Neuro-Inflammation in Pediatric Traumatic Brain Injury—from Mechanisms to Inflammatory Networks

Abstract
:1. Introduction
2. Overview of TBI
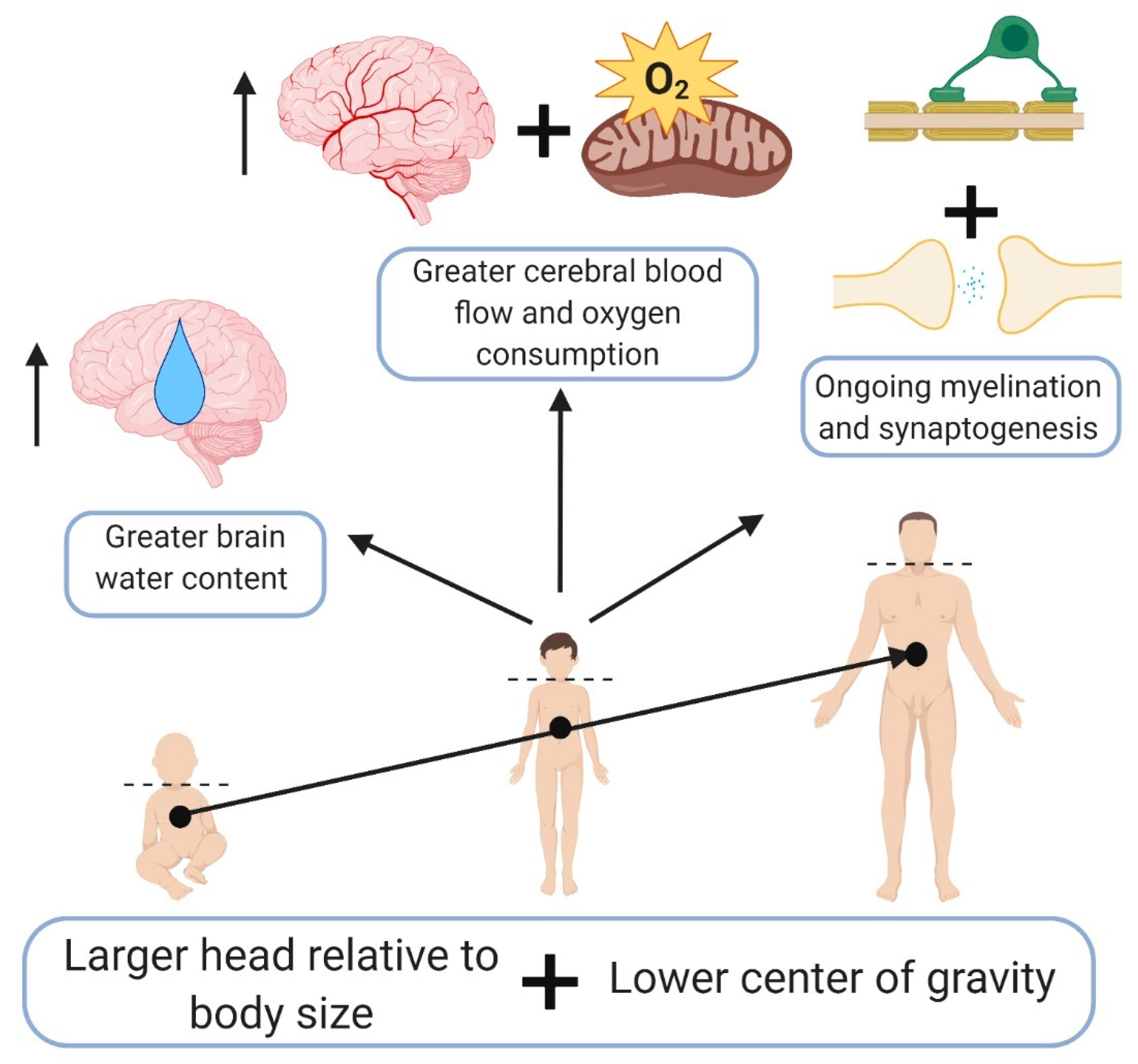
3. Pediatric TBI—Different Than Adults
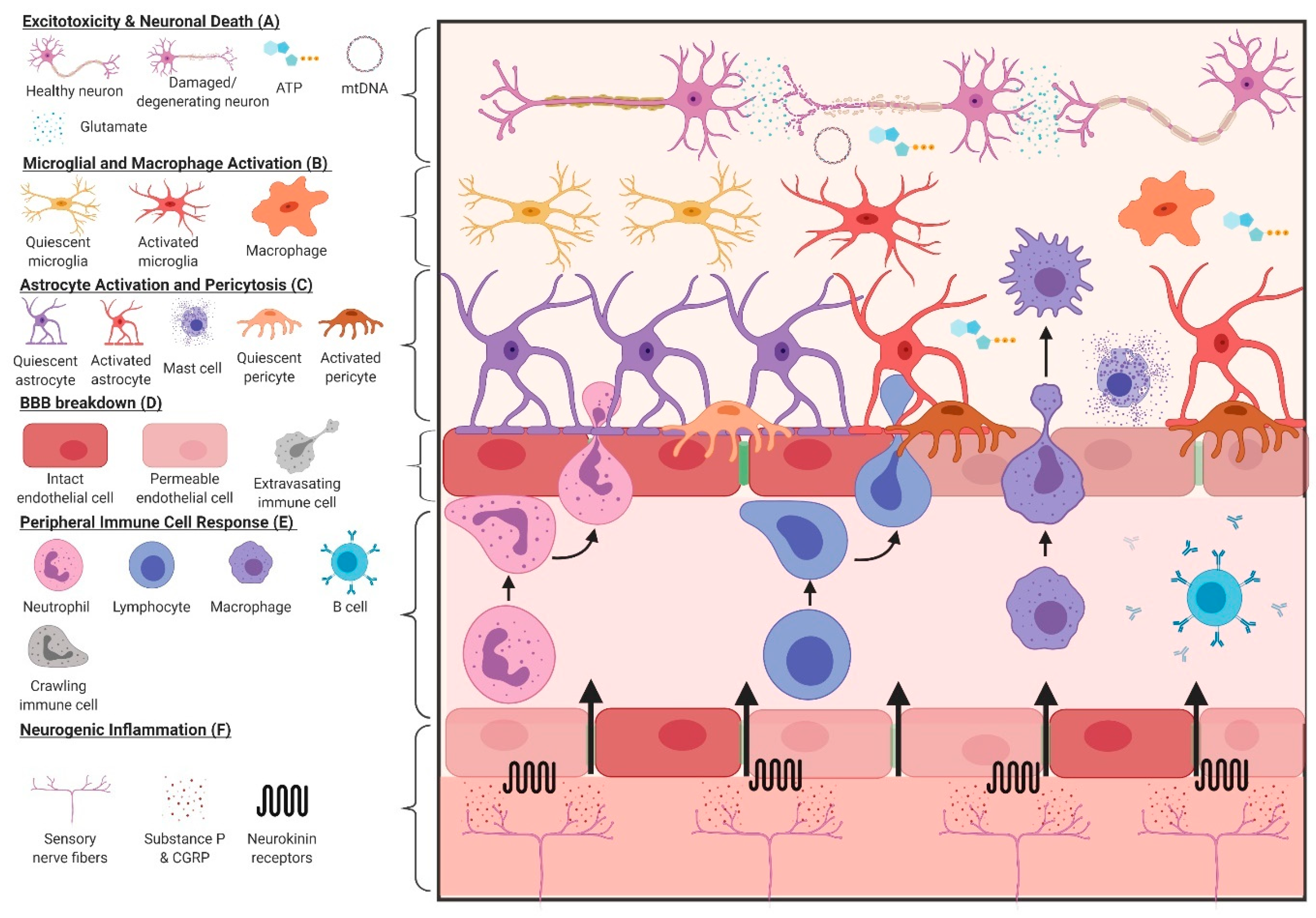
4. Inflammatory Response to TBI
4.1. Astrocytes
4.2. Microglia
4.3. Pericytes
4.4. Mast Cells
4.5. Peripheral Immune Response to TBI
4.6. Neurogenic Inflammation After TBI
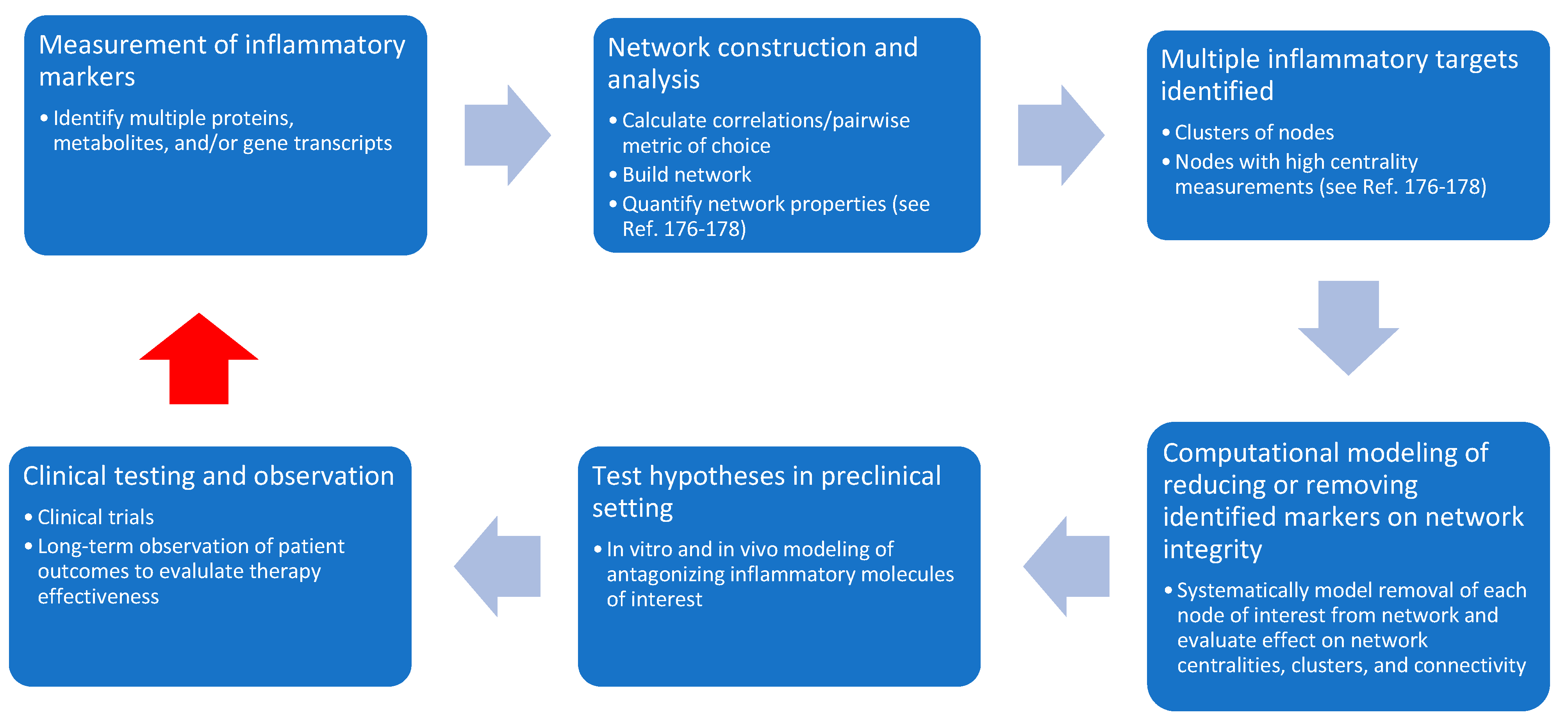
5. Moving Beyond Singular Markers—Taking an Interactive & Dynamic Network Approach to Inflammation
Biological Networks and Relevance to Inflammation after TBI
6. Conclusions and Future Directions for Research and Therapy
Author Contributions
Funding
Conflicts of Interest
References
- Levin, H.S.; Benton, A.L.; Grossman, R.G. Neurobehavioral Consequences of Closed Head Injury; Oxford University Press: Oxford, UK, 1982; p. 6. [Google Scholar]
- Hon, K.L.; Leung, A.K.C.; Torres, A.R. Concussion: A global perspective. Semin. Pediatr. Neurol. 2019, 30, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Emery, C.A.; Barlow, K.M.; Brooks, B.L.; Max, J.E.; Villavicencio-Requis, A.; Gnanakumar, V.; Robertson, H.L.; Schneider, K.; Yeates, K.O. A systematic review of psychiatric, psychological, and behavioural outcomes following mild traumatic brain injury in children and adolescents. Can. J. Psychiatry 2016, 61, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths; Centers for Disease Control and Prevention, U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Gennarelli, T.A.; Thibault, L.E.; Tomei, G.; Wiser, R.; Graham, D.; Adams, J. Directional Dependence of Axonal Brain Injury due to Centroidal and Non-Centroidal Acceleration, Proceedings of the 31st Stapp Car Crash Conference, New Orleans, LA, USA, 9–11 November 1987; Society of Automotive Engineers: Warrendale, PA, USA, 1987; SAE Technical. Paper 872197. [Google Scholar]
- Kleiven, S. Why most traumatic brain injuries are Not Caused by Linear Acceleration but Skull Fractures are. Front Bioeng. Biotechnol. 2013, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Malec, J.F.; Brown, A.W.; Leibson, C.L.; Flaada, J.T.; Mandrekar, J.N.; Diehl, N.N.; Perkins, P.K. The mayo classification system for traumatic brain injury severity. J. Neurotrauma 2007, 24, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Rimel, R.W.; Giordani, B.; Barth, J.T.; Jane, J.A. Moderate head injury: Completing the clinical spectrum of brain trauma. Neurosurgery 1982, 11, 344–351. [Google Scholar] [CrossRef]
- Nakase-Richardson, R.; Sherer, M.; Seel, R.T.; Hart, T.; Hanks, R.; Arango-Lasprilla, J.C.; Yablon, S.A.; Sander, A.M.; Barnett, S.D.; Walker, W.C.; et al. Utility of post-traumatic amnesia in predicting 1-year productivity following traumatic brain injury: Comparison of the Russell and Mississippi PTA classification intervals. J. Neurol. Neurosurg. Psychiatry 2011, 82, 494–499. [Google Scholar] [CrossRef]
- Johnson, A.M.; Nishijima, D.K.; Kuppermann, N. The Association of Glasgow coma scale score with clinically important traumatic brain injuries in children. Pediatric. Emerg. Care 2019. (published ahead of print). [Google Scholar] [CrossRef]
- Drews, J.D.; Shi, J.; Papandria, D.; Wheeler, K.K.; Sribnick, E.A.; Thakkar, R.K. Prehospital versus trauma center Glasgow coma scale in pediatric traumatic brain injury patients. J. Surg. Res. 2019, 241, 112–118. [Google Scholar] [CrossRef]
- Lieh-Lai, M.W.; Theodorou, A.A.; Sarnaik, A.P.; Meert, K.L.; Moylan, P.M.; Canady, A.I. Limitations of the Glasgow Coma Scale in predicting outcome in children with traumatic brain injury. J. Pediatr. 1992, 120, 195–199. [Google Scholar] [CrossRef]
- Rutgers, D.R.; Toulgoat, F.; Cazejust, J.; Fillard, P.; Lasjaunias, P.; Ducreux, D. White matter abnormalities in mild traumatic brain injury: A diffusion tensor imaging study. AJNR Am. J. Neuroradiol. 2008, 29, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Sorg, S.F.; Schiehser, D.M.; Bondi, M.W.; Luc, N.; Clark, A.L.; Jacobson, M.W.; Frank, L.R.; Delano-Wood, L. White Matter Microstructural Compromise Is Associated With Cognition But Not Posttraumatic Stress Disorder Symptoms in Military Veterans With Traumatic Brain Injury. J. Head Trauma Rehabil. 2016, 31, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Mallott, J.M.; Palacios, E.M.; Maruta, J.; Ghajar, J.; Mukherjee, P. Disrupted white matter microstructure of the cerebellar peduncles in scholastic athletes after concussion. Front Neurol. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.J.; Bockhorst, K.; Kondraganti, S.; Stertz, L.; Quevedo, J.; Narayana, P.A. Acute white matter tract damage after frontal mild traumatic brain injury. J. Neurotrauma 2017, 34, 291–299. [Google Scholar] [CrossRef]
- Marion, C.M.; Radomski, K.L.; Cramer, N.P.; Galdzicki, Z.; Armstrong, R.C. Experimental traumatic brain injury identifies distinct early and late phase axonal conduction deficits of white matter pathophysiology, and reveals intervening recovery. J. Neurosci. 2018, 38, 8723–8736. [Google Scholar] [CrossRef]
- Kokiko-Cochran, O.N.; Godbout, J.P. The Inflammatory continuum of traumatic brain injury and Alzheimer’s disease. Front Immunol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Giza, C.C.; Mink, R.B.; Madikians, A. Pediatric traumatic brain injury: Not just little adults. Curr. Opin. Crit. Care 2007, 13, 143–152. [Google Scholar] [CrossRef]
- Figaji, A.A. Anatomical and physiological differences between children and adults relevant to traumatic brain injury and the implications for clinical assessment and care. Front. Neurol 2017, 8, 685. [Google Scholar] [CrossRef]
- Wu, C.; Honarmand, A.R.; Schnell, S.; Kuhn, R.; Schoeneman, S.E.; Ansari, S.A.; Carr, J.; Markl, M.; Shaibani, A. Age-related changes of normal cerebral and cardiac blood flow in children and adults aged 7 months to 61 years. J. Am. Heart Assoc. 2016, 5, e002657. [Google Scholar] [CrossRef]
- Takahashi, T.; Shirane, R.; Sato, S.; Yoshimoto, T. Developmental changes of cerebral blood flow and oxygen metabolism in children. AJNR Am. J. Neuroradiol. 1999, 20, 917–922. [Google Scholar]
- Prins, M.L.; Hovda, D.A. Developing experimental models to address traumatic brain injury in children. J. Neurotrauma 2003, 20, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Prange, M.T.; Margulies, S.S. Regional, directional, and age-dependent properties of the brain undergoing large deformation. J. Biomech. Eng. 2002, 124, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Huelke, D. An overview of anatomical considerations of infants and children in the adult world of automobile safety design. Proceedings of the 42nd Annual Proceedings of the Association for the Advancement of Automotive Medicine, Charlottesville, VA, USA, 5–6 October 1998; Association for the Advancement of Automotive Medicine (AAAM): Chicago, IL, USA, 1998; pp. 93–113. [Google Scholar]
- Charles, S. Stepchild of American pediatrics: Child transportation safety. Pediatr. Ann. 1977, 6, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Wallisch, J.S.; Bayır, H.; Clark, R.S.B. Pre-clinical models in pediatric traumatic brain injury-challenges and lessons learned. Childs Nerv. Syst. 2017, 33, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M. Age at puberty and first litter size in early and late paired rats. Biol. Reprod. 1986, 34, 322–326. [Google Scholar] [CrossRef]
- Sengupta, P. The Laboratory Rat: Relating Its Age With Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Kinder, H.A.; Baker, E.W.; West, F.D. The pig as a preclinical traumatic brain injury model: Current models, functional outcome measures, and translational detection strategies. Neural. Regen. Res. 2019, 14, 413–424. [Google Scholar]
- Mychasiuk, R.; Farran, A.; Angoa-Perez, M.; Briggs, D.; Kuhn, D.; Esser, M.J. A novel model of mild traumatic brain injury for juvenile rats. J. Vis. Exp. 2014. [Google Scholar] [CrossRef]
- Mychasiuk, R.; Farran, A.; Esser, M.J. Assessment of an experimental rodent model of pediatric mild traumatic brain injury. J. Neurotrauma 2014, 31, e51820. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Li, J.; Wu, H.; Peng, Y.; Fan, L.; Chen, J.; Gu, C.; Yan, F.; Wang, L.; et al. The polarization states of microglia in TBI: A new paradigm for pharmacological intervention. Neural. Plast. 2017, 2017, 5405104. [Google Scholar] [CrossRef] [PubMed]
- Walko, T.D.; Bola, R.A.; Hong, J.D.; Au, A.K.; Bell, M.J.; Kochanek, P.M.; Clark, R.S.; Aneja, R.K. Cerebrospinal fluid mitochondrial DNA: A novel DAMP in pediatric traumatic brain injury. Shock 2014, 41, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Farfara, D.; Frenkel, D. Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: Towards future therapeutic application. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef]
- Rao, V.L.; Başkaya, M.K.; Doğan, A.; Rothstein, J.D.; Dempsey, R.J. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem 1998, 70, 2020–2027. [Google Scholar]
- Nichols, J.; Bjorklund, G.R.; Newbern, J.; Anderson, T. Parvalbumin fast-spiking interneurons are selectively altered by paediatric traumatic brain injury. J. Physiol. 2018, 596, 1277–1293. [Google Scholar] [CrossRef]
- Almeida-Suhett, C.P.; Prager, E.M.; Pidoplichko, V.; Figueiredo, T.H.; Marini, A.M.; Li, Z.; Eiden, L.E.; Braga, M.F. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp. Neurol. 2015, 273, 11–23. [Google Scholar] [CrossRef]
- Jeyaraju, D.V.; Cisbani, G.; Pellegrini, L. Calcium regulation of mitochondria motility and morphology. Biochim. Biophys. Acta 2009, 1787, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Hroudová, J.; Fišar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural. Regen. Res. 2013, 8, 363–375. [Google Scholar]
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J.I.; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem 2013, 127, 221–232. [Google Scholar] [CrossRef]
- Sánchez-Alegría, K.; Flores-León, M.; Avila-Muñoz, E.; Rodríguez-Corona, N.; Arias, C. PI3K signaling in neurons: A central node for the control of multiple functions. Int. J. Mol. Sci 2018, 19, 3725. [Google Scholar] [CrossRef] [PubMed]
- Pagé, E.L.; Chan, D.A.; Giaccia, A.J.; Levine, M.; Richard, D.E. Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: Role of oxidation and intracellular ascorbate depletion. Mol. Biol. Cell 2008, 19, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Tsao, J.W.; Stanfill, A.G. The current state of biomarkers of mild traumatic brain injury. JCI Insight 2018, 3, e97105. [Google Scholar] [CrossRef]
- Saatman, K.E.; Bozyczko-Coyne, D.; Marcy, V.; Siman, R.; McIntosh, T.K. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J. Neuropathol. Exp. Neurol 1996, 55, 850–860. [Google Scholar] [CrossRef]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef]
- Honda, S.; Sasaki, Y.; Ohsawa, K.; Imai, Y.; Nakamura, Y.; Inoue, K.; Kohsaka, S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 2001, 21, 1975–1982. [Google Scholar] [CrossRef]
- Ohsawa, K.; Irino, Y.; Nakamura, Y.; Akazawa, C.; Inoue, K.; Kohsaka, S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia 2007, 55, 604–616. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Shibata, K.; Yoshida, K.; Shigetomi, E.; Gachet, C.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y. Cell Rep. 2017, 19, 1151–1164. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Rancan, M.; Bye, N.; Otto, V.I.; Trentz, O.; Kossmann, T.; Frentzel, S.; Morganti-Kossmann, M.C. The chemokine fractalkine in patients with severe traumatic brain injury and a mouse model of closed head injury. J. Cereb Blood Flow Metab. 2004, 24, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Zanier, E.R.; Marchesi, F.; Ortolano, F.; Perego, C.; Arabian, M.; Zoerle, T.; Sammali, E.; Pischiutta, F.; De Simoni, M.G. Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J. Neurotrauma 2016, 33, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Febinger, H.Y.; Thomasy, H.E.; Pavlova, M.N.; Ringgold, K.M.; Barf, P.R.; George, A.M.; Grillo, J.N.; Bachstetter, A.D.; Garcia, J.A.; Cardona, A.E.; et al. Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J. Neuroinflamm. 2015, 12, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulthard, M.G.; Morgan, M.; Woodruff, T.M.; Arumugam, T.V.; Taylor, S.M.; Carpenter, T.C.; Lackmann, M.; Boyd, A.W. Eph/Ephrin signaling in injury and inflammation. Am. J. Pathol 2012, 181, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef]
- De Souza, D.F.; Leite, M.C.; Quincozes-Santos, A.; Nardin, P.; Tortorelli, L.S.; Rigo, M.M.; Gottfried, C.; Leal, R.B.; Gonçalves, C.A. S100B secretion is stimulated by IL-1beta in glial cultures and hippocampal slices of rats: Likely involvement of MAPK pathway. J. Neuroimmunol. 2009, 206, 52–57. [Google Scholar] [CrossRef]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef]
- Miller, S.J. Astrocyte Heterogeneity in the Adult Central Nervous System. Front. Cell Neurosci. 2018, 12, 401. [Google Scholar] [CrossRef] [Green Version]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Scemes, E.; Spray, D.C. The astrocytic syncytium. Adv. Mol. Cell Biol. 2004, 31, 165–179. [Google Scholar]
- Matsuzawa, K.; Kosako, H.; Inagaki, N.; Shibata, H.; Mukai, H.; Ono, Y.; Amano, M.; Kaibuchi, K.; Matsuura, Y.; Azuma, I.; et al. Domain-specific phosphorylation of vimentin and glial fibrillary acidic protein by PKN. Biochem. Biophys. Res. Commun. 1997, 234, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Nakamura, Y.; Takeda, M.; Nishimura, T.; Inagaki, N. Glial fibrillary acidic protein: Dynamic property and regulation by phosphorylation. Brain Pathol. 1994, 4, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, C.; Valentim, L.; Salbego, C.; Karl, J.; Wofchuk, S.T.; Rodnight, R. Regulation of protein phosphorylation in astrocyte cultures by external calcium ions: Specific effects on the phosphorylation of glial fibrillary acidic protein (GFAP), vimentin and heat shock protein 27 (HSP27). Brain Res. 1999, 833, 142–149. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Van Landeghem, F.K.; Weiss, T.; Oehmichen, M.; von Deimling, A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J. Neurotrauma 2006, 23, 1518–1528. [Google Scholar] [CrossRef]
- Frugier, T.; Conquest, A.; McLean, C.; Currie, P.; Moses, D.; Goldshmit, Y. Expression and activation of EphA4 in the human brain after traumatic injury. J. Neuropathol. Exp. Neurol. 2012, 71, 242–250. [Google Scholar] [CrossRef]
- Moreno-Flores, M.T.; Wandosell, F. Up-regulation of Eph tyrosine kinase receptors after excitotoxic injury in adult hippocampus. Neuroscience 1999, 91, 193–201. [Google Scholar] [CrossRef]
- Gesteira, T.F.; Coulson-Thomas, Y.M.; Coulson-Thomas, V.J. Anti-inflammatory properties of the glial scar. Neural. Regen Res. 2016, 11, 1742–1743. [Google Scholar]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Stowell, R.D.; Wong, E.L.; Batchelor, H.N.; Mendes, M.S.; Lamantia, C.E.; Whitelaw, B.S.; Majewska, A.K. Cerebellar microglia are dynamically unique and survey Purkinje neurons in vivo. Dev. Neurobiol. 2018, 78, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Byrnes, K.R. Role of microglia in neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef]
- Raes, G.; Van den Bergh, R.; De Baetselier, P.; Ghassabeh, G.H.; Scotton, C.; Locati, M.; Mantovani, A.; Sozzani, S. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J. Immunol 2005, 174, 6561. [Google Scholar] [CrossRef]
- Giannoni, P.; Badaut, J.; Dargazanli, C.; De Maudave, A.F.; Klement, W.; Costalat, V.; Marchi, N. The pericyte-glia interface at the blood-brain barrier. Clin. Sci (Lond) 2018, 132, 361–374. [Google Scholar] [CrossRef]
- Dore-Duffy, P.; Owen, C.; Balabanov, R.; Murphy, S.; Beaumont, T.; Rafols, J.A. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res. 2000, 60, 55–69. [Google Scholar] [CrossRef]
- Zehendner, C.M.; Sebastiani, A.; Hugonnet, A.; Bischoff, F.; Luhmann, H.J.; Thal, S.C. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci Rep. 2015, 5, 13497. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev. Oncol Hematol 2006, 59, 15–26. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Blixt, J.; Svensson, M.; Gunnarson, E.; Wanecek, M. Aquaporins and blood–brain barrier permeability in early edema development after traumatic brain injury. Brain Res. 2015, 1611, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lei, X.-Y.; Hu, H.; He, Z.-P. Relationship between AQP4 expression and structural damage to the blood-brain barrier at early stages of traumatic brain injury in rats. Chin. Med. J. 2013, 126, 4316–4321. [Google Scholar]
- Zhang, C.; Chen, J.; Lu, H. Expression of aquaporin-4 and pathological characteristics of brain injury in a rat model of traumatic brain injury. Mol. Med. Rep. 2015, 12, 7351–7357. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.M.; Pop, V.; Spagnoli, D.; Ashwal, S.; Obenaus, A.; Badaut, J. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: Possible role in edema resolution? Neuroscience 2012, 222, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, V.; Badaut, J. A Neurovascular Perspective for Long-Term Changes After Brain Trauma. Transl. Stroke Res. 2011, 2, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadou, V.; Rouleau, A.; Trung Tuong, M.D.; Newlands, G.J.; Miller, H.R.; Luffau, G.; Schwartz, J.C.; Garbarg, M. Functional relationships between sensory nerve fibers and mast cells of dura mater in normal and inflammatory conditions. Neuroscience 1997, 77, 829–839. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Stewart, J.M.; Panagiotidou, S.; Melamed, I. Mast cells, brain inflammation and autism. Eur. J. Pharm. 2016, 778, 96–102. [Google Scholar] [CrossRef]
- Theoharides, T.C. Mast cells: The immune gate to the brain. Life Sci 1990, 46, 607–617. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Dong, H.; Xu, Y.; Zhang, S. Induction of Microglial Activation by Mediators Released from Mast Cells. Cell Physiol. Biochem. 2016, 38, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, Y.; Zhang, X.; Qian, Y.; Ding, H.; Zhang, S. Stabilization of Brain Mast Cells Alleviates LPS-Induced Neuroinflammation by Inhibiting Microglia Activation. Front. Cell Neurosci 2019, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, X.; Wang, Y.; Zhou, X.; Qian, Y.; Zhang, S. Suppression of Brain Mast Cells Degranulation Inhibits Microglial Activation and Central Nervous System Inflammation. Mol. Neurobiol 2017, 54, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Lozada, A.; Maegele, M.; Stark, H.; Neugebauer, E.M.; Panula, P. Traumatic brain injury results in mast cell increase and changes in regulation of central histamine receptors. Neuropathol. Appl. Neurobiol. 2005, 31, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Stokely, M.E.; Orr, E.L. Acute effects of calvarial damage on dural mast cells, pial vascular permeability, and cerebral cortical histamine levels in rats and mice. J. Neurotrauma 2008, 25, 52–61. [Google Scholar] [CrossRef]
- Bree, D.; Levy, D. Intact mast cell content during mild head injury is required for development of latent pain sensitization: Implications for mechanisms underlying post-traumatic headache. Pain 2019, 160, 1050–1058. [Google Scholar] [CrossRef]
- Levy, D.; Edut, S.; Baraz-Goldstein, R.; Rubovitch, V.; Defrin, R.; Bree, D.; Gariepy, H.; Zhao, J.; Pick, C.G. Responses of dural mast cells in concussive and blast models of mild traumatic brain injury in mice: Potential implications for post-traumatic headache. Cephalalgia 2016, 36, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Chhor, V.; Bettati, D.; Banino, E.; De Lucia, S.; Le Charpentier, T.; Lebon, S.; Schwendimann, L.; Pansiot, J.; Rasika, S.; et al. Contribution of mast cells to injury mechanisms in a mouse model of pediatric traumatic brain injury. J. Neurosci Res. 2016, 94, 1546–1560. [Google Scholar] [CrossRef] [Green Version]
- Aldrimer, M.; Ridefelt, P.; Rödöö, P.; Niklasson, F.; Gustafsson, J.; Hellberg, D. Population-based pediatric reference intervals for hematology, iron and transferrin. Scand. J. Clin. Lab. Invest. 2013, 73, 253–261. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khy, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Povlishock, J.T.; Becker, D.P.; Sullivan, H.G.; Miller, J.D. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 1978, 153, 223–239. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Vink, R.; Gabrielian, L.; Thornton, E. The Role of Substance P in Secondary Pathophysiology after Traumatic Brain Injury. Front. Neurol. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, S.J.; Nakamura, T.; Miyazaki, D.; Ohbayashi, M.; Dawson, M.; Toda, M. Chemokines: Roles in leukocyte development, trafficking, and effector function. J. Allergy Clin. Immunol. 2003, 111, 1185–1199. [Google Scholar] [CrossRef]
- Semple, B.D.; Trivedi, A.; Gimlin, K.; Noble-Haeusslein, L.J. Neutrophil elastase mediates acute pathogenesis and is a determinant of long-term behavioral recovery after traumatic injury to the immature brain. Neurobiol. Dis. 2015, 74, 263–280. [Google Scholar] [CrossRef]
- Rancan, M.; Otto, V.I.; Hans, V.H.; Gerlach, I.; Jork, R.; Trentz, O.; Kossmann, T.; Morganti-Kossmann, M.C. Upregulation of ICAM-1 and MCP-1 but not of MIP-2 and sensorimotor deficit in response to traumatic axonal injury in rats. J. Neurosci. Res. 2001, 63, 438–446. [Google Scholar] [CrossRef]
- Clark, R.S.; Schiding, J.K.; Kaczorowski, S.L.; Marion, D.W.; Kochanek, P.M. Neutrophil accumulation after traumatic brain injury in rats: Comparison of weight drop and controlled cortical impact models. J. Neurotrauma 1994, 11, 499–506. [Google Scholar] [CrossRef]
- Adelson, P.D.; Robichaud, P.; Hamilton, R.L.; Kochanek, P.M. A model of diffuse traumatic brain injury in the immature rat. J. Neurosurg. 1996, 85, 877–884. [Google Scholar] [CrossRef]
- Adelson, P.D.; Whalen, M.J.; Kochanek, P.M.; Robichaud, P.; Carlos, T.M. Blood brain barrier permeability and acute inflammation in two models of traumatic brain injury in the immature rat: A preliminary report. Acta Neurochir Suppl 1998, 71, 104–106. [Google Scholar]
- Trahanas, D.M.; Cuda, C.M.; Perlman, H.; Schwulst, S.J. Differential Activation of Infiltrating Monocyte-Derived Cells After Mild and Severe Traumatic Brain Injury. Shock 2015, 43, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beschorner, R.; Nguyen, T.D.; Gözalan, F.; Pedal, I.; Mattern, R.; Schluesener, H.J.; Meyermann, R.; Schwab, J.M. CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol. 2002, 103, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.D.; Zarruk, J.G.; Healy, L.M.; Baskar Jesudasan, S.J.; Jhelum, P.; Salmon, C.K.; Formanek, A.; Russo, M.V.; Antel, J.P.; McGavern, D.B.; et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. 2018, 16, e2005264. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci 2009, 29, 13435–13444. [Google Scholar] [CrossRef]
- Doran, S.J.; Ritzel, R.M.; Glaser, E.P.; Henry, R.J.; Faden, A.I.; Loane, D.J. Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. J. Neurotrauma 2019, 36, 1040–1053. [Google Scholar] [CrossRef]
- Makinde, H.M.; Cuda, C.M.; Just, T.B.; Perlman, H.R.; Schwulst, S.J. Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J. Immunol. 2017, 199, 3583–3591. [Google Scholar] [CrossRef] [Green Version]
- Ingersoll, M.A.; Spanbroek, R.; Lottaz, C.; Gautier, E.L.; Frankenberger, M.; Hoffmann, R.; Lang, R.; Haniffa, M.; Collin, M.; Tacke, F.; et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 2010, 115, e10–e19. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. Blood Monocytes and Their Subsets: Established Features and Open Questions. Front. Immunol. 2015, 6, 423. [Google Scholar] [CrossRef]
- Yegin, O. Chemotaxis in childhood. Pediatr. Res. 1983, 17, 183–187. [Google Scholar] [CrossRef]
- Klein, R.B.; Fischer, T.J.; Gard, S.E.; Biberstein, M.; Rich, K.C.; Stiehm, E.R. Decreased mononuclear and polymorphonuclear chemotaxis in human newborns, infants, and young children. Pediatrics 1977, 60, 467–472. [Google Scholar]
- Cox, A.L.; Coles, A.J.; Nortje, J.; Bradley, P.G.; Chatfield, D.A.; Thompson, S.J.; Menon, D.K. An investigation of auto-reactivity after head injury. J. Neuroimmunol. 2006, 174, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Yang, Z.; Yue, J.K.; Zhang, Z.; Winkler, E.A.; Puccio, A.M.; Diaz-Arrastia, R.; Lingsma, H.F.; Yuh, E.L.; Mukherjee, P.; et al. Plasma anti-glial fibrillary acidic protein autoantibody levels during the acute and chronic phases of traumatic brain injury: A transforming research and clinical knowledge in traumatic brain injury pilot study. J. Neurotrauma 2016, 33, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Bazarian, J.J.; Puvenna, V.; Janigro, M.; Ghosh, C.; Zhong, J.; Zhu, T.; Blackman, E.; Stewart, D.; Ellis, J.; et al. Consequences of repeated blood-brain barrier disruption in football players. PLoS ONE 2013, 8, e56805. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Dupont, G.; Schmidt, C.; Yilmaz, E.; Oskouian, R.J.; Macchi, V.; de Caro, R.; Tubbs, R.S. Our current understanding of the lymphatics of the brain and spinal cord. Clin. Anat 2019, 32, 117–121. [Google Scholar] [CrossRef]
- Moalem, G.; Leibowitz-Amit, R.; Yoles, E.; Mor, F.; Cohen, I.R.; Schwartz, M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat. Med. 1999, 5, 49–55. [Google Scholar] [CrossRef]
- Schwulst, S.J.; Trahanas, D.M.; Saber, R.; Perlman, H. Traumatic brain injury-induced alterations in peripheral immunity. J. Trauma Acute Care Surg. 2013, 75, 780–788. [Google Scholar] [CrossRef]
- Mrakovcic-Sutic, I.; Tokmadzic, V.S.; Laskarin, G.; Mahmutefendic, H.; Lucin, P.; Zupan, Z.; Sustic, A. Early changes in frequency of peripheral blood lymphocyte subpopulations in severe traumatic brain-injured patients. Scand J. Immunol. 2010, 72, 57–65. [Google Scholar] [CrossRef]
- Ortega, S.B.; Torres, V.O.; Tian, F.; Poinsatte, K.; Tweed, J.; Greenwell, C.; Windsor, J.; Raman, L.; Miles, D.; Stowe, A. Adaptive immune cell activation in acute pediatric traumatic brain injury. J. Immunol. 2019, 202, 182.55. [Google Scholar]
- Hazeldine, J.; Lord, J.M.; Belli, A. Traumatic Brain Injury and Peripheral Immune Suppression: Primer and Prospectus. Front. Neurol 2015, 6, 235. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Li, J.P.; Song, Y.L.; Zhao, Q.H. Humoral and Cellular Immunity Changed after Traumatic Brain Injury in Human Patients. Ann. Clin. Lab. Sci. 2017, 47, 10–16. [Google Scholar] [PubMed]
- Javidi, E.; Magnus, T. Autoimmunity After Ischemic Stroke and Brain Injury. Front. Immunol 2019, 10, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankeny, D.P.; Popovich, P.G. B cells and autoantibodies: Complex roles in CNS injury. Trends Immunol. 2010, 31, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Goryunova, A.V.; Bazarnaya, N.A.; Sorokina, E.G.; Semenova, N.Y.; Globa, O.V.; Semenova, Z.B.; Pinelis, V.G.; Roshal’, L.M.; Maslova, O.I. Glutamate receptor autoantibody concentrations in children with chronic post-traumatic headache. Neurosci. Behav. Physiol. 2007, 37, 761–764. [Google Scholar] [CrossRef]
- Needham, E.J.; Helmy, A.; Zanier, E.R.; Jones, J.L.; Coles, A.J.; Menon, D.K. The immunological response to traumatic brain injury. J. Neuroimmunol. 2019, 332, 112–125. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef]
- Parenti, A.; De Logu, F.; Geppetti, P.; Benemei, S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br. J. Pharm. 2016, 173, 953–969. [Google Scholar] [CrossRef]
- Brain, S.D.; Williams, T.J.; Tippins, J.R.; Morris, H.R.; MacIntyre, I. Calcitonin gene-related peptide is a potent vasodilator. Nature 1985, 313, 54–56. [Google Scholar] [CrossRef]
- Brain, S.D.; Williams, T.J. Substance P regulates the vasodilator activity of calcitonin gene-related peptide. Nature 1988, 335, 73–75. [Google Scholar] [CrossRef]
- Lundberg, J.M.; Franco-Cereceda, A.; Hua, X.; Hökfelt, T.; Fischer, J.A. Co-existence of substance P and calcitonin gene-related peptide-like immunoreactivities in sensory nerves in relation to cardiovascular and bronchoconstrictor effects of capsaicin. Eur J. Pharm. 1985, 108, 315–319. [Google Scholar] [CrossRef]
- Quartara, L.; Maggi, C.A. The tachykinin NK1 receptor. Part II: Distribution and pathophysiological roles. Neuropeptides 1998, 32, 1–49. [Google Scholar] [CrossRef]
- Burmeister, A.R.; Johnson, M.B.; Chauhan, V.S.; Moerdyk-Schauwecker, M.J.; Young, A.D.; Cooley, I.D.; Martinez, A.N.; Ramesh, G.; Philipp, M.T.; Marriott, I. Human microglia and astrocytes constitutively express the neurokinin-1 receptor and functionally respond to substance P. J. Neuroinflamm. 2017, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, K.L.; Naik, S.M.; Cannon, G.; Armstrong, C.A.; Bunnett, N.W.; Ansel, J.C.; Caughman, S.W. Substance P activates coincident NF-AT- and NF-kappa B-dependent adhesion molecule gene expression in microvascular endothelial cells through intracellular calcium mobilization. J. Immunol. 1999, 163, 5656–5665. [Google Scholar] [PubMed]
- Quinlan, K.L.; Song, I.S.; Naik, S.M.; Letran, E.L.; Olerud, J.E.; Bunnett, N.W.; Armstrong, C.A.; Caughman, S.W.; Ansel, J.C. VCAM-1 expression on human dermal microvascular endothelial cells is directly and specifically up-regulated by substance P. J. Immunol. 1999, 162, 1656–1661. [Google Scholar]
- Johnson, M.B.; Young, A.D.; Marriott, I. The Therapeutic Potential of Targeting Substance P/NK-1R Interactions in Inflammatory CNS Disorders. Front. Cell Neurosci. 2016, 10, 296. [Google Scholar] [CrossRef]
- Li, W.W.; Guo, T.Z.; Liang, D.Y.; Sun, Y.; Kingery, W.S.; Clark, J.D. Substance P signaling controls mast cell activation, degranulation, and nociceptive sensitization in a rat fracture model of complex regional pain syndrome. Anesthesiology 2012, 116, 882–895. [Google Scholar] [CrossRef]
- Marriott, I.; Bost, K.L. Substance P receptor mediated macrophage responses. Adv. Exp. Med. Biol 2001, 493, 247–254. [Google Scholar]
- Suzuki, H.; Miura, S.; Liu, Y.Y.; Tsuchiya, M.; Ishii, H. Substance P induces degranulation of mast cells and leukocyte adhesion to venular endothelium. Peptides 1995, 16, 1447–1452. [Google Scholar] [CrossRef]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell Mol. Life Sci 2016, 73, 4249–4264. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Takahashi, K.; Matsubara, Y.; Inuzuka, E.; Nakashima, F.; Takahashi, N.; Kozai, D.; Mori, Y.; Uchida, K. Identification of a prostaglandin D2 metabolite as a neuritogenesis enhancer targeting the TRPV1 ion channel. Sci Rep. 2016, 6, 21261. [Google Scholar] [CrossRef]
- Ohshima, M.; Miyake, M.; Takeda, M.; Muto, T.; Ueda, N.; Ito, K.; Sakamoto, T. Development of mechanisms associated with neurogenic-mediated skin inflammation during the growth of rats. Pediatr. Res. 2010, 67, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, A.J.; Cernak, I.; Heath, D.L.; Hu, X.; Bennett, C.J.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Almeida, T.; Pérez-Cejas, A.; Ramos, L.; Argueso, M.; Riaño-Ruiz, M.; Solé-Violán, J.; Hernández, M. Serum levels of substance P and mortality in patients with a severe acute Ischemic stroke. Int. J. Mol. Sci. 2016, 17, 991. [Google Scholar] [CrossRef]
- Blume, H.K.; Vavilala, M.S.; Jaffe, K.M.; Koepsell, T.D.; Wang, J.; Temkin, N.; Durbin, D.; Dorsch, A.; Rivara, F.P. Headache after pediatric traumatic brain injury: A cohort study. Pediatrics 2012, 129, e31–e39. [Google Scholar] [CrossRef]
- Kwan, V.; Vo, M.; Noel, M.; Yeates, K. A scoping review of pain in children after traumatic brain injury: Is there more than headache? J. Neurotrauma 2018, 35, 877–888. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef]
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926. [Google Scholar] [CrossRef]
- Edvinsson, L. Role of CGRP in Migraine. Handb. Exp. Pharm. 2019, 255, 121–130. [Google Scholar]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; McFadden, G. Cytokine synergy: An underappreciated contributor to innate anti-viral immunity. Cytokine 2013, 63, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuteri, A.; Orru, M.; Morrell, C.; Piras, M.G.; Taub, D.; Schlessinger, D.; Uda, M.; Lakatta, E.G. Independent and additive effects of cytokine patterns and the metabolic syndrome on arterial aging in the SardiNIA Study. Atherosclerosis 2011, 215, 459–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F.A.; Yaron, I.; Yaron, M. Synergistic, additive, and antagonistic effects of interleukin-1 beta, tumor necrosis factor alpha, and gamma-interferon on prostaglandin E, hyaluronic acid, and collagenase production by cultured synovial fibroblasts. Arthritis Rheum. 1990, 33, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Cavaillon, J.M. Pro- versus anti-inflammatory cytokines: Myth or reality. Cell Mol. Biol. (Noisy-Le-Grand) 2001, 47, 695–702. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Constantine, G.; Rubin, J.; Csete, M.; Voit, E.O.; An, G. Mechanistic simulations of inflammation: Current state and future prospects. Math. Biosci. 2009, 217, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Ballina, M.; Tracey, K.J. The neurology of the immune system: Neural reflexes regulate immunity. Neuron. 2009, 64, 28–32. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Mathew, S.; Bartels, J.; Banerjee, I.; Vodovotz, Y. Global sensitivity analysis of a mathematical model of acute inflammation identifies nonlinear dependence of cumulative tissue damage on host interleukin-6 responses. J. Biol. 2014, 358, 132–148. [Google Scholar] [CrossRef] [Green Version]
- De Siqueira Santos, S.; Takahashi, D.Y.; Nakata, A.; Fujita, A. A comparative study of statistical methods used to identify dependencies between gene expression signals. Brief. Bioinform. 2014, 15, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Scardoni, G.; Petterlini, M.; Laudanna, C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25, 2857–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe: Centralities and experimental dataset integration. F1000 Res. 2014, 3, 139. [Google Scholar] [CrossRef]
- Jalili, M.; Salehzadeh-Yazdi, A.; Asgari, Y.; Arab, S.S.; Yaghmaie, M.; Ghavamzadeh, A.; Alimoghaddam, K. CentiServer: A comprehensive resource, web-based application and R package for centrality analysis. PLoS ONE 2015, 10, e0143111. [Google Scholar] [CrossRef]
- Subramanian, N.; Torabi-Parizi, P.; Gottschalk, R.A.; Germain, R.N.; Dutta, B. Network representations of immune system complexity. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 13–38. [Google Scholar] [CrossRef]
- Albert, R. Scale-free networks in cell biology. J. Cell Sci. 2005, 118 Pt 21, 4947–4957. [Google Scholar] [CrossRef] [Green Version]
- Przytycka, T.M.; Singh, M.; Slonim, D.K. Toward the dynamic interactome: it’s about time. Brief. Bioinform. 2010, 11, 15–29. [Google Scholar] [CrossRef]
- Rowland, B.; Savarraj, J.P.J.; Karri, J.; Zhang, X.; Cardenas, J.; Choi, H.A.; Holcomb, J.B.; Wade, C.E. Acute inflammation in traumatic brain injury and polytrauma patients using network analysis. Shock 2019. (published ahead of print). [Google Scholar] [CrossRef]
- McKee, C.A.; Lukens, J.R. Emerging Roles for the Immune System in Traumatic Brain Injury. Front. Immunol. 2016, 7, 556. [Google Scholar] [CrossRef] [Green Version]
- Appavu, B.; Foldes, S.T.; Adelson, P.D. Clinical trials for pediatric traumatic brain injury: Definition of insanity? J. Neurosurg. Pediatr. 2019, 23, 661–669. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Severity | Clinical Criteria | Glasgow Coma Scale (GCS) Score |
|---|---|---|
| Symptomatic (Possible) | - Blurred Vision - Confusion - Dazed - Dizziness - Focal neurologic symptoms - Headache - Nausea | Mild: 13–15 |
| Mild (Probable) | - LOC < 30 min - Post-traumatic anterograde amnesia < 24 h - Depressed, basilar, or linear skull fracture (dura intact) | |
| Moderate–Severe (Definite) | - LOC > 30 min - PTA ≥ 24 h - GCS < 13 - One or more of ICH, subdural/epidural hematoma, cerebral contusion, hemorrhagic contusion, penetrating TBI, SAH, brain stem injury | Moderate: 9–12 Severe: 3–8 |
| Biological Mechanism | Connection to Inflammation |
|---|---|
| Excitotoxicity | - As glutamate is known to be a co-stimulator of T cells and a potent gliotransmitter, decreased uptake of glutamate, via downregulation of excitatory amino acid transporters on astrocytes [39] and alterations of GABAergic interneurons [40,41], reduces inhibition of neighbouring excitatory circuits and can activate immune cells (astrocytes, microglia, etc.) |
| Mitochondrial Dysfunction & Metabolic Disruption | - Increased Ca2+ influx can overload the mitochondria, promoting network fission [42]. Mitochondrial network fission increases reactive oxygen species (ROS) production, reduces oxidative phosphorylation [43], and has been shown to be required for the activation of microglia in vitro [44] - A shift to glycolysis within neurons is enhanced by cytokines produced by nearby immune cells, notably through activation of the PI3K-mTOR pathway [45]. In combination with mitochondrial dysfunction and increased ROS, this can lead to neuronal degeneration and increase levels of damage-associated molecular patterns (DAMPs) in the surrounding tissue |
| Increased Oxidative Stress | - Stabilizes HIF1α [46] and promotes NLRP3 [47] inflammasome formation necessary for the production of inflammatory mediators |
| Weakened BBB Integrity | - Increased leakage of DAMPs, including GFAP, NFL, p-tau, and UCH-L1, into the bloodstream and extravasation of peripheral immune cells into the brain [48] |
| Cytoskeletal Breakdown/Protein Aggregation | - Increased Ca2+ influx can activate Ca2+-dependent enzymes, such as calpains [49], leading to cytoskeletal breakdown. These broken down proteins can then leak into the peripheral circulation and/or aggregate into plaques within the CNS, leading to further inflammation |
| Cerebral Blood Flow Dysregulation | - Hypoxia or ischemia can kill cells, causing them to release their internal contents and activate surrounding immune cells via DAMPs and PRRs - Hypoxia/ischemia can stabilize HIF1α in an ROS-independent manner [50] to increase production of cytokines |
| Edema (Vasogenic) | - Facilitated by neurogenic inflammation (release of Substance P and neurokinins) - Increases ability of immune cells to extravasate into the brain - Increases transmission of DAMPs from the brain into the blood to recruit peripheral immune cells |
| Edema (Cytotoxic) | - Influx of water into the cell can lead to swelling and membrane and organelle disruption, leading to cell death and release of DAMPs into the extracellular space |
| Glial Cell Activation | - Injury to the CNS activates astrocytes and microglia, which reciprocally signal to activate (and de-activate) gliosis. These signals include an initial burst of purinergic substrates, such as ATP from astrocytes, which activate the P2Y12R and P2X4R purinergic receptors [51,52], leading to microglial process extension towards the injury site - Microglia signal to astrocytes to convert them to a neuroprotective phenotype via downregulation of the P2Y1R receptor on the astrocyte surface using TNFα, IL-1β, and IL-6 [53]. The converse can also occur, with microglia inducing a toxic astrocyte phenotype through secreting TNFα, IL-1β, and C1q [54] - CX3CR1 on microglia exhibits a time dependent effect on outcome after injury, playing a key role in inflammation (accumulation of leukocytes [55]), but is required for proper recovery in severe [56] and mild [57] TBI - Formation of a glial scar using Eph/ephrin signaling, namely EphA4 and CSPGs in the CNS [58], can affect vascular permeability and enhance immune cell migration into the injured CNS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraunberger, E.; Esser, M.J. Neuro-Inflammation in Pediatric Traumatic Brain Injury—from Mechanisms to Inflammatory Networks. Brain Sci. 2019, 9, 319. https://doi.org/10.3390/brainsci9110319
Fraunberger E, Esser MJ. Neuro-Inflammation in Pediatric Traumatic Brain Injury—from Mechanisms to Inflammatory Networks. Brain Sciences. 2019; 9(11):319. https://doi.org/10.3390/brainsci9110319
Chicago/Turabian StyleFraunberger, Erik, and Michael J. Esser. 2019. "Neuro-Inflammation in Pediatric Traumatic Brain Injury—from Mechanisms to Inflammatory Networks" Brain Sciences 9, no. 11: 319. https://doi.org/10.3390/brainsci9110319
APA StyleFraunberger, E., & Esser, M. J. (2019). Neuro-Inflammation in Pediatric Traumatic Brain Injury—from Mechanisms to Inflammatory Networks. Brain Sciences, 9(11), 319. https://doi.org/10.3390/brainsci9110319