Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation

 ,
,  , , , and
, , , and 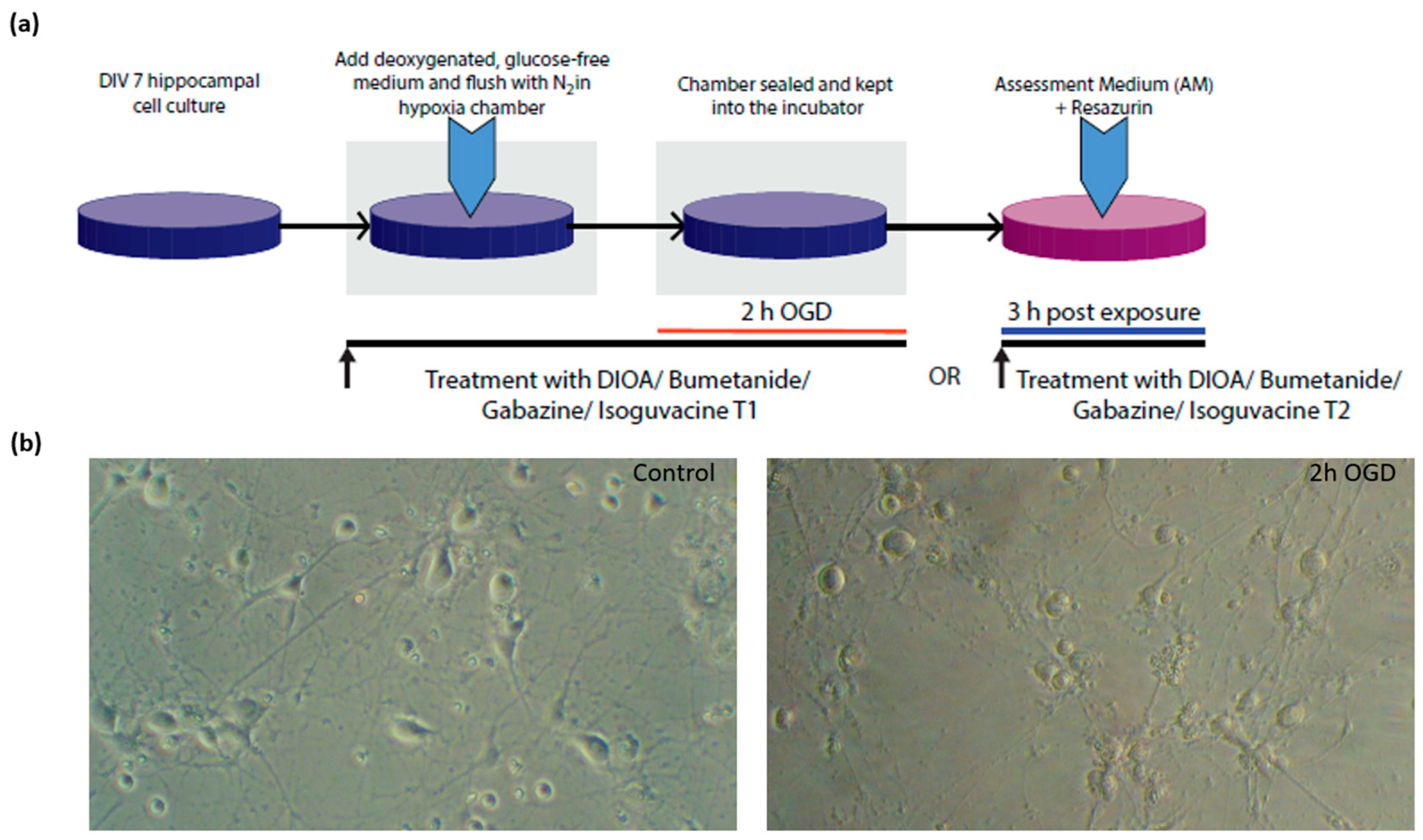{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Cultures of Hippocampal Neurons
2.2. Exposure to Oxygen-Glucose Deprivation (OGD)
2.3. Assessment of Cellular Metabolism and Viability
2.4. Treatment with Chloride Membrane Transport Blockers
2.5. Data Analysis
3. Results
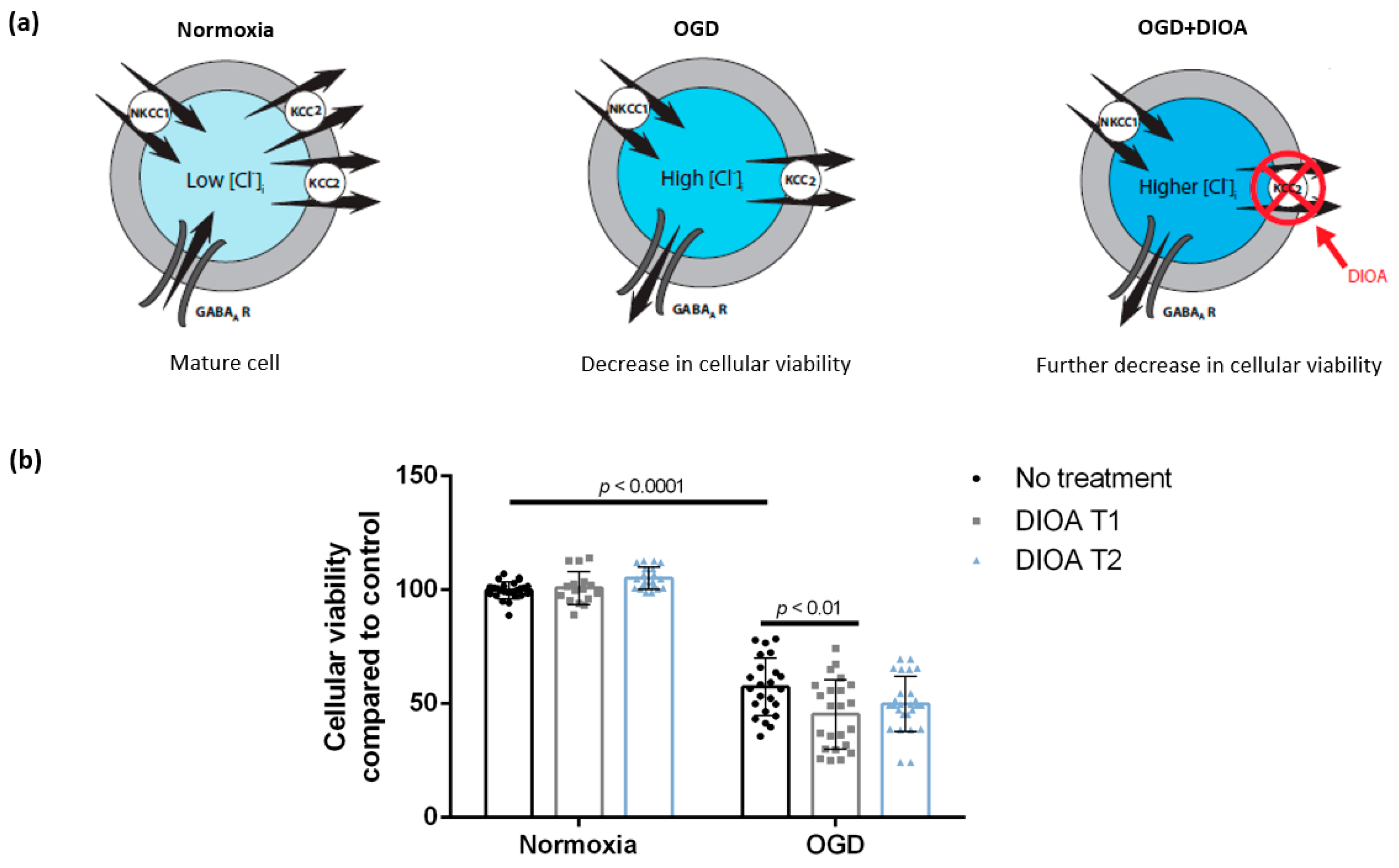
3.1. DIOA Treatment during Oxygen-Glucose Deprivation Decreases Cellular Viability of DIV 7 Hippocampal Cell Cultures
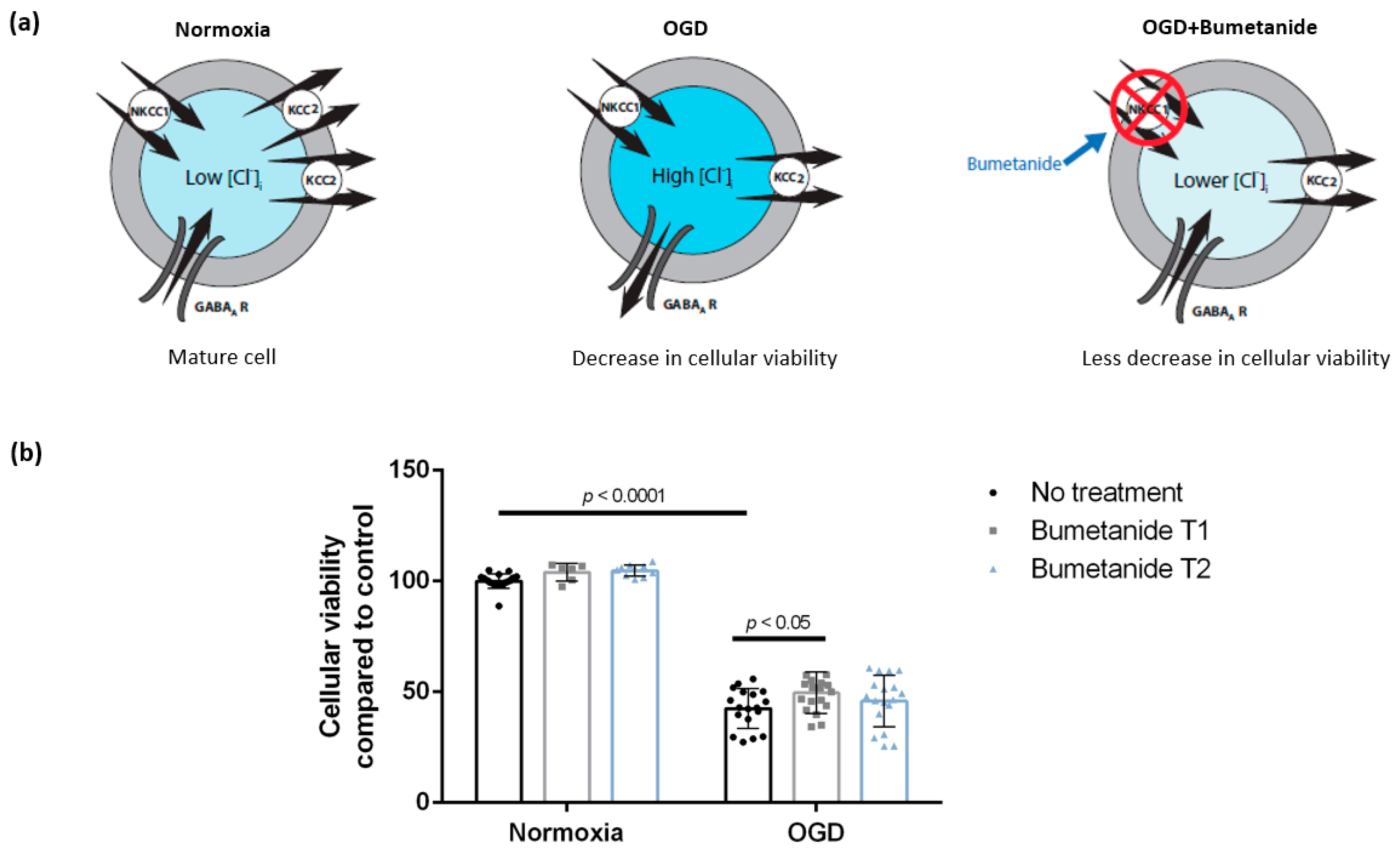
3.2. Treatment with Bumetanide during Oxygen-Glucose Deprivation Is Associated with Increased Cellular Viability of DIV 7 Hippocampal Cells
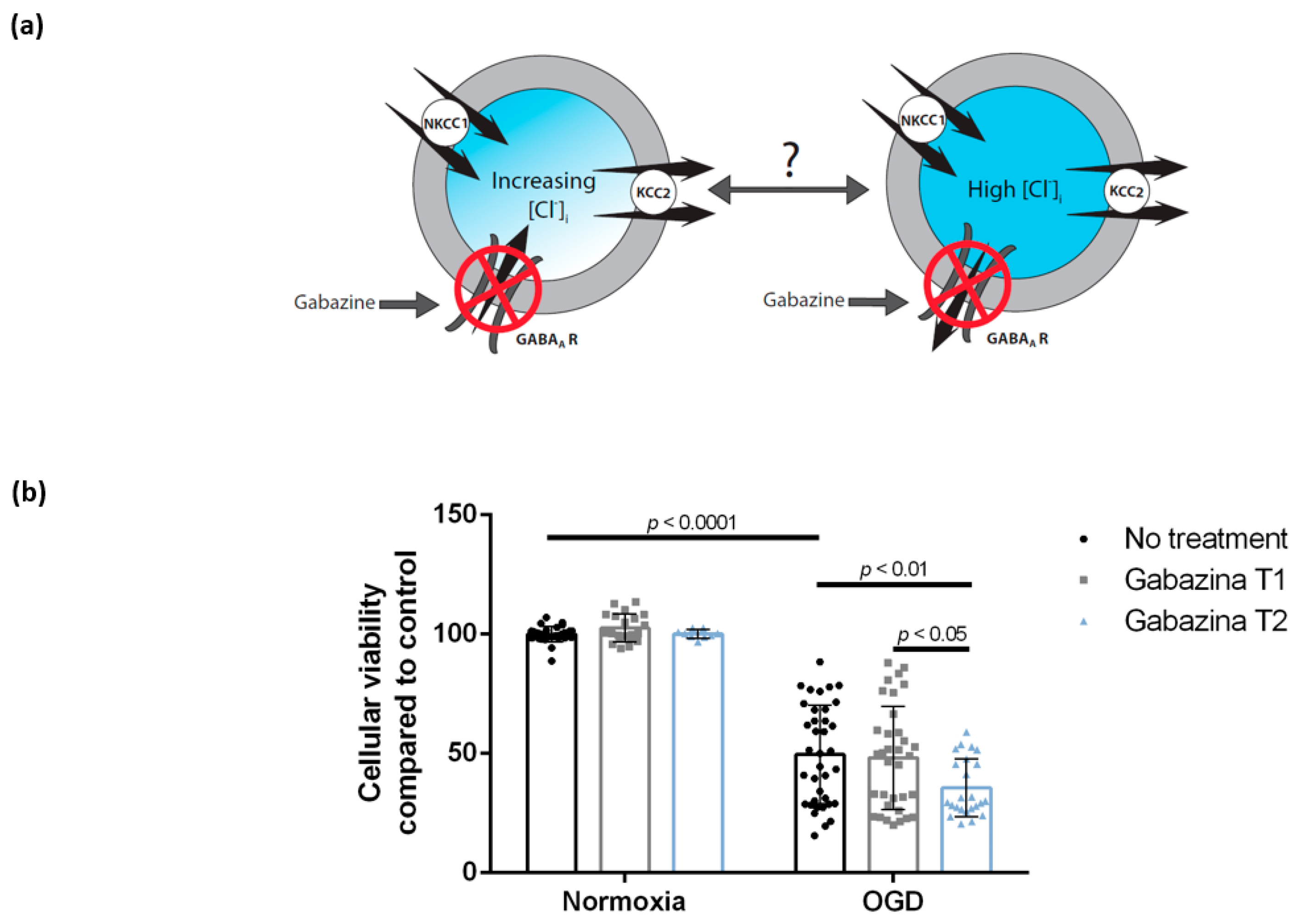
3.3. Blocking the GABAA Receptor Using Gabazine Post-Exposure, but Not during Oxygen-Glucose Deprivation, Is Associated with Decreased Cellular Viability of DIV 7 Hippocampal Cells
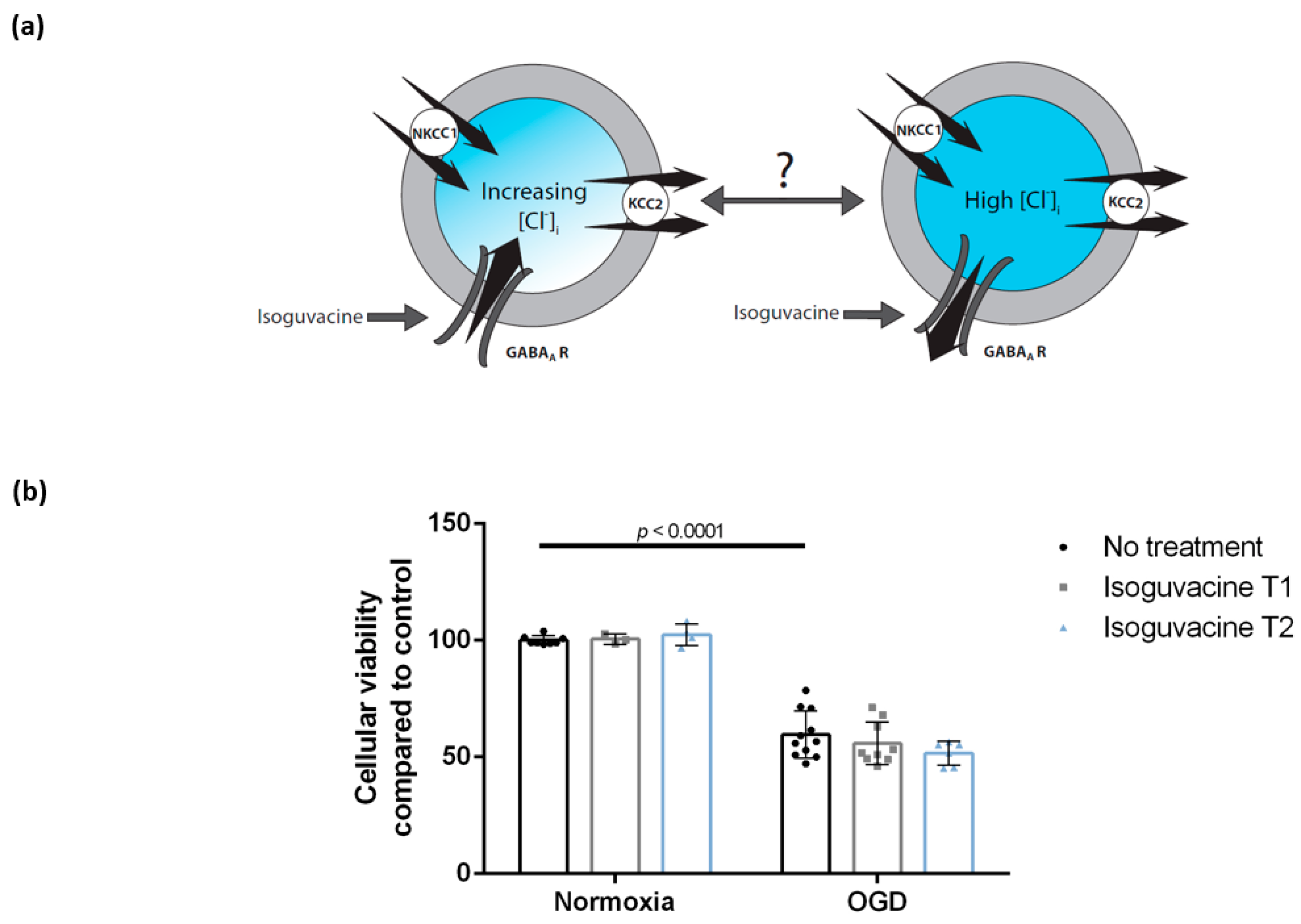
3.4. Enhancing GABAA Activation Using Isoguvacine Does Not Influence Cellular Viability Either during Oxygen-Glucose Deprivation or Post-Exposure
4. Discussion
4.1. Treatment with Bumetanide Is Associated with Increased Cellular Viability of DIV 7 Hippocampal Cells after Oxygen-Glucose Deprivation
4.2. Treatment with DIOA Is Associated with Decreased Cellular Viability of DIV 7 Hippocampal Cell Cultures after Oxygen-Glucose Deprivation
4.3. Blocking of the GABAA Receptor Using Gabazine during Reoxygenation, but Not during Oxygen-Glucose Deprivation, Is Associated with Decreased Cellular Viability of DIV 7 Hippocampal Cells
4.4. Enhancing GABAA Activation Using Isoguvacine Does Not Influence Cellular Viability Either during Oxygen-Glucose Deprivation or Post-Exposure
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J.L. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Rha, J.-H.; Saver, J.L. The impact of recanalization on ischemic stroke outcome: A meta-analysis. Stroke 2007, 38, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Thomalla, G.; Simonsen, C.Z.; Boutitie, F.; Andersen, G.; Berthezene, Y.; Cheng, B.; Cheripelli, B.; Cho, T.-H.; Fazekas, F.; Fiehler, J.; et al. MRI-Guided Thrombolysis for Stroke with Unknown Time of Onset. N. Engl. J. Med. 2018, 379, 611–622. [Google Scholar] [CrossRef]
- Mendez, A.A.; Samaniego, E.A.; Sheth, S.A.; Dandapat, S.; Hasan, D.M.; Limaye, K.S.; Hindman, B.J.; Derdeyn, C.P.; Ortega-Gutierrez, S. Update in the Early Management and Reperfusion Strategies of Patients with Acute Ischemic Stroke. Crit. Care Res. Pract. 2018, 2018, 9168731. [Google Scholar] [CrossRef]
- Oddo, M.; Crippa, I.A.; Mehta, S.; Menon, D.; Payen, J.-F.; Taccone, F.S.; Citerio, G. Optimizing sedation in patients with acute brain injury. Crit. Care 2016, 20, 128. [Google Scholar] [CrossRef]
- Huang, L.-Q.; Zhu, G.-F.; Deng, Y.-Y.; Jiang, W.-Q.; Fang, M.; Chen, C.-B.; Cao, W.; Wen, M.-Y.; Han, Y.-L.; Zeng, H.-K. Hypertonic saline alleviates cerebral edema by inhibiting microglia-derived TNF-α and IL-1β-induced Na-K-Cl Cotransporter up-regulation. J. Neuroinflamm. 2014, 11, 102. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Shen, F.C.; Xie, D.; Han, Q.P.; Fang, M.; Chen, C.B.; Zeng, H.K. Progress in Drug Treatment of Cerebral Edema. Mini Rev. Med. Chem. 2016, 16, 917–925. [Google Scholar] [CrossRef]
- Mayor, D.; Tymianski, M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 2018, 134, 178–188. [Google Scholar] [CrossRef]
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-Receptor Trafficking and Synaptic Dysfunction in Brain Disorders. Front. Cell. Neurosci. 2019, 13, 77. [Google Scholar] [CrossRef]
- Smith, K.R.; Muir, J.; Rao, Y.; Browarski, M.; Gruenig, M.C.; Sheehan, D.F.; Haucke, V.; Kittler, J.T. Stabilization of GABA(A) receptors at endocytic zones is mediated by an AP2 binding motif within the GABA(A) receptor β3 subunit. J. Neurosci. 2012, 32, 2485–2498. [Google Scholar] [CrossRef] [PubMed]
- Mielke, J.G.; Wang, Y.T. Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABA receptors following oxygen-glucose deprivation in cultured cortical neurons. J. Neurochem. 2005, 92, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Dzhala, V.I.; Talos, D.M.; Sdrulla, D.A.; Brumback, A.C.; Mathews, G.C.; Benke, T.A.; Delpire, E.; Jensen, F.E.; Staley, K.J. NKCC1 transporter facilitates seizures in the developing brain. Nat. Med. 2005, 11, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA Excitatory/Inhibitory Shift in Brain Maturation and Neurological Disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef]
- Wu, H.; Che, X.; Tang, J.; Ma, F.; Pan, K.; Zhao, M.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. The K(+)-Cl(−) Cotransporter KCC2 and Chloride Homeostasis: Potential Therapeutic Target in Acute Central Nervous System Injury. Mol. Neurobiol. 2016, 53, 2141–2151. [Google Scholar] [CrossRef]
- Jaenisch, N.; Witte, O.W.; Frahm, C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 2010, 41, e151–e159. [Google Scholar] [CrossRef]
- Galeffi, F.; Sah, R.; Pond, B.B.; George, A.; Schwartz-Bloom, R.D. Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: Effect of diazepam. J. Neurosci. 2004, 24, 4478–4488. [Google Scholar] [CrossRef]
- Coull, J.A.M.; Boudreau, D.; Bachand, K.; Prescott, S.A.; Nault, F.; Sík, A.; De Koninck, P.; De Koninck, Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 2003, 424, 938–942. [Google Scholar] [CrossRef]
- Bonislawski, D.P.; Schwarzbach, E.P.; Cohen, A.S. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol. Dis. 2007, 25, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Pond, B.B.; Berglund, K.; Kuner, T.; Feng, G.; Augustine, G.J.; Schwartz-Bloom, R.D. The chloride transporter Na(+)-K(+)-Cl- cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J. Neurosci. 2006, 26, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.-T.; Huang, T.-C.; Wang, J.-Y.; You, Y.-S.; Chou, J.-L.; Chan, M.W.Y.; Wo, P.Y.Y.; Amstislavskaya, T.G.; Tikhonova, M.A.; Yang, Y.-L. NKCC1 mediates traumatic brain injury-induced hippocampal neurogenesis through CREB phosphorylation and HIF-1α expression. Pflug. Arch. 2015, 467, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Ceanga, M.; Spataru, A.; Zagrean, A.-M. Oxytocin is neuroprotective against oxygen–glucose deprivation and reoxygenation in immature hippocampal cultures. Neurosci. Lett. 2010, 477, 15–18. [Google Scholar] [CrossRef]
- Panaitescu, A.M.; Isac, S.; Pavel, B.; Ilie, A.; Ceanga, M.; Totan, A.; Zagrean, L.; Peltecu, G.; Zagrean, A.-M. Oxytocin Reduces Seizure Burden and Hippocampal Injury in a Rat Model of Perinatal Asphyxia. Acta Endocrinol. 2018, 14, 315–319. [Google Scholar] [CrossRef]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef]
- Pellegrino, C.; Gubkina, O.; Schaefer, M.; Becq, H.; Ludwig, A.; Mukhtarov, M.; Chudotvorova, I.; Corby, S.; Salyha, Y.; Salozhin, S.; et al. Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J. Physiol. 2011, 589, 2475–2496. [Google Scholar] [CrossRef]
- Voytenko, L.P.; Lushnikova, I.V.; Savotchenko, A.V.; Isaeva, E.V.; Skok, M.V.; Lykhmus, O.Y.; Patseva, M.A.; Skibo, G.G. Hippocampal GABAergic interneurons coexpressing alpha7-nicotinic receptors and connexin-36 are able to improve neuronal viability under oxygen-glucose deprivation. Brain Res. 2015, 1616, 134–145. [Google Scholar] [CrossRef]
- Brewer, G.J.; Torricelli, J.R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2007, 2, 1490–1498. [Google Scholar] [CrossRef]
- Valeeva, G.; Valiullina, F.; Khazipov, R. Excitatory actions of GABA in the intact neonatal rodent hippocampus in vitro. Front. Cell. Neurosci. 2013, 7, 20. [Google Scholar] [CrossRef]
- Leonzino, M.; Busnelli, M.; Antonucci, F.; Verderio, C.; Mazzanti, M.; Chini, B. The Timing of the Excitatory-to-Inhibitory GABA Switch Is Regulated by the Oxytocin Receptor via KCC2. Cell Rep. 2016, 15, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Le Duc, D.; Spataru, A.; Ceanga, M.; Zagrean, L.; Schöneberg, T.; Toescu, E.C.; Zagrean, A.-M. Developmental exposure to ethanol increases the neuronal vulnerability to oxygen-glucose deprivation in cerebellar granule cell cultures. Brain Res. 2015, 1614, 1–13. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; DiCaprio, M.J.; Greenberg, D.A. Assessment of neuronal viability with Alamar blue in cortical and granule cell cultures. J. Neurosci. Methods 1996, 70, 195–200. [Google Scholar] [CrossRef]
- Spataru, A.; Le Duc, D.; Zagrean, L.; Zagrean, A.-M. Ethanol exposed maturing rat cerebellar granule cells show impaired energy metabolism and increased cell death after oxygen-glucose deprivation. Neural Regen. Res. 2019, 14, 485–490. [Google Scholar]
- Beck, J.; Lenart, B.; Kintner, D.B.; Sun, D. Na-K-Cl cotransporter contributes to glutamate-mediated excitotoxicity. J. Neurosci. 2003, 23, 5061–5068. [Google Scholar] [CrossRef]
- Blauwblomme, T.; Dzhala, V.; Staley, K. Transient ischemia facilitates neuronal chloride accumulation and severity of seizures. Ann. Clin. Transl. Neurol. 2018, 5, 1048–1061. [Google Scholar] [CrossRef]
- Rothman, S.M. The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J. Neurosci. 1985, 5, 1483–1489. [Google Scholar] [CrossRef]
- Allen, N.J.; Rossi, D.J.; Attwell, D. Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J. Neurosci. 2004, 24, 3837–3849. [Google Scholar] [CrossRef]
- Glykys, J.; Dzhala, V.; Egawa, K.; Kahle, K.T.; Delpire, E.; Staley, K. Chloride Dysregulation, Seizures, and Cerebral Edema: A Relationship with Therapeutic Potential. Trends Neurosci. 2017, 40, 276–294. [Google Scholar] [CrossRef]
- Sun, D.; Murali, S.G. Stimulation of Na+-K+-2Cl- cotransporter in neuronal cells by excitatory neurotransmitter glutamate. Am. J. Physiol. 1998, 275, C772–C779. [Google Scholar] [CrossRef]
- Su, G.; Haworth, R.A.; Dempsey, R.J.; Sun, D. Regulation of Na(+)-K(+)-Cl(−) cotransporter in primary astrocytes by dibutyryl cAMP and high [K(+)](o). Am. J. Physiol. Cell Physiol. 2000, 279, C1710–C1721. [Google Scholar] [CrossRef]
- Schomberg, S.L.; Su, G.; Haworth, R.A.; Sun, D. Stimulation of Na-K-2Cl cotransporter in neurons by activation of Non-NMDA ionotropic receptor and group-I mGluRs. J. Neurophysiol. 2001, 85, 2563–2575. [Google Scholar] [CrossRef]
- Begum, G.; Yuan, H.; Kahle, K.T.; Li, L.; Wang, S.; Shi, Y.; Shmukler, B.E.; Yang, S.-S.; Lin, S.-H.; Alper, S.L.; et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke 2015, 46, 1956–1965. [Google Scholar] [CrossRef]
- Tyzio, R.; Cossart, R.; Khalilov, I.; Minlebaev, M.; Hübner, C.A.; Represa, A.; Ben-Ari, Y.; Khazipov, R. Maternal Oxytocin Triggers a Transient Inhibitory Switch in GABA Signaling in the Fetal Brain During Delivery. Science 2006, 314, 1788–1792. [Google Scholar] [CrossRef]
- Karadsheh, M.F.; Delpire, E. Neuronal restrictive silencing element is found in the KCC2 gene: Molecular basis for KCC2-specific expression in neurons. J. Neurophysiol. 2001, 85, 995–997. [Google Scholar] [CrossRef]
- Uvarov, P.; Pruunsild, P.; Timmusk, T.; Airaksinen, M.S. Neuronal K+/Cl- co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J. Neurochem. 2005, 95, 1144–1155. [Google Scholar] [CrossRef]
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+]o regulation. Am. J. Physiol. 1997, 273, C1516–C1525. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- DeFazio, R.A.; Keros, S.; Quick, M.W.; Hablitz, J.J. Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J. Neurosci. 2000, 20, 8069–8076. [Google Scholar] [CrossRef]
- Babot, Z.; Cristòfol, R.; Suñol, C. Excitotoxic death induced by released glutamate in depolarized primary cultures of mouse cerebellar granule cells is dependent on GABAA receptors and niflumic acid-sensitive chloride channels. Eur. J. Neurosci. 2005, 21, 103–112. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Oomura, Y.; Yakushiji, T.; Akaike, N. Intracellular calcium ions decrease the affinity of the GABA receptor. Nature 1986, 324, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, A.; Shi, H. Impairment of GABAA receptor function by N-methyl-D-aspartate-mediated calcium influx in isolated CA1 pyramidal cells. Neuroscience 1994, 62, 813–828. [Google Scholar] [CrossRef]
- Schwartz-Bloom, R.D.; Sah, R. gamma-Aminobutyric acid(A) neurotransmission and cerebral ischemia. J. Neurochem. 2001, 77, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Michelson, H.B.; Wong, R.K. Excitatory synaptic responses mediated by GABAA receptors in the hippocampus. Science 1991, 253, 1420–1423. [Google Scholar] [CrossRef] [PubMed]
- Staley, K.J.; Mody, I. Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J. Neurophysiol. 1992, 68, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Staley, K.J.; Soldo, B.L.; Proctor, W.R. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 1995, 269, 977–981. [Google Scholar] [CrossRef]
- Gulledge, A.T.; Stuart, G.J. Excitatory actions of GABA in the cortex. Neuron 2003, 37, 299–309. [Google Scholar] [CrossRef]
- Abramowicz, A.E.; Kass, I.S.; Chambers, G.; Cottrell, J.E. Midazolam improves electrophysiologic recovery after anoxia and reduces the changes in ATP levels and calcium influx during anoxia in the rat hippocampal slice. Anesthesiology 1991, 74, 1121–1128. [Google Scholar] [CrossRef]
- Schwartz, R.D.; Yu, X.; Katzman, M.R.; Hayden-Hixson, D.M.; Perry, J.M. Diazepam, given postischemia, protects selectively vulnerable neurons in the rat hippocampus and striatum. J. Neurosci. 1995, 15, 529–539. [Google Scholar] [CrossRef]
- Galeffi, F.; Sinnar, S.; Schwartz-Bloom, R.D. Diazepam promotes ATP recovery and prevents cytochrome c release in hippocampal slices after in vitro ischemia. J. Neurochem. 2000, 75, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Jennum, P.; Baandrup, L.; Iversen, H.K.; Ibsen, R.; Kjellberg, J. Mortality and use of psychotropic medication in patients with stroke: A population-wide, register-based study. BMJ Open 2016, 6, e010662. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B. Potential effects of common drugs on stroke recovery. Arch. Neurol. 1998, 55, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, X.; Kang, F.; Chen, Z.; Meng, Y.; Dai, M. Neuroprotective effects of midazolam on focal cerebral ischemia in rats through anti‑apoptotic mechanisms. Int. J. Mol. Med. 2019, 43, 443–451. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagrean, A.-M.; Grigoras, I.-F.; Iesanu, M.I.; Ionescu, R.-B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain Sci. 2019, 9, 360. https://doi.org/10.3390/brainsci9120360
Zagrean A-M, Grigoras I-F, Iesanu MI, Ionescu R-B, Chitimus DM, Haret RM, Ianosi B, Ceanga M, Zagrean L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain Sciences. 2019; 9(12):360. https://doi.org/10.3390/brainsci9120360
Chicago/Turabian StyleZagrean, Ana-Maria, Ioana-Florentina Grigoras, Mara Ioana Iesanu, Rosana-Bristena Ionescu, Diana Maria Chitimus, Robert Mihai Haret, Bogdan Ianosi, Mihai Ceanga, and Leon Zagrean. 2019. "Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation" Brain Sciences 9, no. 12: 360. https://doi.org/10.3390/brainsci9120360
APA StyleZagrean, A.-M., Grigoras, I.-F., Iesanu, M. I., Ionescu, R.-B., Chitimus, D. M., Haret, R. M., Ianosi, B., Ceanga, M., & Zagrean, L. (2019). Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain Sciences, 9(12), 360. https://doi.org/10.3390/brainsci9120360