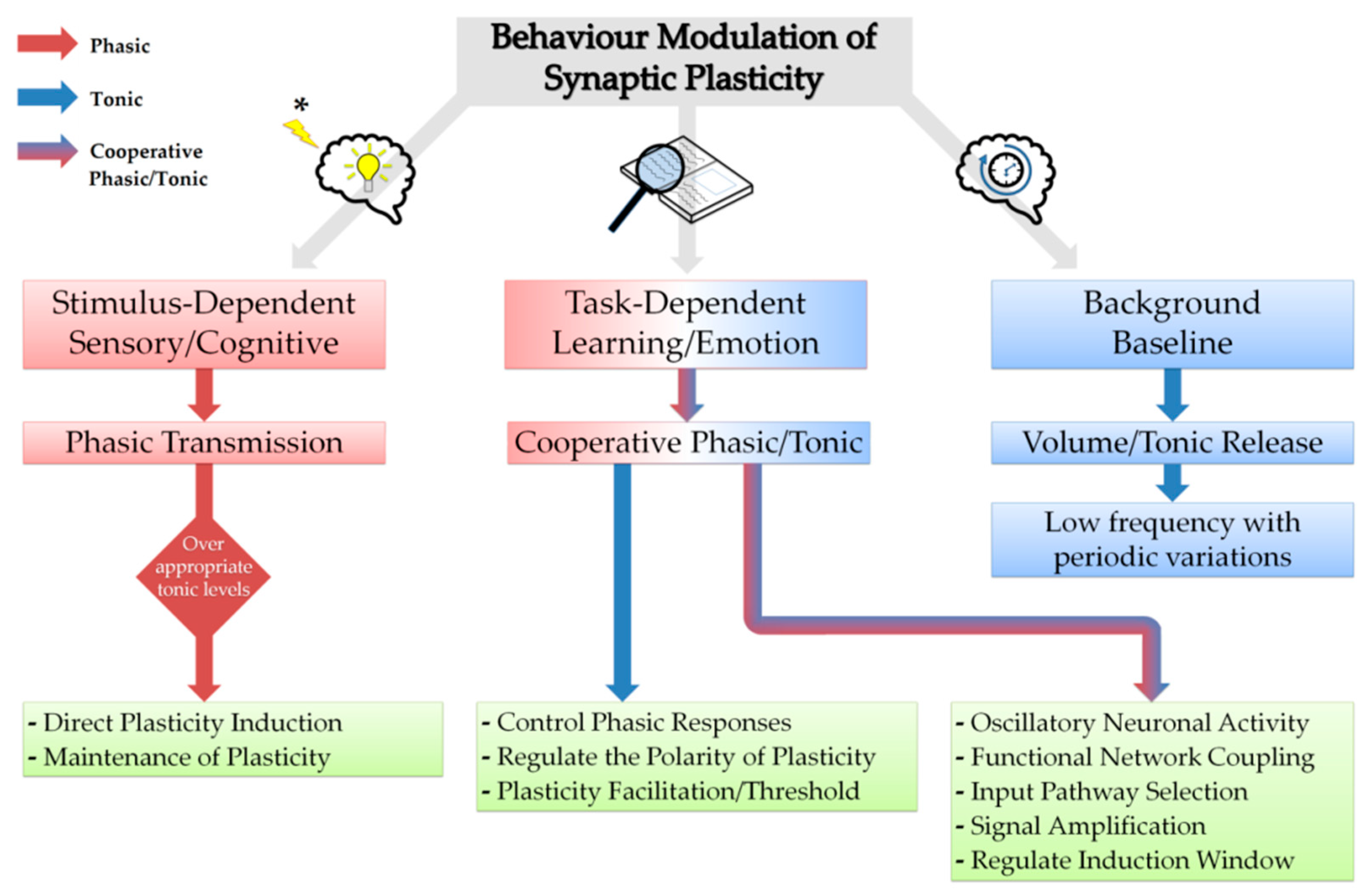
3. Neuromodulators in Synchronisation and Synaptic Plasticity
4. Transmission System-Based GPCR Modulation of Plasticity
4.1. Dopaminergic Transmission
4.1.1. Mesocortical Pathway
4.1.2. Nigrostriatal Pathway
4.1.3. Mesolimbic Pathway
4.1.4. Hippocampal Dopamine
4.2. Cholinergic Transmission in the Hippocampus
4.3. Adrenergic Transmission
4.4. Serotonergic Transmission
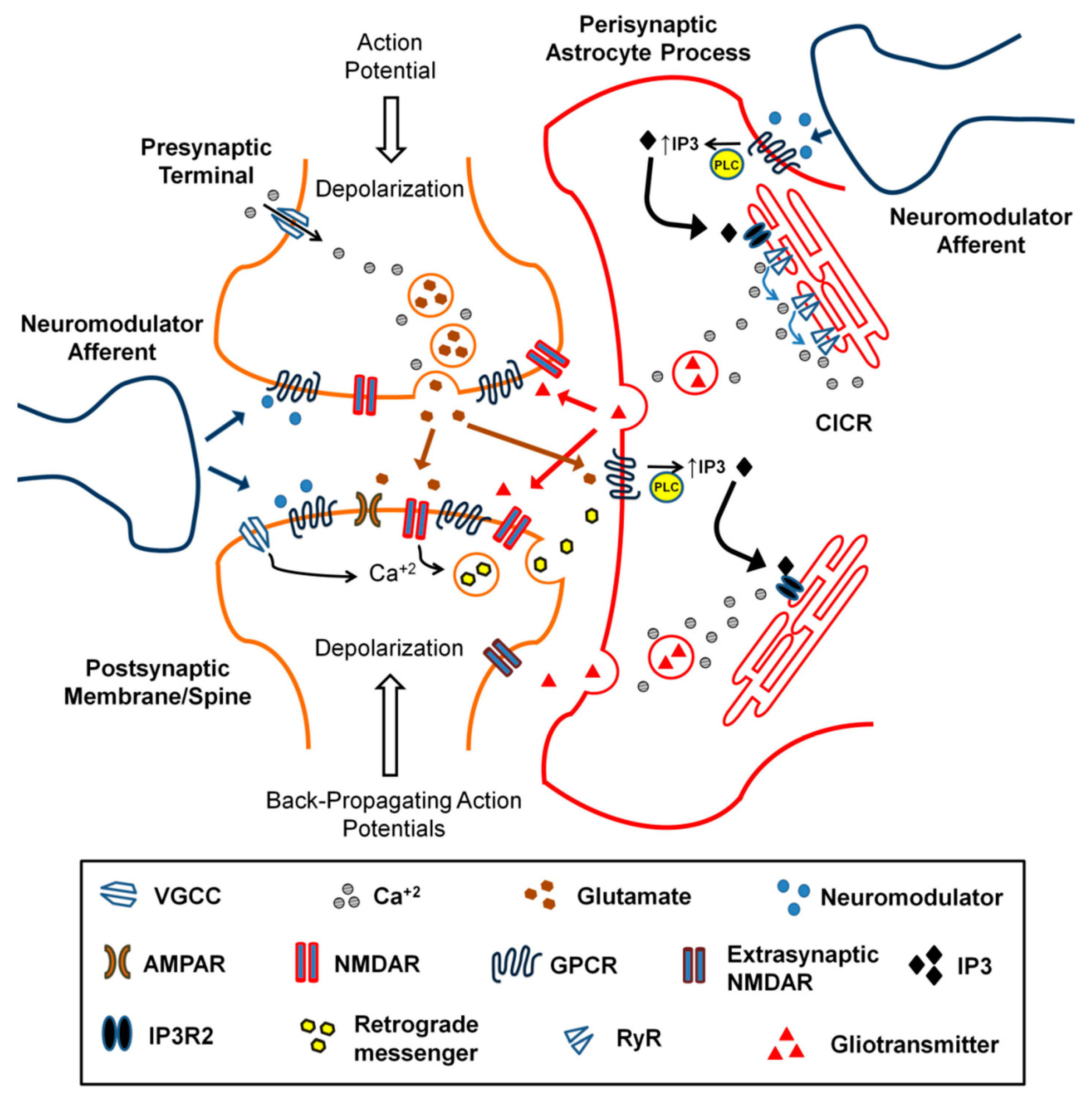
5. Astrocytes and Neuromodulators
6. Neuromodulators Steer Glutamatergic Transmission and Plasticity
Author Contributions
Conflicts of Interest
References
- Bliss, T.V.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Holtmaat, A.; Caroni, P. Functional and structural underpinnings of neuronal assembly formation in learning. Nat. Neurosci. 2016, 19, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Caroni, P.; Donato, F.; Muller, D. Structural plasticity upon learning: Regulation and functions. Nat. Rev. Neurosci. 2012, 13, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.C. How long will long-term potentiation last? Phil. Trans. R. Soc. Lond. B 2003, 358, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Reymann, K.G.; Frey, J.U. The late maintenance of hippocampal LTP: Requirements, phases,’synaptic tagging’,’late-associativity’and implications. Neuropharmacology 2007, 52, 24–40. [Google Scholar] [CrossRef]
- Wang, H.; Feng, R.; Wang, L.P.; Li, F.; Cao, X.; Tsien, J.Z. CaMKII activation state underlies synaptic labile phase of LTP and short-term memory formation. Curr. Biol. 2008, 18, 1546–1554. [Google Scholar] [CrossRef]
- Rossetti, T.; Banerjee, S.; Kim, C.; Leubner, M.; Lamar, C.; Gupta, P.; Lee, B.; Neve, R.; Lisman, J. Memory erasure experiments indicate a critical role of CaMKII in memory storage. Neuron 2017, 96, 207–216. [Google Scholar] [CrossRef]
- Juárez-Muñoz, Y.; Ramos-Languren, L.E.; Escobar, M.L. CaMKII Requirement for in Vivo Insular Cortex LTP Maintenance and CTA Memory Persistence. Front. Pharmacol. 2017, 8, 822. [Google Scholar] [CrossRef]
- Sanhueza, M.; Lisman, J. The CaMKII/NMDAR complex as a molecular memory. Mol. Brain 2013, 6, 10. [Google Scholar] [CrossRef]
- Chen, H.X.; Otmakhov, N.; Strack, S.; Colbran, R.J.; Lisman, J.E. Is persistent activity of calcium/calmodulin-dependent kinase required for the maintenance of LTP? J. Neurophysiol. 2001, 85, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Shin, M.E.; Parra-Bueno, P.; Szatmari, E.M.; Shibata, A.C.; Yasuda, R. Kinetics of endogenous CaMKII required for synaptic plasticity revealed by optogenetic kinase inhibitor. Neuron 2017, 94, 37–47. [Google Scholar] [CrossRef]
- Buard, I.; Coultrap, S.J.; Freund, R.K.; Lee, Y.S.; Dell’Acqua, M.L.; Silva, A.J.; Bayer, K.U. CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J. Neurosci. 2010, 30, 8214–8220. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, T.C.; Fenton, A.A. What does LTP tell us about the roles of CaMKII and PKMζ in memory? Mol. Brain 2018, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.R.; Barondes, S.H. Anisomycin, like other inhibitors of cerebral protein synthesis, impairs ‘long-term’memory of a discrimination task. Brain Res. 1974, 66, 301–308. [Google Scholar] [CrossRef]
- Frey, U.; Krug, M.; Reymann, K.G.; Matthies, H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988, 452, 57–65. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Li, X.C.; Kandel, E.R. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell 1994, 79, 69–79. [Google Scholar] [CrossRef]
- Nguyen, P.V.; Kandel, E.R. A macromolecular synthesis-dependent late phase of long-term potentiation requiring cAMP in the medial perforant pathway of rat hippocampal slices. J. Neurosci. 1996, 16, 3189–3198. [Google Scholar] [CrossRef]
- Abate, G.; Colazingari, S.; Accoto, A.; Conversi, D.; Bevilacqua, A. Dendritic spine density and EphrinB2 levels of hippocampal and anterior cingulate cortex neurons increase sequentially during formation of recent and remote fear memory in the mouse. Behav. Brain Res. 2018, 344, 120–131. [Google Scholar] [CrossRef]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Bramham, C.R.; Alme, M.N.; Bittins, M.; Kuipers, S.D.; Nair, R.R.; Pai, B.; Panja, D.; Schubert, M.; Soule, J.; Tiron, A.; et al. The Arc of synaptic memory. Exp. Brain Res. 2010, 200, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, B.E.; Huber, K.M. Current advances in local protein synthesis and synaptic plasticity. J. Neurosci. 2006, 26, 7147–7150. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R. Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol. 2008, 18, 524–531. [Google Scholar] [CrossRef]
- Park, S.; Park, J.M.; Kim, S.; Kim, J.A.; Shepherd, J.D.; Smith-Hicks, C.L.; Chowdhury, S.; Kaufmann, W.; Kuhl, D.; Ryazanov, A.G.; et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3. 1 essential for mGluR-LTD. Neuron 2008, 59, 70–83. [Google Scholar] [CrossRef]
- Dore, K.; Aow, J.; Malinow, R. The emergence of NMDA receptor metabotropic function: Insights from imaging. Front. Synaptic Neurosci. 2016, 8, 20. [Google Scholar] [CrossRef]
- Grover, L.M.; Teyler, T.J. Two components of long-term potentiation induced by different patterns of afferent activation. Nature 1990, 347, 477–479. [Google Scholar] [CrossRef]
- Pigott, B.M.; Garthwaite, J. Nitric oxide is required for L-type Ca2+ channel-dependent long-term potentiation in the hippocampus. Front. Synaptic Neurosci. 2016, 8, 17. [Google Scholar] [CrossRef]
- Wang, Y.; Rowan, M.J.; Anwyl, R. Induction of LTD in the dentate gyrus in vitro is NMDA receptor independent, but dependent on Ca2+ influx via low-voltage–activated Ca2+ channels and release of Ca2+ from intracellular stores. J. Neurophysiol. 1997, 77, 812–825. [Google Scholar] [CrossRef]
- Wang, H.; Ardiles, A.O.; Yang, S.; Tran, T.; Posada-Duque, R.; Valdivia, G.; Baek, M.; Chuang, Y.A.; Palacios, A.G.; Gallagher, M.; et al. Metabotropic glutamate receptors induce a form of LTP controlled by translation and arc signaling in the hippocampus. J. Neurosci. 2016, 36, 1723–1729. [Google Scholar] [CrossRef]
- Daniel, H.; Hemart, N.; Jaillard, D.; Crepel, F. Coactivation of metabotropic glutamate receptors and of voltage-gated calcium channels induces long-term depression in cerebellar Purkinje cells in vitro. Exp. Brain Res. 1992, 90, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.G.; Kang, S.J.; Shi, T.Y.; Koga, K.; Zhang, M.M.; Collingridge, G.L.; Kaang, B.K.; Zhuo, M. Long-term potentiation of synaptic transmission in the adult mouse insular cortex: Multielectrode array recordings. J. Neurophysiol. 2013, 110, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.V.; Woo, N.H. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog. Neurobiol. 2003, 71, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Sossin, W.S. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn. Mem. 2007, 14, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeffer, C.A.; Klann, E. mTOR signaling: At the crossroads of plasticity, memory and disease. Trends Neurosci. 2010, 33, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 2004, 14, 311–317. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- De Pittà, M.; Brunel, N.; Volterra, A. Astrocytes: Orchestrating synaptic plasticity? Neuroscience 2016, 323, 43–61. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Konnerth, A. Stores not just for storage: Intracellular calcium release and synaptic plasticity. Neuron 2001, 31, 519–522. [Google Scholar] [CrossRef]
- O’Riordan, K.J.; Hu, N.W.; Rowan, M.J. Physiological activation of mGlu5 receptors supports the ion channel function of NMDA receptors in hippocampal LTD induction in vivo. Sci. Rep. 2018, 8, 4391. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.; Gerber, U.; Ster, J. Activation of group II metabotropic glutamate receptors promotes LTP induction at schaffer collateral-CA1 pyramidal cell synapses by priming NMDA receptors. J. Neurosci. 2016, 36, 11521–11531. [Google Scholar] [CrossRef] [PubMed]
- Pöschel, B.; Wroblewska, B.; Heinemann, U.; Manahan-Vaughan, D. The metabotropic glutamate receptor mGluR3 is critically required for hippocampal long-term depression and modulates long-term potentiation in the dentate gyrus of freely moving rats. Cereb. Cortex 2005, 15, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Altinbilek, B.; Manahan-Vaughan, D. Antagonism of group III metabotropic glutamate receptors results in impairment of LTD but not LTP in the hippocampal CA1 region, and prevents long-term spatial memory. Eur. J. Neurosci. 2007, 26, 1166–1172. [Google Scholar] [CrossRef]
- Jung, S.J.; Kim, S.J.; Park, Y.K.; Oh, S.B.; Cho, K.; Kim, J. Group I mGluR regulates the polarity of spike-timing dependent plasticity in substantia gelatinosa neurons. Biochem. Biophys. Res. Commun. 2006, 347, 509–516. [Google Scholar] [CrossRef]
- Fujii, S.; Sasaki, H.; Mikoshiba, K.; Kuroda, Y.; Yamazaki, Y.; Taufiq, A.M.; Kato, H. A chemical LTP induced by co-activation of metabotropic and N-methyl-D-aspartate glutamate receptors in hippocampal CA1 neurons. Brain Res. 2004, 999, 20–28. [Google Scholar] [CrossRef]
- Takeuchi, T.; Duszkiewicz, A.J.; Morris, R.G. The synaptic plasticity and memory hypothesis: Encoding, storage and persistence. Phil. Trans. R. Soc. B. 2014, 369, 20130288. [Google Scholar] [CrossRef]
- McGann, J.P. Associative learning and sensory neuroplasticity: How does it happen and what is it good for? Learn. Mem. 2015, 22, 567–576. [Google Scholar] [CrossRef]
- Kida, H.; Mitsushima, D. Mechanisms of motor learning mediated by synaptic plasticity in rat primary motor cortex. Neurosci. Res. 2018, 128, 14–18. [Google Scholar] [CrossRef]
- Howland, J.G.; Wang, Y.T. Synaptic plasticity in learning and memory: Stress effects in the hippocampus. Prog. Brain. Res. 2008, 169, 145–158. [Google Scholar]
- Hong, I.; Kim, J.; Lee, J.; Park, S.; Song, B.; Kim, J.; An, B.; Park, K.; Lee, H.W.; Lee, S.; et al. Reversible plasticity of fear memory-encoding amygdala synaptic circuits even after fear memory consolidation. PLoS ONE 2011, 6, e24260. [Google Scholar] [CrossRef] [PubMed]
- Luchkina, N.V.; Bolshakov, V.Y. Mechanisms of fear learning and extinction: Synaptic plasticity–fear memory connection. Psychopharmacology 2019, 236, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Bocchio, M.; Nabavi, S.; Capogna, M. Synaptic plasticity, engrams, and network oscillations in amygdala circuits for storage and retrieval of emotional memories. Neuron 2017, 94, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Christoffel, D.J.; Golden, S.A.; Russo, S.J. Structural and synaptic plasticity in stress-related disorders. Rev. Neurosci. 2011, 22, 535–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pereira, A. Neuromodulation, emotional feelings and affective disorders. Mens Sana Monogr. 2016, 14, 5–29. [Google Scholar]
- Albensi, B.C.; Oliver, D.R.; Toupin, J.; Odero, G. Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability: Are they effective or relevant? Exp. Neurol. 2007, 204, 1–13. [Google Scholar] [CrossRef]
- Thut, G.; Miniussi, C.; Gross, J. The functional importance of rhythmic activity in the brain. Curr. Biol. 2012, 22, R658–R663. [Google Scholar] [CrossRef]
- Noh, N.A. Exploring cortical plasticity and oscillatory brain dynamics via transcranial magnetic stimulation and resting-state electroencephalogram. Malays. J. Med. Sci. 2016, 23, 5–16. [Google Scholar] [CrossRef]
- Winson, J. Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science 1978, 201, 160–163. [Google Scholar] [CrossRef]
- Köster, M.; Finger, H.; Graetz, S.; Kater, M.; Gruber, T. Theta-gamma coupling binds visual perceptual features in an associative memory task. Sci. Rep. 2018, 8, 17688. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Ravel, N. Beta and gamma oscillatory activities associated with olfactory memory tasks: Different rhythms for different functional networks? Front. Behav. Neurosci. 2014, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Seidenbecher, T.; Laxmi, T.R.; Stork, O.; Pape, H.C. Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science 2003, 301, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, D.S.; Kang, E.; Kang, H.; Hahm, J.; Kim, J.S.; Chung, C.K.; Jiang, H.; Gross, J.; Jensen, O. Formation of visual memories controlled by gamma power phase-locked to alpha oscillations. Sci. Rep. 2016, 6, 28092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.T.; Schuman, E.M. Frequency-dependent signal transmission and modulation by neuromodulators. Front. Neurosci. 2008, 2, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Alexander, G.M.; Dudek, S.M.; Yakel, J.L. Hippocampus and entorhinal cortex recruit cholinergic and NMDA receptors separately to generate hippocampal theta oscillations. Cell Rep. 2017, 21, 3585–3595. [Google Scholar] [CrossRef]
- Barkus, C.; Line, S.J.; Huber, A.; Capitao, L.; Lima, J.; Jennings, K.; Lowry, J.; Sharp, T.; Bannerman, D.M.; McHugh, S.B. Variation in serotonin transporter expression modulates fear-evoked hemodynamic responses and theta-frequency neuronal oscillations in the amygdala. Biol. Psychiat. 2014, 75, 901–908. [Google Scholar] [CrossRef]
- Narayanan, V.; Heiming, R.S.; Jansen, F.; Lesting, J.; Sachser, N.; Pape, H.C.; Seidenbecher, T. Social defeat: Impact on fear extinction and amygdala-prefrontal cortical theta synchrony in 5-HTT deficient mice. PLoS ONE 2011, 6, e22600. [Google Scholar] [CrossRef]
- Walling, S.G.; Brown, R.A.; Milway, J.S.; Earle, A.G.; Harley, C.W. Selective tuning of hippocampal oscillations by phasic locus coeruleus activation in awake male rats. Hippocampus 2011, 21, 1250–1262. [Google Scholar] [CrossRef]
- Lemon, N.; Aydin-Abidin, S.; Funke, K.; Manahan-Vaughan, D. Locus coeruleus activation facilitates memory encoding and induces hippocampal LTD that depends on β-adrenergic receptor activation. Cereb. Cortex 2009, 19, 2827–2837. [Google Scholar] [CrossRef]
- Andersson, R.H.; Johnston, A.; Herman, P.A.; Winzer-Serhan, U.H.; Karavanova, I.; Vullhorst, D.; Fisahn, A.; Buonanno, A. Neuregulin and dopamine modulation of hippocampal gamma oscillations is dependent on dopamine D4 receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 13118–13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemercier, C.E.; Schulz, S.B.; Heidmann, K.E.; Kovács, R.; Gerevich, Z. Dopamine D3 receptors inhibit hippocampal gamma oscillations by disturbing CA3 pyramidal cell firing synchrony. Front. Pharmacol. 2016, 6, 297. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, C.M.; Martin, S.J.; Sandin, J.; Frenguelli, B.; Morris, R.G. Dopaminergic modulation of the persistence of one-trial hippocampus-dependent memory. Learn. Mem. 2006, 13, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tononi, G.; Cirelli, C. Sleep function and synaptic homeostasis. Sleep Med. Rev. 2006, 10, 49–62. [Google Scholar] [CrossRef]
- Fellin, T.; Halassa, M.M.; Terunuma, M.; Succol, F.; Takano, H.; Frank, M.; Moss, S.J.; Haydon, P.G. Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 15037–15042. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.L.; Hardy, N.F.; Jimenez, D.V.; Maynard, K.R.; Kardian, A.S.; Pollock, C.J.; Schloesser, R.J.; Martinowich, K. Loss of promoter IV-driven BDNF expression impacts oscillatory activity during sleep, sensory information processing and fear regulation. Transl. Psychiat. 2016, 6, e873. [Google Scholar] [CrossRef]
- Shiromani, P.J.; Basheer, R.; Thakkar, J.; Wagner, D.; Greco, M.A.; Charness, M.E. Sleep and wakefulness in c-fos and fos B gene knockout mice. Mol. Brain Res. 2000, 80, 75–87. [Google Scholar] [CrossRef]
- Nieoullon, A. Dopamine and the regulation of cognition and attention. Prog. Neurobiol. 2002, 67, 53–83. [Google Scholar] [CrossRef]
- Jaber, M.; Robinson, S.W.; Missale, C.; Caron, M.G. Dopamine receptors and brain function. Neuropharmacology 1996, 35, 1503–1519. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef]
- Fox, M.E.; Mikhailova, M.A.; Bass, C.E.; Takmakov, P.; Gainetdinov, R.R.; Budygin, E.A.; Wightman, R.M. Cross-hemispheric dopamine projections have functional significance. Proc. Natl. Acad. Sci. USA 2016, 113, 6985–6990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, J.I. Co-transmission of dopamine and glutamate. J. Gen. Physiol. 2012, 139, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Roeper, J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 2013, 36, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.G.; Jang, S.H. Differences in neural connectivity between the substantia nigra and ventral tegmental area in the human brain. Front. Hum. Neurosci. 2014, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Decot, H.K.; Namboodiri, V.M.; Gao, W.; McHenry, J.A.; Jennings, J.H.; Lee, S.H.; Kantak, P.A.; Kao, Y.C.; Das, M.; Witten, I.B.; et al. Coordination of brain-wide activity dynamics by dopaminergic neurons. Neuropsychopharmacology 2017, 42, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.; Rose, J.; Schmidt, R.; Freund, N. Dopamine modulation of learning and memory in the prefrontal cortex: Insights from studies in primates, rodents, and birds. Front. Neural Circuit 2014, 8, 93. [Google Scholar] [CrossRef]
- Gao, M.; Liu, C.L.; Yang, S.; Jin, G.Z.; Bunney, B.S.; Shi, W.X. Functional coupling between the prefrontal cortex and dopamine neurons in the ventral tegmental area. J. Neurosci. 2007, 27, 5414–5421. [Google Scholar] [CrossRef]
- Sheynikhovich, D.; Otani, S.; Arleo, A. The role of tonic and phasic dopamine for long-term synaptic plasticity in the prefrontal cortex: A computational model. J. Physiol. Paris 2011, 105, 45–52. [Google Scholar] [CrossRef]
- Sheynikhovich, D.; Otani, S.; Arleo, A. Dopaminergic control of long-term depression/long-term potentiation threshold in prefrontal cortex. J. Neurosci. 2013, 33, 13914–13926. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Simpson, E.; Kellendonk, C.; Kandel, E.R. Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 3236–3241. [Google Scholar] [CrossRef] [Green Version]
- Gurden, H.; Takita, M.; Jay, T.M. Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J. Neurosci. 2000, 20, RC106. [Google Scholar] [CrossRef] [PubMed]
- Gurden, H.; Tassin, J.P.; Jay, T.M. Integrity of the mesocortical dopaminergic system is necessary for complete expression of in vivo hippocampal–prefrontal cortex long-term potentiation. Neuroscience 1999, 94, 1019–1027. [Google Scholar] [CrossRef]
- Law-Tho, D.; Desce, J.M.; Crepel, F. Dopamine favours the emergence of long-term depression versus long-term potentiation in slices of rat prefrontal cortex. Neurosci. Lett. 1995, 188, 125–128. [Google Scholar] [CrossRef]
- Matsuda, Y.; Marzo, A.; Otani, S. The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J. Neurosci. 2006, 26, 4803–4810. [Google Scholar] [CrossRef]
- Otani, S.; Auclair, N.; Desce, J.M.; Roisin, M.P.; Crépel, F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J. Neurosci. 1999, 19, 9788–9802. [Google Scholar] [CrossRef]
- Xu, T.X.; Yao, W.D. D1 and D2 dopamine receptors in separate circuits cooperate to drive associative long-term potentiation in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 16366–16371. [Google Scholar] [CrossRef] [Green Version]
- Laroche, S.; Davis, S.; Jay, T.M. Plasticity at hippocampal to prefrontal cortex synapses: Dual roles in working memory and consolidation. Hippocampus 2000, 10, 438–446. [Google Scholar] [CrossRef]
- Banks, P.J.; Burroughs, A.C.; Barker, G.R.; Brown, J.T.; Warburton, E.C.; Bashir, Z.I. Disruption of hippocampal–prefrontal cortex activity by dopamine D2R-dependent LTD of NMDAR transmission. Proc. Natl. Acad. Sci. USA 2015, 112, 11096–11101. [Google Scholar] [CrossRef]
- Seamans, J.K.; Durstewitz, D.; Christie, B.R.; Stevens, C.F.; Sejnowski, T.J. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 301–306. [Google Scholar] [CrossRef]
- Gao, C.; Wolf, M.E. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J. Neurochem. 2008, 106, 2489–2501. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhao, Y.; Wolf, M.E. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 2005, 25, 7342–7351. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.S.; Prémont, J.; Krebs, M.O.; Jay, T.M. Interaction of dopamine D1 with NMDA NR1 receptors in rat prefrontal cortex. Eur. Neuropsychopharmacol. 2009, 19, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Yuen, E.Y.; Yan, Z. Dopamine D4 receptors regulate AMPA receptor trafficking and glutamatergic transmission in GABAergic interneurons of prefrontal cortex. J. Neurosci. 2009, 29, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Yan, Z. Distinct physiological effects of dopamine D4 receptors on prefrontal cortical pyramidal neurons and fast-spiking interneurons. Cereb. Cortex 2014, 26, 180–191. [Google Scholar] [CrossRef]
- Xu, T.X.; Ma, Q.; Spealman, R.D.; Yao, W.D. Amphetamine modulation of long-term potentiation in the prefrontal cortex: Dose dependency, monoaminergic contributions, and paradoxical rescue in hyperdopaminergic mutant. J. Neurochem. 2010, 115, 1643–1654. [Google Scholar] [CrossRef]
- Bai, J.; Blot, K.; Tzavara, E.; Nosten-Bertrand, M.; Giros, B.; Otani, S. Inhibition of dopamine transporter activity impairs synaptic depression in rat prefrontal cortex through over-stimulation of D1 receptors. Cereb. Cortex 2012, 24, 945–955. [Google Scholar] [CrossRef]
- Zhang, B.; Guo, F.; Ma, Y.; Song, Y.; Lin, R.; Shen, F.Y.; Jin, G.Z.; Li, Y.; Liu, Z.Q. Activation of D1R/PKA/mTOR signaling cascade in medial prefrontal cortex underlying the antidepressant effects of l-SPD. Sci. Rep. 2017, 7, 3809. [Google Scholar] [CrossRef] [Green Version]
- Molina-Luna, K.; Pekanovic, A.; Röhrich, S.; Hertler, B.; Schubring-Giese, M.; Rioult-Pedotti, M.S.; Luft, A.R. Dopamine in motor cortex is necessary for skill learning and synaptic plasticity. PLoS ONE 2009, 4, e7082. [Google Scholar] [CrossRef]
- Hosp, J.A.; Pekanovic, A.; Rioult-Pedotti, M.S.; Luft, A.R. Dopaminergic projections from midbrain to primary motor cortex mediate motor skill learning. J. Neurosci. 2011, 31, 2481–2487. [Google Scholar] [CrossRef]
- Rioult-Pedotti, M.S.; Pekanovic, A.; Atiemo, C.O.; Marshall, J.; Luft, A.R. Dopamine promotes motor cortex plasticity and motor skill learning via PLC activation. PLoS ONE 2015, 10, e0124986. [Google Scholar] [CrossRef]
- Hosp, J.A.; Luft, A.R. Dopaminergic meso-cortical projections to M1: Role in motor learning and motor cortex plasticity. Front. Neurol. 2013, 4, 145. [Google Scholar] [CrossRef] [PubMed]
- Hosp, J.A.; Molina-Luna, K.; Hertler, B.; Atiemo, C.O.; Luft, A.R. Dopaminergic modulation of motor maps in rat motor cortex: An in vivo study. Neuroscience 2009, 159, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ko, H.; Qian, Z.M.; Yan, L.Y.; Chan, D.C.; Arbuthnott, G.; Ke, Y.; Yung, W.H. Refinement of learned skilled movement representation in motor cortex deep output layer. Nat. Commun. 2017, 8, 15834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balleine, B.W.; Delgado, M.R.; Hikosaka, O. The role of the dorsal striatum in reward and decision-making. J. Neurosci. 2007, 27, 8161–8165. [Google Scholar] [CrossRef] [PubMed]
- Tepper, J.M.; Tecuapetla, F.; Koós, T.; Ibáñez-Sandoval, O. Heterogeneity and diversity of striatal GABAergic interneurons. Front. Neuroanat. 2010, 4, 150. [Google Scholar] [CrossRef]
- Routtenberg, A.; Holzman, N. Memory disruption by electrical stimulation of substantia nigra, pars compacta. Science 1973, 181, 83–86. [Google Scholar] [CrossRef]
- Da Cunha, C.; Wietzikoski, S.; Wietzikoski, E.C.; Miyoshi, E.; Ferro, M.M.; Anselmo-Franci, J.A.; Canteras, N.S. Evidence for the substantia nigra pars compacta as an essential component of a memory system independent of the hippocampal memory system. Neurobiol. Learn. Mem. 2003, 79, 236–242. [Google Scholar] [CrossRef]
- Ramayya, A.G.; Misra, A.; Baltuch, G.H.; Kahana, M.J. Microstimulation of the human substantia nigra alters reinforcement learning. J. Neurosci. 2014, 34, 6887–6895. [Google Scholar] [CrossRef]
- de Berker, A.O.; Rutledge, R.B. A role for the human substantia nigra in reinforcement learning. J. Neurosci. 2014, 34, 12947–12949. [Google Scholar] [CrossRef]
- Beal, M.F. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 325–332. [Google Scholar] [CrossRef]
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Suppa, A.; Marsili, L.; Belvisi, D.; Conte, A.; Iezzi, E.; Modugno, N.; Fabbrini, G.; Berardelli, A. Lack of LTP-like plasticity in primary motor cortex in Parkinson’s disease. Exp. Neurol. 2011, 227, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Centonze, D.; Bernardi, G.; Calabresi, P. Striatal synaptic plasticity: Implications for motor learning and Parkinson’s disease. Mov. Disord. 2005, 20, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Paillé, V.; Picconi, B.; Bagetta, V.; Ghiglieri, V.; Sgobio, C.; Di Filippo, M.; Viscomi, M.T.; Giampà, C.; Fusco, F.R.; Gardoni, F.; et al. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J. Neurosci. 2010, 30, 14182–14193. [Google Scholar]
- Rylander, D.; Bagetta, V.; Pendolino, V.; Zianni, E.; Grealish, S.; Gardoni, F.; Di Luca, M.; Calabresi, P.; Cenci, M.A.; Picconi, B. Region-specific restoration of striatal synaptic plasticity by dopamine grafts in experimental parkinsonism. Proc. Natl. Acad. Sci. USA 2013, 110, E4375–E4384. [Google Scholar] [CrossRef]
- Kishore, A.; Joseph, T.; Velayudhan, B.; Popa, T.; Meunier, S. Early, severe and bilateral loss of LTP and LTD-like plasticity in motor cortex (M1) in de novo Parkinson’s disease. Clin. Neurophysiol. 2012, 123, 822–828. [Google Scholar] [CrossRef]
- Morgante, F.; Espay, A.J.; Gunraj, C.; Lang, A.E.; Chen, R. Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 2006, 129, 1059–1069. [Google Scholar] [CrossRef]
- Thiele, S.L.; Chen, B.; Lo, C.; Gertler, T.S.; Warre, R.; Surmeier, J.D.; Brotchie, J.M.; Nash, J.E. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiol. Dis. 2014, 71, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Hawes, S.L.; Gillani, F.; Evans, R.C.; Benkert, E.A.; Blackwell, K.T. Sensitivity to theta-burst timing permits LTP in dorsal striatal adult brain slice. J. Neurophysiol. 2013, 110, 2027–2036. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.N.; Wickens, J.R. Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro. J. Neurophysiol. 2001, 85, 117–124. [Google Scholar] [CrossRef]
- Calabresi, P.; Gubellini, P.; Centonze, D.; Picconi, B.; Bernardi, G.; Chergui, K.; Svenningsson, P.; Fienberg, A.A.; Greengard, P. Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J. Neurosci. 2000, 20, 8443–8451. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Perez, S.; Cornil, A.; Detraux, B.; Prokin, I.; Cui, Y.; Degos, B.; Berry, H.; de Kerchove d’Exaerde, A.; Venance, L. Dopamine–endocannabinoid interactions mediate spike-timing-dependent potentiation in the striatum. Nat. Commun. 2018, 9, 4118. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Kim, J.I.; Tawfik, V.L.; Lalchandani, R.R.; Scherrer, G.; Ding, J.B. Input-and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 2015, 10, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, V.; Kerr, J.N. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J. Neurosci. 2008, 28, 2435–2446. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Surmeier, D.J. Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef]
- Cerovic, M.; d’Isa, R.; Tonini, R.; Brambilla, R. Molecular and cellular mechanisms of dopamine-mediated behavioral plasticity in the striatum. Neurobiol. Learn. Mem. 2013, 105, 63–80. [Google Scholar] [CrossRef]
- Colelli, V.; Fiorenza, M.T.; Conversi, D.; Orsini, C.; Cabib, S. Strain-specific proportion of the two isoforms of the dopamine D2 receptor in the mouse striatum: Associated neural and behavioral phenotypes. Genes Brain Behav. 2010, 9, 703–711. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Ding, J.; Day, M.; Wang, Z.; Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007, 30, 228–235. [Google Scholar] [CrossRef]
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50. [Google Scholar] [CrossRef]
- Setlow, B. The nucleus accumbens and learning and memory. J. Neurosci. Res. 1997, 49, 515–521. [Google Scholar] [CrossRef]
- Salamone, J.D.; Correa, M. Motivational views of reinforcement: Implications for understanding the behavioral functions of nucleus accumbens dopamine. Behav. Brain Res. 2002, 137, 3–25. [Google Scholar] [CrossRef]
- Brady, A.M.; O’Donnell, P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J. Neurosci. 2004, 24, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Floresco, S.B.; Blaha, C.D.; Yang, C.R.; Phillips, A.G. Modulation of hippocampal and amygdalar-evoked activity of nucleus accumbens neurons by dopamine: Cellular mechanisms of input selection. J. Neurosci. 2001, 21, 2851–2860. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Jankowski, M.; Sesack, S.R. Projections from the paraventricular nucleus of the thalamus to the rat prefrontal cortex and nucleus accumbens shell: Ultrastructural characteristics and spatial relationships with dopamine afferents. J. Comp. Neurol. 2003, 459, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Britt, J.P.; Benaliouad, F.; McDevitt, R.A.; Stuber, G.D.; Wise, R.A.; Bonci, A. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 2012, 76, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wienecke, C.F.; Nachtrab, G.; Chen, X. A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 2016, 530, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Stuber, G.D.; Sparta, D.R.; Stamatakis, A.M.; van Leeuwen, W.A.; Hardjoprajitno, J.E.; Cho, S.; Tye, K.M.; Kempadoo, K.A.; Zhang, F.; Deisseroth, K.; et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature 2011, 475, 377–380. [Google Scholar] [CrossRef]
- Schacter, G.B.; Yang, C.R.; Innis, N.K.; Mogenson, G.J. The role of the hippocampal-nucleus accumbens pathway in radial-arm maze performance. Brain Res. 1989, 494, 339–349. [Google Scholar] [CrossRef]
- LeGates, T.A.; Kvarta, M.D.; Tooley, J.R.; Francis, T.C.; Lobo, M.K.; Creed, M.C.; Thompson, S.M. Reward behaviour is regulated by the strength of hippocampus–nucleus accumbens synapses. Nature 2018, 564, 258–262. [Google Scholar] [CrossRef]
- Luís, C.; Cannella, N.; Spanagel, R.; Köhr, G. Persistent strengthening of the prefrontal cortex–nucleus accumbens pathway during incubation of cocaine-seeking behavior. Neurobiol. Learn. Mem. 2017, 138, 281–290. [Google Scholar] [CrossRef]
- Qi, J.; Zhang, S.; Wang, H.L.; Barker, D.J.; Miranda-Barrientos, J.; Morales, M. VTA glutamatergic inputs to nucleus accumbens drive aversion by acting on GABAergic interneurons. Nat. Neurosci. 2016, 19, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.J.; Totterdell, S. Hippocampal and prefrontal cortical inputs monosynaptically converge with individual projection neurons of the nucleus accumbens. J. Comp. Neurol. 2002, 446, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Hayen, A. Opposing roles of nucleus accumbens core and shell dopamine in the modulation of limbic information processing. J. Neurosci. 2011, 31, 6001–6007. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Canciani, B.; Pennartz, C.M. Dopamine D1-receptors modulate lateral inhibition between principal cells of the nucleus accumbens. J. Neurophysiol. 2005, 93, 1816–1819. [Google Scholar] [CrossRef] [PubMed]
- Mele, A.; Avena, M.; Roullet, P.; De Leonibus, E.; Mandillo, S.; Sargolini, F.; Coccurello, R.; Oliverio, A. Nucleus accumbens dopamine receptors in the consolidation of spatial memory. Behav. Pharmacol. 2004, 15, 423–431. [Google Scholar] [CrossRef]
- Smith-Roe, S.L.; Kelley, A.E. Coincident activation of NMDA and dopamine D1Receptors within the nucleus accumbens core is required for appetitive instrumental learning. J. Neurosci. 2000, 20, 7737–7742. [Google Scholar] [CrossRef]
- Fadok, J.P.; Darvas, M.; Dickerson, T.M.; Palmiter, R.D. Long-term memory for pavlovian fear conditioning requires dopamine in the nucleus accumbens and basolateral amygdala. PLoS ONE 2010, 5, e12751. [Google Scholar] [CrossRef]
- Yawata, S.; Yamaguchi, T.; Danjo, T.; Hikida, T.; Nakanishi, S. Pathway-specific control of reward learning and its flexibility via selective dopamine receptors in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 2012, 109, 12764–12769. [Google Scholar] [CrossRef] [Green Version]
- Ng, E.; Varaschin, R.K.; Su, P.; Browne, C.J.; Hermainski, J.; Le Foll, B.; Pongs, O.; Liu, F.; Trudeau, L.E.; Roder, J.C.; et al. Neuronal calcium sensor-1 deletion in the mouse decreases motivation and dopamine release in the nucleus accumbens. Behav. Brain Res. 2016, 301, 213–225. [Google Scholar] [CrossRef]
- Chao, S.Z.; Ariano, M.A.; Peterson, D.A.; Wolf, M.E. D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J. Neurochem. 2002, 83, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Chao, S.Z.; Lu, W.; Lee, H.K.; Huganir, R.L.; Wolf, M.E. D1 dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J. Neurochem. 2002, 81, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiavacchi, S.; Wolf, M.E. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 2004, 88, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.E. Regulation of AMPA receptor trafficking in the nucleus accumbens by dopamine and cocaine. Neurotox. Res. 2010, 18, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, S.; Hayashi-Takagi, A.; Ellis-Davies, G.C.; Urakubo, H.; Ishii, S.; Kasai, H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science 2014, 345, 1616–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Goto, A.; Nakahara, I.; Yawata, S.; Hikida, T.; Matsuda, M.; Funabiki, K.; Nakanishi, S. Role of PKA signaling in D2 receptor-expressing neurons in the core of the nucleus accumbens in aversive learning. Proc. Natl. Acad. Sci. USA 2015, 112, 11383–11388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schotanus, S.M.; Chergui, K. Dopamine D1 receptors and group I metabotropic glutamate receptors contribute to the induction of long-term potentiation in the nucleus accumbens. Neuropharmacology 2008, 54, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.J.; Dietz, D.M.; Dumitriu, D.; Morrison, J.H.; Malenka, R.C.; Nestler, E.J. The addicted synapse: Mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010, 33, 267–276. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef]
- Neves, G.; Cooke, S.F.; Bliss, T.V. Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat. Rev. Neurosci. 2008, 9, 65–75. [Google Scholar] [CrossRef]
- Kempadoo, K.A.; Mosharov, E.V.; Choi, S.J.; Sulzer, D.; Kandel, E.R. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc. Natl. Acad. Sci. USA 2016, 113, 14835–14840. [Google Scholar] [CrossRef] [Green Version]
- Shohamy, D.; Adcock, R.A. Dopamine and adaptive memory. Trends. Cogn. Sci. 2010, 14, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Duszkiewicz, A.J.; McNamara, C.G.; Takeuchi, T.; Genzel, L. Novelty and dopaminergic modulation of memory persistence: A tale of two systems. Trends Neurosci. 2019, 42, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cullen, W.K.; Anwyl, R.; Rowan, M.J. Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat. Neurosci. 2003, 6, 526–531. [Google Scholar] [CrossRef]
- Takeuchi, T.; Duszkiewicz, A.J.; Sonneborn, A.; Spooner, P.A.; Yamasaki, M.; Watanabe, M.; Smith, C.C.; Fernández, G.; Deisseroth, K.; Greene, R.W.; et al. Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 2016, 537, 357–362. [Google Scholar] [CrossRef]
- Heath, F.C.; Jurkus, R.; Bast, T.; Pezze, M.A.; Lee, J.L.; Voigt, J.P.; Stevenson, C.W. Dopamine D1-like receptor signalling in the hippocampus and amygdala modulates the acquisition of contextual fear conditioning. Psychopharmacology 2015, 232, 2619–2629. [Google Scholar] [CrossRef]
- Nyberg, L.; Karalija, N.; Salami, A.; Andersson, M.; Wåhlin, A.; Kaboovand, N.; Köhncke, Y.; Axelsson, J.; Rieckmann, A.; Papenberg, G.; et al. Dopamine D2 receptor availability is linked to hippocampal–caudate functional connectivity and episodic memory. Proc. Natl. Acad. Sci. USA 2016, 113, 7918–7923. [Google Scholar] [CrossRef]
- Smith, W.B.; Starck, S.R.; Roberts, R.W.; Schuman, E.M. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron 2005, 45, 765–779. [Google Scholar] [CrossRef]
- Frey, U.; Matthies, H.; Reymann, K.G.; Matthies, H. The effect of dopaminergic D1 receptor blockade during tetanization on the expression of long-term potentiation in the rat CA1 region in vitro. Neurosci. Lett. 1991, 129, 111–114. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Kandel, E.R. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc. Natl. Acad. Sci. USA 1995, 92, 2446–2450. [Google Scholar] [CrossRef]
- Navakkode, S.; Sajikumar, S.; Frey, J.U. Synergistic requirements for the induction of dopaminergic D1/D5-receptor-mediated LTP in hippocampal slices of rat CA1 in vitro. Neuropharmacology 2007, 52, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Otmakhova, N.A.; Lisman, J.E. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J. Neurosci. 1996, 16, 7478–7486. [Google Scholar] [CrossRef] [PubMed]
- Shetty, M.S.; Sajikumar, S. Differential involvement of Ca2+/calmodulin-dependent protein kinases and mitogen-activated protein kinases in the dopamine D1/D5 receptor-mediated potentiation in hippocampal CA1 pyramidal neurons. Neurobiol. Learn. Mem. 2017, 138, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lemon, N.; Manahan-Vaughan, D. Dopamine D1/D5 receptors gate the acquisition of novel information through hippocampal long-term potentiation and long-term depression. J. Neurosci. 2006, 26, 7723–7729. [Google Scholar] [CrossRef]
- Rocchetti, J.; Isingrini, E.; Dal Bo, G.; Sagheby, S.; Menegaux, A.; Tronche, F.; Levesque, D.; Moquin, L.; Gratton, A.; Wong, T.P.; et al. Presynaptic D2 dopamine receptors control long-term depression expression and memory processes in the temporal hippocampus. Biol. Psychiat. 2015, 77, 513–525. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Wang, F.; Cai, F.; Hu, Z.L.; Yang, Y.J.; Chen, J.; Chen, J.G. Phosphatidylinositol-linked novel D1 dopamine receptor facilitates long-term depression in rat hippocampal CA1 synapses. Neuropharmacology 2009, 57, 164–171. [Google Scholar] [CrossRef]
- Swant, J.; Wagner, J.J. Dopamine transporter blockade increases LTP in the CA1 region of the rat hippocampus via activation of the D3 dopamine receptor. Learn. Mem. 2006, 13, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Kwon, O.B.; Paredes, D.; Gonzalez, C.M.; Neddens, J.; Hernandez, L.; Vullhorst, D.; Buonanno, A. Neuregulin-1 regulates LTP at CA1 hippocampal synapses through activation of dopamine D4 receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 15587–15592. [Google Scholar] [CrossRef] [Green Version]
- Hasselmo, M.E. The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 2006, 16, 710–715. [Google Scholar] [CrossRef] [Green Version]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Therapeut. 2013, 137, 22–54. [Google Scholar] [CrossRef]
- Parri, H.R.; Hernandez, C.M.; Dineley, K.T. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem. Pharmacol. 2011, 82, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Kos, T.; Potasiewicz, A.; Popik, P. Positive allosteric modulation of alpha 7 nicotinic acetylcholine receptors enhances recognition memory and cognitive flexibility in rats. Eur. Neuropsychopharmacol. 2015, 25, 1300–1313. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli, L.A.; Levey, A.I. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 2004, 145, 59–66. [Google Scholar] [PubMed]
- Záborszky, L.; Gombkoto, P.; Varsanyi, P.; Gielow, M.R.; Poe, G.; Role, L.W.; Ananth, M.; Rajebhosale, P.; Talmage, D.A.; Hasselmo, M.E.; et al. Specific Basal Forebrain–Cortical Cholinergic Circuits Coordinate Cognitive Operations. J. Neurosci. 2018, 38, 9446–9458. [Google Scholar]
- Mori, F.; Okada, K.I.; Nomura, T.; Kobayashi, Y. The pedunculopontine tegmental nucleus as a motor and cognitive interface between the cerebellum and basal ganglia. Front. Neuroanat. 2016, 10, 109. [Google Scholar] [CrossRef]
- Deffains, M.; Bergman, H. Striatal cholinergic interneurons and cortico-striatal synaptic plasticity in health and disease. Mov. Disord. 2015, 30, 1014–1025. [Google Scholar] [CrossRef]
- Easton, A.; Douchamps, V.; Eacott, M.; Lever, C. A specific role for septohippocampal acetylcholine in memory? Neuropsychologia 2012, 50, 3156–3168. [Google Scholar] [CrossRef] [Green Version]
- Bland, B.H.; Oddie, S.D.; Colom, L.V. Mechanisms of neural synchrony in the septohippocampal pathways underlying hippocampal theta generation. J. Neurosci. 1999, 19, 3223–3237. [Google Scholar] [CrossRef]
- Cataldi, M.; Vigliotti, C. The evolving concept of the intrinsic hippocampal theta/gamma oscillator. Front. Biosci. 2018, 10, 143–165. [Google Scholar] [CrossRef]
- Li, S.; Topchiy, I.; Kocsis, B. The effect of atropine administered in the medial septum or hippocampus on high-and low-frequency theta rhythms in the hippocampus of urethane anesthetized rats. Synapse 2007, 61, 412–419. [Google Scholar] [CrossRef]
- Fornari, R.V.; Moreira, K.M.; Oliveira, M.G. Effects of the selective M1 muscarinic receptor antagonist dicyclomine on emotional memory. Learn. Mem. 2000, 7, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.A.; Mash, D.C.; de Toledo-Morrell, L. Spatial memory in aged rats is related to PKCγ-dependent G-protein coupling of the M1 receptor. Neurobiol. Aging 2005, 26, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Patricio, R.R.; Soares, J.C.; Oliveira, M.G. M1 muscarinic receptors are necessary for retrieval of remote context fear memory. Physiol. Behav. 2017, 169, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Carey, G.J.; Billard, W.; Binch, H., III; Cohen-Williams, M.; Crosby, G.; Grzelak, M.; Guzik, H.; Kozlowski, J.A.; Lowe, D.B.; Pond, A.J.; et al. SCH 57790, a selective muscarinic M2 receptor antagonist, releases acetylcholine and produces cognitive enhancement in laboratory animals. Eur. J. Pharmacol. 2001, 431, 189–200. [Google Scholar] [CrossRef]
- Rowe, W.B.; O’Donnell, J.P.; Pearson, D.; Rose, G.M.; Meaney, M.J.; Quirion, R. Long-term effects of BIBN-99, a selective muscarinic M2 receptor antagonist, on improving spatial memory performance in aged cognitively impaired rats. Behav. Brain Res. 2003, 145, 171–178. [Google Scholar] [CrossRef]
- Bainbridge, N.K.; Koselke, L.R.; Jeon, J.; Bailey, K.R.; Wess, J.; Crawley, J.N.; Wrenn, C.C. Learning and memory impairments in a congenic C57BL/6 strain of mice that lacks the M2 muscarinic acetylcholine receptor subtype. Behav. Brain Res. 2008, 190, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, B.; Butcher, A.; McWilliams, P.; Bourgognon, J.M.; Pawlak, R.; Kong, K.C.; Bottrill, A.; Mistry, S.; Wess, J.; Rosethorne, E.M.; et al. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc. Natl. Acad. Sci. USA 2010, 107, 9440–9445. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Fürstenau, L.; Blanco, C.; Kornisiuk, E.; Sánchez, G.; Daroit, D.; e Silva, M.C.; Cerveñansky, C.; Jerusalinsky, D.; Quillfeldt, J.A. Role of hippocampal M1 and M4 muscarinic receptor subtypes in memory consolidation in the rat. Pharmacol. Biochem. Behav. 2003, 74, 411–415. [Google Scholar] [CrossRef]
- Mulugeta, E.; Karlsson, E.; Islam, A.; Kalaria, R.; Mangat, H.; Winblad, B.; Adem, A. Loss of muscarinic M4 receptors in hippocampus of Alzheimer patients. Brain Res. 2003, 960, 259–262. [Google Scholar] [CrossRef]
- Foster, D.J.; Choi, D.L.; Conn, P.J.; Rook, J.M. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsych. Dis. Treat. 2014, 10, 183–191. [Google Scholar]
- Dannenberg, H.; Young, K.; Hasselmo, M. Modulation of hippocampal circuits by muscarinic and nicotinic receptors. Front. Neural Circuit. 2017, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Teles-Grilo Ruivo, L.; Mellor, J. Cholinergic modulation of hippocampal network function. Front. Synaptic Neurosci. 2013, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaderbrand, K.; Chen, H.J.; Corcoran, K.A.; Guedea, A.L.; Jovasevic, V.; Wess, J.; Radulovic, J. Muscarinic acetylcholine receptors act in synergy to facilitate learning and memory. Learn. Mem. 2016, 23, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoe, T.; Matsui, M.; Taketo, M.M.; Manabe, T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J. Neurosci. 2005, 25, 11194–11200. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, K.A.; Petrovic, M.M.; Chamberlain, S.E.; Marrion, N.V.; Mellor, J.R. Facilitation of long-term potentiation by muscarinic M1 receptors is mediated by inhibition of SK channels. Neuron 2010, 68, 948–963. [Google Scholar] [CrossRef]
- Dennis, S.H.; Pasqui, F.; Colvin, E.M.; Sanger, H.; Mogg, A.J.; Felder, C.C.; Broad, L.M.; Fitzjohn, S.M.; Isaac, J.T.; Mellor, J.R. Activation of muscarinic M1 acetylcholine receptors induces long-term potentiation in the hippocampus. Cereb. Cortex 2015, 26, 414–426. [Google Scholar] [CrossRef]
- Anisuzzaman, A.S.; Uwada, J.; Masuoka, T.; Yoshiki, H.; Nishio, M.; Ikegaya, Y.; Takahashi, N.; Matsuki, N.; Fujibayashi, Y.; Yonekura, Y.; et al. Novel contribution of cell surface and intracellular M1-muscarinic acetylcholine receptors to synaptic plasticity in hippocampus. J. Neurochem. 2013, 126, 360–371. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; Anwyl, R.; Rowan, M.J. Endogenous acetylcholine lowers the threshold for long-term potentiation induction in the CA1 area through muscarinic receptor activation: In vivo study. Eur. J. Neurosci. 2004, 20, 1267–1275. [Google Scholar] [CrossRef]
- Le Duigou, C.; Savary, E.; Kullmann, D.M.; Miles, R. Induction of anti-Hebbian LTP in CA1 stratum oriens interneurons: Interactions between group I metabotropic glutamate receptors and M1 muscarinic receptors. J. Neurosci. 2015, 35, 13542–13554. [Google Scholar] [CrossRef]
- Kamsler, A.; McHugh, T.J.; Gerber, D.; Huang, S.Y.; Tonegawa, S. Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 1618–1623. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, B.A.; Jo, J.; Seok, H.; Son, G.H.; Whitcomb, D.J.; Davies, C.H.; Sheng, M.; Collingridge, G.L.; Cho, K. A novel mechanism of hippocampal LTD involving muscarinic receptor-triggered interactions between AMPARs, GRIP and liprin-α. Mol. Brain 2009, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Son, G.H.; Winters, B.L.; Kim, M.J.; Whitcomb, D.J.; Dickinson, B.A.; Lee, Y.B.; Futai, K.; Amici, M.; Sheng, M.; et al. Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin, AP2 and PSD-95. Nat. Neurosci. 2010, 13, 1216–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Chen, W.H.; Wang, M.; Zhu, D.M.; She, J.Q.; Ruan, D.Y. Modulation of long-term potentiation by individual subtypes of muscarinic acetylcholine receptor in the rat dentate gyrus. Hippocampus 2008, 18, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Prokopova, I. Noradrenaline and behavior. Cesk Fysiol. 2010, 59, 51–58. [Google Scholar]
- Borodovitsyna, O.; Flamini, M.; Chandler, D. Noradrenergic modulation of cognition in health and disease. Neural Plast. 2017, 2017, 6031478. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Hutchinson, D.S.; Summers, R.J. Noradrenaline release in the locus coeruleus modulates memory formation and consolidation; roles for α-and β-adrenergic receptors. Neuroscience 2010, 170, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Kukolja, J.; Klingmüller, D.; Maier, W.; Fink, G.R.; Hurlemann, R. Noradrenergic-glucocorticoid modulation of emotional memory encoding in the human hippocampus. Psychol. Med. 2011, 41, 2167–2176. [Google Scholar] [CrossRef]
- Gazarini, L.; Stern, C.A.; Carobrez, A.P.; Bertoglio, L.J. Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting α1-and β-adrenergic receptors. Learn. Mem. 2013, 20, 210–219. [Google Scholar] [CrossRef]
- Coradazzi, M.; Gulino, R.; Fieramosca, F.; Falzacappa, L.V.; Riggi, M.; Leanza, G. Selective noradrenaline depletion impairs working memory and hippocampal neurogenesis. Neurobiol. Aging 2016, 48, 93–102. [Google Scholar] [CrossRef]
- Conversi, D.; Cruciani, F.; Accoto, A.; Cabib, S. Positive emotional arousal increases duration of memory traces: Different role of dopamine D1 receptor and β-adrenoceptor activation. Pharmacol. Biochem. Behav. 2014, 122, 158–163. [Google Scholar] [CrossRef]
- Sara, S.J. Locus coeruleus in time with the making of memories. Curr. Opin. Neurobiol. 2015, 35, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Cuevas-Olguin, R.; Esquivel-Rendon, E.; Garcia-Oscos, F.; Salgado-Delgado, R.C.; Saderi, N.; Miranda-Morales, M.; Treviño, M.; Pineda, J.C.; Salgado, H. Locus ceruleus norepinephrine release: A central regulator of CNS spatio-temporal activation? Front. Synaptic Neurosci. 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Puumala, T.; Greijus, S.; Narinen, K.; Haapalinna, A.; Riekkinen Sr, P.; Sirviö, J. Stimulation of alpha-1 adrenergic receptors facilitates spatial learning in rats. Eur. Neuropsychopharmacol. 1998, 8, 17–26. [Google Scholar] [CrossRef]
- Doze, V.A.; Papay, R.S.; Goldenstein, B.L.; Gupta, M.K.; Collette, K.M.; Nelson, B.W.; Lyons, M.J.; Davis, B.A.; Luger, E.J.; Wood, S.G.; et al. Long-term α1A-adrenergic receptor stimulation improves synaptic plasticity, cognitive function, mood, and longevity. Mol. Pharmacol. 2011, 80, 747–758. [Google Scholar] [CrossRef]
- Petrasek, T.; Doulames, V.; Prokopova, I.; Vales, K.; Stuchlik, A. Combined administration of alpha1-adrenoceptor antagonist prazosin and beta-blocker propranolol impairs spatial avoidance learning on a dry arena. Behav. Brain Res. 2010, 208, 402–407. [Google Scholar] [CrossRef]
- Haggerty, D.C.; Glykos, V.; Adams, N.E.; LeBeau, F.E. Bidirectional modulation of hippocampal gamma (20–80 Hz) frequency activity in vitro via alpha (α)-and beta (β)-adrenergic receptors (AR). Neuroscience 2013, 253, 142–154. [Google Scholar] [CrossRef]
- Lazzaro, S.C.; Hou, M.; Cunha, C.; LeDoux, J.E.; Cain, C.K. Antagonism of lateral amygdala alpha1-adrenergic receptors facilitates fear conditioning and long-term potentiation. Learn. Mem. 2010, 17, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Ko, I.G.; Kim, S.E.; Shin, M.S.; Kang, Y.H.; Cho, J.W.; Shin, K.M.; Kim, C.J.; Lim, B.V.; Kim, K.H. Alpha1-adrenoceptor antagonists improve memory by activating N-methyl-D-aspartate-induced ion currents in the rat hippocampus. Int. Neurourol. J. 2015, 19, 228–236. [Google Scholar] [CrossRef]
- Scheiderer, C.L.; Dobrunz, L.E.; McMahon, L.L. Novel form of long-term synaptic depression in rat hippocampus induced by activation of α1 adrenergic receptors. J. Neurophysiol. 2004, 91, 1071–1077. [Google Scholar] [CrossRef]
- Li, C.J.; Zhou, M.; Li, H.G.; Lv, Q.; Xu, X.L.; Guo, L.J. Clonidine suppresses the induction of long-term potentiation by inhibiting HCN channels at the Schaffer collateral–CA1 synapse in anesthetized adult rats. Cell. Mol. Neurobiol. 2013, 33, 1075–1086. [Google Scholar] [CrossRef]
- Lim, E.P.; Dawe, G.S.; Jay, T.M. Activation of beta-and alpha-2-adrenoceptors in the basolateral amygdala has opposing effects on hippocampal-prefrontal long-term potentiation. Neurobiol. Learn. Mem. 2017, 137, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Papaleonidopoulos, V.; Papatheodoropoulos, C. β-adrenergic receptors reduce the threshold for induction and stabilization of LTP and enhance its magnitude via multiple mechanisms in the ventral but not the dorsal hippocampus. Neurobiol. Learn. Mem. 2018, 151, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Jenson, D.; Yang, K.; Acevedo-Rodriguez, A.; Levine, A.; Broussard, J.I.; Tang, J.; Dani, J.A. Dopamine and norepinephrine receptors participate in methylphenidate enhancement of in vivo hippocampal synaptic plasticity. Neuropharmacology 2015, 90, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Connor, S.A.; Maity, S.; Roy, B.; Ali, D.W.; Nguyen, P.V. Conversion of short-term potentiation to long-term potentiation in mouse CA1 by coactivation of β-adrenergic and muscarinic receptors. Learn. Mem. 2012, 19, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Matt, L.; Zhang, M.; Nguyen, M.; Patriarchi, T.; Koval, O.M.; Anderson, M.E.; He, K.; Lee, H.K.; Hell, J.W. β2-Adrenergic receptor supports prolonged theta tetanus-induced LTP. J. Neurophysiol. 2012, 107, 2703–2712. [Google Scholar] [CrossRef]
- Zhou, H.C.; Sun, Y.Y.; Cai, W.; He, X.T.; Yi, F.; Li, B.M.; Zhang, X.H. Activation of β2-adrenoceptor enhances synaptic potentiation and behavioral memory via cAMP-PKA signaling in the medial prefrontal cortex of rats. Learn. Mem. 2013, 20, 274–284. [Google Scholar] [CrossRef]
- Havekes, R.; Canton, D.A.; Park, A.J.; Huang, T.; Nie, T.; Day, J.P.; Guercio, L.A.; Grimes, Q.; Luczak, V.; Gelman, I.H.; et al. Gravin orchestrates protein kinase A and β2-adrenergic receptor signaling critical for synaptic plasticity and memory. J. Neurosci. 2012, 32, 18137–18149. [Google Scholar] [CrossRef]
- Chay, A.; Zamparo, I.; Koschinski, A.; Zaccolo, M.; Blackwell, K.T. Control of βAR-and N-methyl-D-aspartate (NMDA) receptor-dependent cAMP dynamics in hippocampal neurons. PLoS Comput. Biol. 2016, 12, e1004735. [Google Scholar] [CrossRef]
- Qian, H.; Patriarchi, T.; Price, J.L.; Matt, L.; Lee, B.; Nieves-Cintrón, M.; Buonarati, O.R.; Chowdhury, D.; Nanou, E.; Nystoriak, M.A.; et al. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1. 2 by the β2-adrenergic receptor in neurons. Sci. Signal. 2017, 10, eaaf9659. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, L.; Schwarz, M.K.; Dong, Y.; Schlüter, O.M. Adrenergic gate release for spike timing-dependent synaptic potentiation. Neuron 2017, 93, 394–408. [Google Scholar] [CrossRef]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Nichols, D.E.; Nichols, C.D. Serotonin receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.P.; Muzerelle, A.; Scotto-Lomassese, S.; Barik, J.; Gruart, A.; Delgado-García, J.M.; Gaspar, P. Constitutive and acquired serotonin deficiency alters memory and hippocampal synaptic plasticity. Neuropsychopharmacology 2017, 42, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Baratta, M.V.; Kodandaramaiah, S.B.; Monahan, P.E.; Yao, J.; Weber, M.D.; Lin, P.A.; Gisabella, B.; Petrossian, N.; Amat, J.; Kim, K.; et al. Stress enables reinforcement-elicited serotonergic consolidation of fear memory. Biol. Psychiat. 2016, 79, 814–822. [Google Scholar] [CrossRef]
- Darcet, F.; Gardier, A.M.; David, D.J.; Guilloux, J.P. Chronic 5-HT4 receptor agonist treatment restores learning and memory deficits in a neuroendocrine mouse model of anxiety/depression. Neurosci. Lett. 2016, 616, 197–203. [Google Scholar] [CrossRef]
- Lo, A.C.; De Maeyer, J.H.; Vermaercke, B.; Callaerts-Vegh, Z.; Schuurkes, J.A.; D’Hooge, R. SSP-002392, a new 5-HT4 receptor agonist, dose-dependently reverses scopolamine-induced learning and memory impairments in C57Bl/6 mice. Neuropharmacology 2014, 85, 178–189. [Google Scholar] [CrossRef]
- Quiedeville, A.; Boulouard, M.; Hamidouche, K.; Costa-Aze, V.D.; Nee, G.; Rochais, C.; Dallemagne, P.; Fabis, F.; Freret, T.; Bouet, V. Chronic activation of 5-HT4 receptors or blockade of 5-HT6 receptors improve memory performances. Behav. Brain Res. 2015, 293, 10–17. [Google Scholar] [CrossRef]
- Zareifopoulos, N.; Papatheodoropoulos, C. Effects of 5-HT-7 receptor ligands on memory and cognition. Neurobiol. Learn. Mem. 2016, 136, 204–209. [Google Scholar] [CrossRef]
- Chen, A.; Hough, C.J.; Li, H. Serotonin type II receptor activation facilitates synaptic plasticity via N-methyl-D-aspartate-mediated mechanism in the rat basolateral amygdala. Neuroscience 2003, 119, 53–63. [Google Scholar] [CrossRef]
- Barre, A.; Berthoux, C.; De Bundel, D.; Valjent, E.; Bockaert, J.; Marin, P.; Bécamel, C. Presynaptic serotonin 2A receptors modulate thalamocortical plasticity and associative learning. Proc. Natl. Acad. Sci. USA 2016, 113, E1382–E1391. [Google Scholar] [CrossRef] [Green Version]
- Burattini, C.; Battistini, G.; Tamagnini, F.; Aicardi, G. Low-frequency stimulation evokes serotonin release in the nucleus accumbens and induces long-term depression via production of endocannabinoid. J. Neurophysiol. 2013, 111, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Liu, W.; Gu, Z.; Yan, Z. Serotonin facilitates long-term depression induction in prefrontal cortex via p38 MAPK/Rab5-mediated enhancement of AMPA receptor internalization. J. Physiol. 2008, 586, 4465–4479. [Google Scholar] [CrossRef] [PubMed]
- Ciranna, Á. Serotonin as a modulator of glutamate-and GABA-mediated neurotransmission: Implications in physiological functions and in pathology. Curr. Neuropharmacol. 2006, 4, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Pirttimaki, T.M.; Sims, R.E.; Saunders, G.; Antonio, S.A.; Codadu, N.K.; Parri, H.R. Astrocyte-mediated neuronal synchronization properties revealed by false gliotransmitter release. J. Neurosci. 2017, 37, 9859–9870. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.E.; Butcher, J.B.; Parri, H.R.; Glazewski, S. Astrocyte and neuronal plasticity in the somatosensory system. Neural Plast. 2015, 2015, 732014. [Google Scholar] [CrossRef]
- Holtzclaw, L.A.; Pandhit, S.; Bare, D.J.; Mignery, G.A.; Russell, J.T. Astrocytes in adult rat brain express type 2 inositol 1, 4, 5-trisphosphate receptors. Glia 2002, 39, 69–84. [Google Scholar] [CrossRef]
- Newman, E.A. New roles for astrocytes: Regulation of synaptic transmission. Trends Neurosci. 2003, 26, 536–542. [Google Scholar] [CrossRef]
- Verkhratsky, A. Neurotransmitter receptors in astrocytes. In Astrocytes in (Patho) Physiology of the Nervous System, 1st ed.; Parpura, V., Haydon, P.G., Eds.; Springer: Boston, MA, USA, 2009; pp. 49–67. [Google Scholar]
- Brockett, A.T.; Kane, G.A.; Monari, P.K.; Briones, B.A.; Vigneron, P.A.; Barber, G.A.; Bermudez, A.; Dieffenbach, U.; Kloth, A.D.; Buschman, T.J.; et al. Evidence supporting a role for astrocytes in the regulation of cognitive flexibility and neuronal oscillations through the Ca2+ binding protein S100β. PLoS ONE 2018, 13, e0195726. [Google Scholar] [CrossRef]
- Pabst, M.; Braganza, O.; Dannenberg, H.; Hu, W.; Pothmann, L.; Rosen, J.; Mody, I.; van Loo, K.; Deisseroth, K.; Becker, A.J.; et al. Astrocyte intermediaries of septal cholinergic modulation in the hippocampus. Neuron 2016, 90, 853–865. [Google Scholar] [CrossRef]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef]
- Bekar, L.K.; He, W.; Nedergaard, M. Locus coeruleus α-adrenergic–mediated activation of cortical astrocytes in vivo. Cereb. Cortex 2008, 18, 2789–2795. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Perea, G.; de Sevilla, D.F.; Gómez-Gonzalo, M.; Núñez, A.; Martín, E.D.; Araque, A. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Boil. 2012, 10, e1001259. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Mishima, T.; Hisatsune, C.; Nagai, T.; Ebisui, E.; Mikoshiba, K.; Hirase, H. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 2011, 31, 18155–18165. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, Y.; Lalo, U. Role for astroglial α1-adrenoreceptors in gliotransmission and control of synaptic plasticity in the neocortex. Front. Cell. Neurosci. 2015, 9, 230. [Google Scholar] [CrossRef]
- Gao, V.; Suzuki, A.; Magistretti, P.J.; Lengacher, S.; Pollonini, G.; Steinman, M.Q.; Alberini, C.M. Astrocytic β2-adrenergic receptors mediate hippocampal long-term memory consolidation. Proc. Natl. Acad. Sci. USA 2016, 113, 8526–8531. [Google Scholar] [CrossRef]
- Jensen, C.J.; Demol, F.; Bauwens, R.; Kooijman, R.; Massie, A.; Villers, A.; Ris, L.; De Keyser, J. Astrocytic β2 adrenergic receptor gene deletion affects memory in aged mice. PLoS ONE 2016, 11, e0164721. [Google Scholar] [CrossRef]
- Petrelli, F.; Dallérac, G.; Pucci, L.; Calì, C.; Zehnder, T.; Sultan, S.; Lecca, S.; Chicca, A.; Ivanov, A.; Asensio, C.S.; et al. Dysfunction of homeostatic control of dopamine by astrocytes in the developing prefrontal cortex leads to cognitive impairments. Mol. Psychiat. 2018. [Google Scholar] [CrossRef]
- Navarrete, M.; Cuartero, M.I.; Palenzuela, R.; Draffin, J.E.; Konomi, A.; Serra, I.; Colié, S.; Castaño-Castaño, S.; Hasan, M.T.; Nebreda, Á.R.; et al. Astrocytic p38α MAPK drives NMDA receptor-dependent long-term depression and modulates long-term memory. Nat. Commun. 2019, 10, 2968. [Google Scholar] [CrossRef]
- Zhu, J.J.; Qin, Y.; Zhao, M.; Van Aelst, L.; Malinow, R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 2002, 110, 443–455. [Google Scholar] [CrossRef]
- Izumi, Y.; Zorumski, C.F. NMDA receptors, mGluR5, and endocannabinoids are involved in a cascade leading to hippocampal long-term depression. Neuropsychopharmacology 2012, 37, 609–617. [Google Scholar] [CrossRef]
- Nevian, T.; Sakmann, B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 2006, 26, 11001–11013. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Talavera, Y.; Duque-Feria, P.; Paulsen, O.; Rodríguez-Moreno, A. Presynaptic spike timing-dependent long-term depression in the mouse hippocampus. Cereb. Cortex 2016, 26, 3637–3654. [Google Scholar] [CrossRef] [PubMed]
- Min, R.; Nevian, T. Astrocyte signaling controls spike timing–dependent depression at neocortical synapses. Nat. Neurosci. 2012, 15, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, O.; Sejnowski, T.J. Natural patterns of activity and long-term synaptic plasticity. Curr. Opin. Neurobiol. 2000, 10, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Zarnadze, S.; Bäuerle, P.; Santos-Torres, J.; Böhm, C.; Schmitz, D.; Geiger, J.R.; Dugladze, T.; Gloveli, T. Cell-specific synaptic plasticity induced by network oscillations. E-Life 2016, 5, e14912. [Google Scholar] [CrossRef] [PubMed]
- Masquelier, T.; Hugues, E.; Deco, G.; Thorpe, S.J. Oscillations, phase-of-firing coding, and spike timing-dependent plasticity: An efficient learning scheme. J. Neurosci. 2009, 29, 13484–13493. [Google Scholar] [CrossRef]
- Hanslmayr, S.; Staresina, B.P.; Bowman, H. Oscillations and episodic memory: Addressing the synchronization/desynchronization conundrum. Trends Neurosci. 2016, 39, 16–25. [Google Scholar] [CrossRef]
- Timofeev, I.; Bazhenov, M.; Seigneur, J.; Sejnowski, T. Neuronal synchronization and thalamocortical rhythms in sleep, wake and epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; Noebels, J., Avoli, M., Rogawski, M., Olsen, R., Delgado-Escueta, A., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012; pp. 217–245. [Google Scholar]
- Nadim, F.; Bucher, D. Neuromodulation of neurons and synapses. Curr. Opin. Neurobiol. 2014, 29, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, V.; Wickens, J.R.; Kirkwood, A.; Kerr, J.N. Timing is not everything: Neuromodulation opens the STDP gate. Front. Synaptic Neurosci. 2010, 2, 146. [Google Scholar] [CrossRef]
- Foncelle, A.; Mendes, A.; Jędrzejewska-Szmek, J.; Valtcheva, S.; Berry, H.; Blackwell, K.T.; Venance, L. Modulation of spike-timing dependent plasticity: Towards the inclusion of a third factor in computational models. Front. Comput. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef]
- Park, A.J.; Abel, T. PKA Anchoring and Synaptic Tagging and Capture. In Synaptic Tagging and Capture, 1st ed.; Sajikumar, S., Ed.; Springer: New York, NY, USA, 2015; pp. 61–78. [Google Scholar]
- Asok, A.; Leroy, F.; Rayman, J.B.; Kandel, E.R. Molecular mechanisms of the memory trace. Trends Neurosci. 2018, 42, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Jarome, T.J.; Blair, J.; Lubin, F.D.; Nguyen, P.V. Noradrenaline goes nuclear: Epigenetic modifications during long-lasting synaptic potentiation triggered by activation of β-adrenergic receptors. J. Physiol. 2016, 594, 863–881. [Google Scholar] [CrossRef] [PubMed]
- Dayan, P.; Yu, A.J. Phasic norepinephrine: A neural interrupt signal for unexpected events. Netw. Comput. Neural Syst. 2006, 17, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Devilbiss, D.M.; Waterhouse, B.D. Phasic and tonic patterns of locus coeruleus output differentially modulate sensory network function in the awake rat. J. Neurophysiol. 2010, 105, 69–87. [Google Scholar] [CrossRef]
- Howells, F.M.; Stein, D.J.; Russell, V.A. Synergistic tonic and phasic activity of the locus coeruleus norepinephrine (LC-NE) arousal system is required for optimal attentional performance. Metab. Brain Dis. 2012, 27, 267–274. [Google Scholar] [CrossRef]
- Sarter, M.; Parikh, V.; Howe, W.M. Phasic acetylcholine release and the volume transmission hypothesis: Time to move on. Nat. Rev. Neurosci. 2009, 10, 383–390. [Google Scholar] [CrossRef]
- Mattinson, C.E.; Burmeister, J.J.; Quintero, J.E.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A. Tonic and phasic release of glutamate and acetylcholine neurotransmission in sub-regions of the rat prefrontal cortex using enzyme-based microelectrode arrays. J. Neurosci. Meth. 2011, 202, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Ruivo, L.M.; Baker, K.L.; Conway, M.W.; Kinsley, P.J.; Gilmour, G.; Phillips, K.G.; Isaac, J.T.; Lowry, J.P.; Mellor, J.R. Coordinated acetylcholine release in prefrontal cortex and hippocampus is associated with arousal and reward on distinct timescales. Cell Rep. 2017, 18, 905–917. [Google Scholar] [CrossRef]
- Hall, I.C.; Rebec, G.V.; Hurley, L.M. Serotonin in the inferior colliculus fluctuates with behavioral state and environmental stimuli. J. Exp. Biol. 2010, 213, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.Y.; Amoroso, M.W.; Uchida, N. Serotonergic neurons signal reward and punishment on multiple timescales. E-Life 2015, 4, e06346. [Google Scholar] [CrossRef]
- Grace, A.A.; Floresco, S.B.; Goto, Y.; Lodge, D.J. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007, 30, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Lodge, D.J.; Grace, A.A. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology 2006, 31, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, J.K.; Herrik, K.F.; Berg, R.W.; Hounsgaard, J.D. Influence of phasic and tonic dopamine release on receptor activation. J. Neurosci. 2010, 30, 14273–14283. [Google Scholar] [CrossRef] [PubMed]
- Otani, S.; Bai, J.; Blot, K. Dopaminergic modulation of synaptic plasticity in rat prefrontal neurons. Neurosci. Bull. 2015, 31, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cools, R.; D’Esposito, M. Inverted-U–shaped dopamine actions on human working memory and cognitive control. Biol. Psychiat. 2011, 69, e113–e125. [Google Scholar] [CrossRef]
- Smucny, J.; Olincy, A.; Eichman, L.S.; Tregellas, J.R. Neuronal effects of nicotine during auditory selective attention. Psychopharmacology 2015, 232, 2017–2028. [Google Scholar] [CrossRef]
- Cano-Colino, M.; Almeida, R.; Gomez-Cabrero, D.; Artigas, F.; Compte, A. Serotonin regulates performance nonmonotonically in a spatial working memory network. Cereb. Cortex 2013, 24, 2449–2463. [Google Scholar] [CrossRef]
- Devilbiss, D.M. Consequences of tuning network function by tonic and phasic locus coeruleus output and stress: Regulating detection and discrimination of peripheral stimuli. Brain Res. 2019, 1709, 16–27. [Google Scholar] [CrossRef]
- Thiele, A.; Bellgrove, M.A. Neuromodulation of attention. Neuron 2018, 97, 769–785. [Google Scholar] [CrossRef]
- Avery, M.C.; Krichmar, J.L. Neuromodulatory systems and their interactions: A review of models, theories, and experiments. Front. Neural Circuit. 2017, 11, 108. [Google Scholar] [CrossRef]
- Shen, W.; Plotkin, J.L.; Francardo, V.; Ko, W.K.; Xie, Z.; Li, Q.; Fieblinger, T.; Wess, J.; Neubig, R.R.; Lindsley, C.W.; et al. M4 muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L-DOPA-induced dyskinesia. Neuron 2015, 88, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.H.; Yang, Q.; Feng, B.; Liu, S.B.; Zhang, N.; Xing, J.H.; Li, X.Q.; Wu, Y.M.; Gao, G.D.; Zhao, M.G. Group I mGluR antagonist rescues the deficit of D1-induced LTP in a mouse model of fragile X syndrome. Mol. Neurodegener. 2012, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Shi, S.H.; Ule, J.; Ruggiu, M.; Barker, L.A.; Darnell, R.B.; Jan, Y.N.; Jan, L.Y. Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell 2005, 123, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodríguez, I.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. Role of GirK Channels in Long-Term Potentiation of Synaptic Inhibition in an In Vivo Mouse Model of Early Amyloid-β Pathology. Int. J. Mol. Sci. 2019, 20, 1168. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Slesinger, P.A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Di Lucente, J.; Jin, L.W.; Maezawa, I.; Wan, Y.J. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet–induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 2018, 32, 2866–2877. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Chenodeoxycholic Acid Ameliorates AlCl3-Induced Alzheimer’s Disease Neurotoxicity and Cognitive Deterioration via Enhanced Insulin Signaling in Rats. Molecules 2019, 24, 1992. [Google Scholar] [CrossRef]
- Jha, S.K.; Jha, N.K.; Kumar, D.; Sharma, R.; Shrivastava, A.; Ambasta, R.K.; Kumar, P. Stress-induced synaptic dysfunction and neurotransmitter release in Alzheimer’s disease: Can neurotransmitters and neuromodulators be potential therapeutic targets? J. Alzheimers Dis. 2017, 57, 1017–1039. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Pharmacological Interventions to Attenuate Alzheimer’s Disease Progression: The Story So Far. Curr. Alzheimer Res. 2019, 16, 261–277. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzari, A.H.; Parri, H.R. Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective. Brain Sci. 2019, 9, 300. https://doi.org/10.3390/brainsci9110300
Bazzari AH, Parri HR. Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective. Brain Sciences. 2019; 9(11):300. https://doi.org/10.3390/brainsci9110300
Chicago/Turabian StyleBazzari, Amjad H., and H. Rheinallt Parri. 2019. "Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective" Brain Sciences 9, no. 11: 300. https://doi.org/10.3390/brainsci9110300
APA StyleBazzari, A. H., & Parri, H. R. (2019). Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective. Brain Sciences, 9(11), 300. https://doi.org/10.3390/brainsci9110300