Somatic Versus Dynamic Genome
The human genome can be explored in two different dimensions: the somatic genome and the dynamic genome. The somatic genome is the heritable DNA structure of an organism, with mutational heterogeneity that can be either the cause or effect of disease. To investigate hereditable factors and somatic mutations in disease, researchers have explored the somatic genome, using such methods as DNA sequencing technologies, single nucleotide polymorphism arrays, and genome-wide association studies. The results have been biologically informative and have produced a few clear medical and clinical successes, such as BRCA and HER2 testing in breast cancer. However, in general, results have been disappointing [
1]. A fundamental problem with cancer DNA genome studies is that the genetic mutations for any given cancer type discovered by modern sensitive analytical methods number in the hundreds, and the determination of the true somatic mutation(s) driving cancer progression is difficult in the clinical setting for any individual patient [
2]. Furthermore, cancer therapies targeted at a single somatic mutation have proven to have limited effect over time, because of resistance caused by cellular somatic evolution [
3]. As human cancer is usually polypoid, containing subpopulations of multiple aneuploid cancer tumor cells, aneuploidy is also the hallmark of cancer cells in general [
4]. Ploidy sequencing has been proved to be beneficial in revealing the somatic evolution of cancer tumor cells [
5]. However, the clinic application of DNA ploidy assessment is very limited for cancer detection because it is still controversial that aneuploidy is the cause of cancer [
6].
The other approach—exploring the dynamic genome—is one that we believe to be more powerful for clinical applications. This approach investigates not the DNA of an organism, but the transcriptional activity of an organism’s genes. The result of this activity is the transcriptome: the complete set of RNA transcripts present in a cell or tissue at any one time. Although the DNA of a particular cell or tissue, the genome, is uniform throughout the organism and except for infrequent random mutations, essentially unchanging, its transcriptome may vary according to the current physiological status of the cell, tissue or organism. Since mRNA profiles will alter in response to the cellular environment, the transcriptome will always be changing in response to immune factors, drugs, disease onset and progression, and healing [
7].
To date, the dynamic genome has best been interrogated using microarray studies. Microarray chips can provide a snapshot of an organism’s gene expression activity at a given time. Compared to RNA sequencing, microarray is more established and cost-effective in analyzing the expression of defined genes by high throughput methods. Furthermore, microarray data is not as complex as that of RNA sequencing, which make it easier to analyze and apply widely to various fields. However, traditional, tissue-based microarray studies have a number of disadvantages. Invasive biopsies can be obtained only in very late stage disease, at transplant or after death in the case of difficult-to-access organs such as lung, breast, prostate, heart, and brain. For these reasons, tissue-based microarray is less useful for research in early-stage disease, or in the clinic.
One additional limiting factor in any tissue-based technology is the problem of heterogeneity. Diseased cells are not necessarily homogeneously distributed throughout tissue, and in cancer, malignant cells can differ from each other in their mutations [
8]. Thus, analysis based on solid biopsy needs to take these factors into account by taking multiple invasive samples. However, this requirement increases the cost of the test and the test’s inconvenience to patients. To avoid this problem, so called “liquid biopsies” attempt to detect circulating tumor cells from a blood sample. While the presence of circulating tumor cells is a strong prognostic factor for overall survival in certain cancer patients, the clinical significance of circulating tumor cells in most patients is still unknown [
9]. Furthermore since circulating tumor cells are very few in number in early stage cancer, analysis requires extreme analytical sensitivity to detect what very few cells are present and at a cost of many false positives, making testing unreliable.
By contrast, white blood cells in peripheral blood provide a near ideal diagnostic sample. Blood sampling is a long-established and well-accepted procedure for disease diagnosis and monitoring. Whole blood is easy to access, and patients and physicians are accustomed to blood sampling. White blood cells are much more abundant than circulating tumor cells, which eliminates the challenge of analytical sensitivity. Furthermore, because the sample is liquid, the distribution of cells is homogeneous. This reduces or eliminates the need for multiple samples. Another advantage of using whole blood is that the immune cells in blood are biologically affected by disease located elsewhere in the body regardless of the tissue affected [
10,
11]. Thus the requirement for direct biopsy is reduced and even potentially eliminated. For these reasons blood-based transcriptomics has significant advantages over tissue-based biopsy technology. We propose this concept as the Sentinel Principle
®, which employs the circulating blood cells as “sentinels” for detecting and responding to micro-environmental changes in the body.
Blood-based transcriptomics may also come to play an important role in detecting disease at an early stage in which clinically apparent pathological variants have not yet emerged. Blood cells act as transporter cells and as mediators of the immune response and are involved in the pathogenesis of many diseases [
12]. Thus when physiological or pathological insults occur anywhere in the organism, the gene expression profile of the peripheral blood cells will change in order to carry and to transfer information to engage the immune system and maintain physiological homeostasis [
13]. Our previous research has shown that peripheral blood cells respond differently to various pathological changes and thus analysis of differential gene expression can distinguish between and among diseases [
14]. Furthermore, since the interaction between the immune system and disease usually precedes the occurrence of clinically pathological variation, the study of blood cell profiles might make it possible to detect diseases at an early stage. For instance, according to the cancer immunoediting theory, cancer has a long equilibrium phase in which tumor cells survive immune elimination and maintain a state of functional tumor dormancy [
15]. During the equilibrium phase, there is no clinically detectable pathological variation, but the interaction of the immune system against cancer cells occurs covertly in the body. Our hypothesis is that early stage cancer and other diseases can be detected by analysis of variation in the gene expression profiles of peripheral blood.
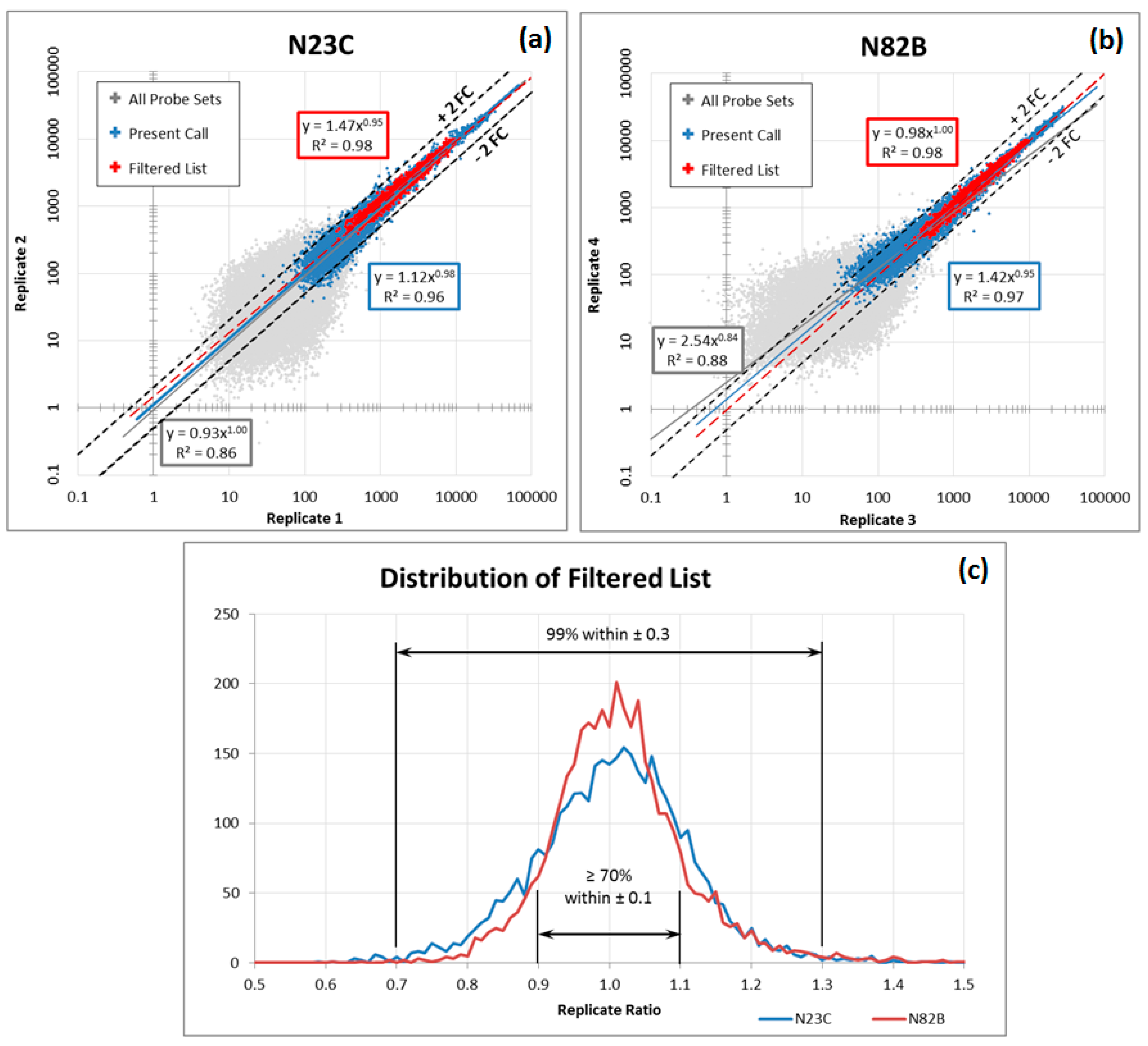
Although blood-based transcriptomics research has great potential in clinical application, it has its own set of limitations. In order optimally to measure the expression levels of messenger RNA in whole blood, several challenges need to be overcome. First, whole blood introduces into sampling the factor of white blood cell population distribution. Second, the dynamic composition of blood cells in response to a constantly changing environment means that even copy numbers normalized to total cell count and sample volume may exhibit variability. Third, sampling technology can introduce additional artefacts, such as differences between EDTA and PAXgene™ (PreAnalytiX) collection tubes and protocols. Moreover, microarrays from different manufacturers each have their own peculiarities and need corrective measures tailored for each. These factors make the analysis of the data more complex and time consuming. A practical solution is required that can identify well-performing gene panels in the face of these challenges.
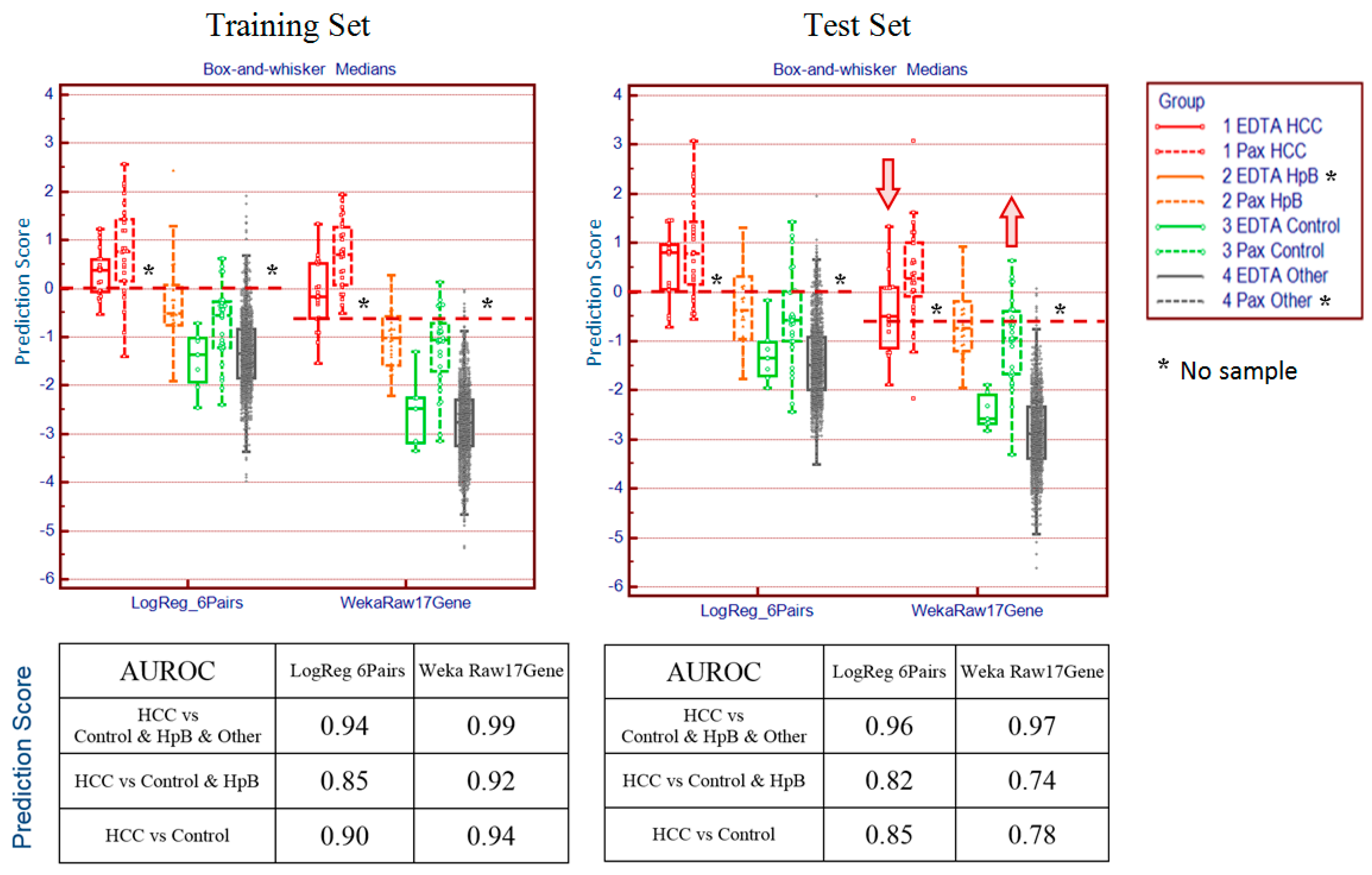
The most common of these limitations, the interference derived from different blood sampling technologies, cannot be ignored in blood-based transcriptomics research. The conventional method for drawing blood uses EDTA collection tubes, which inhibit clotting but do not stabilise intracellular RNA. When EDTA blood collection tubes are used, intracellular RNA needs to be isolated within four hours of collection, as RNA degrades rapidly. Therefore, EDTA collection tubes are not practical for clinical applications when tests involve RNA and the collection sites are far from the laboratory. To overcome this problem, PAXgene tubes contain reagents that stabilise RNA, allowing easy blood collection, storage and transport of blood samples [
16]. However, excessive globin mRNA levels interfere with transcript measurement and increase variability. This difficulty is addressed by the use of specifically designed reagents with different degree of globin signal suppression. Thus, gene expression profiles derived from EDTA and PAXgene blood collection tubes are not completely consistent between samples drawn from the same patient and processed under different protocols, an inconsistency that may lead to confusing or contradictory results.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









