
Glutathione Peroxidase 8 Suppression by Histone Deacetylase Inhibitors Enhances Endoplasmic Reticulum Stress and Cell Death by Oxidative Stress in Hepatocellular Carcinoma Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies, Plasmids, and Chemicals
2.2. Cell Culture and Cell Viability Test and Animal Ethics
2.3. RNA Extraction and RNA-Seq
2.4. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.5. Fluorescence-Activated Cell Sorting (FACS)
2.6. Western Blotting
2.7. Immunofluorescence (IF)
2.8. Chromatin Immunoprecipitation (ChIP)
2.9. Glutathione Assay
2.10. Statistics
3. Results
3.1. HDACi Repressed Expression of GPX8 in the HCC
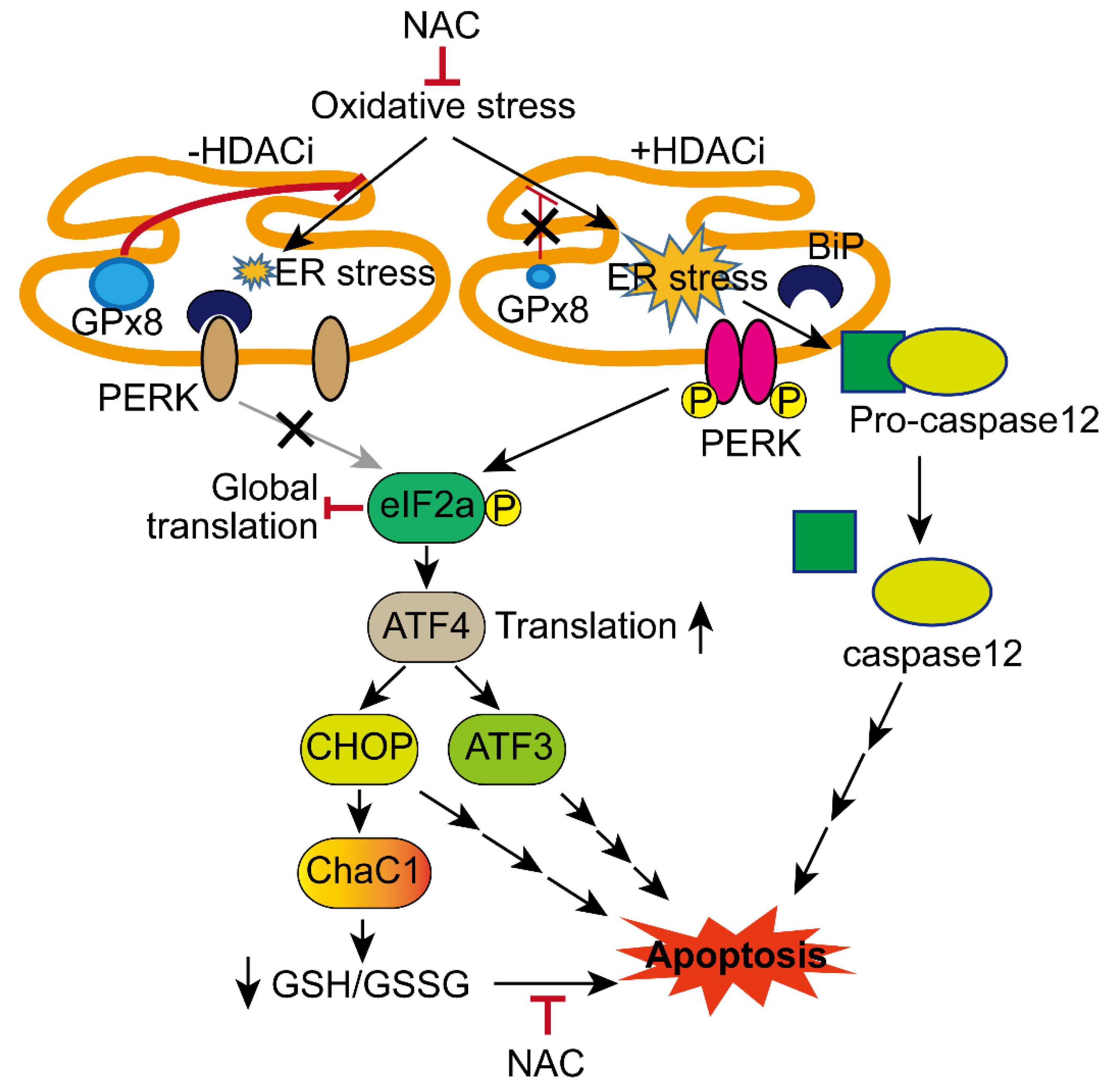
3.2. HDACi Induced Endoplasmic Reticulum Stress (ER Stress)
3.3. Co-Treatment with tBHP and HDACi Enhanced Transcriptional Activity of ATF4 and CHOP
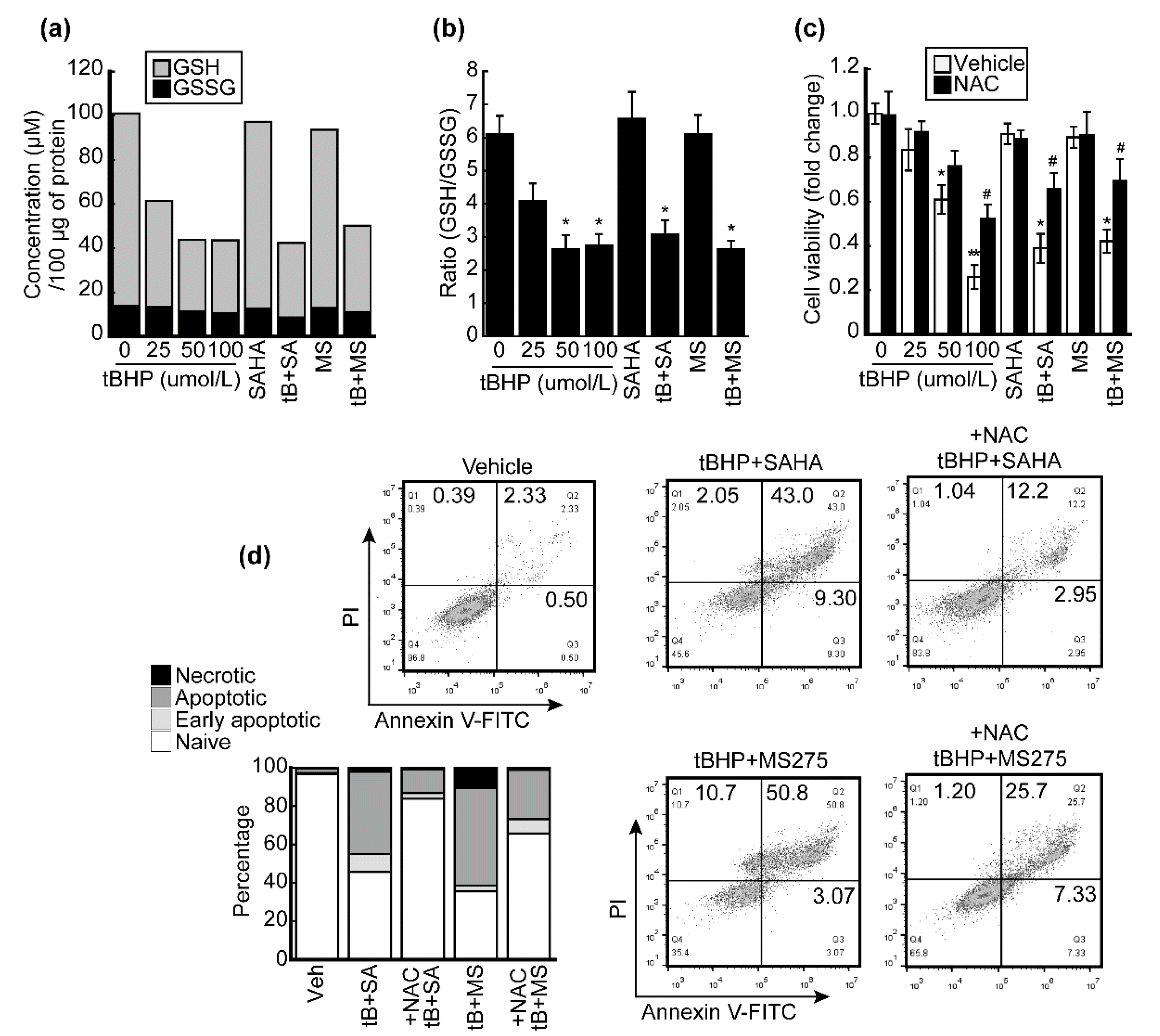
3.4. HDACi Enhanced Oxidative Stress and ER-Stress-Induced Apoptosis of HCC Cells
3.5. HDACi Facilitated Oxidative-Stress-Caused Reduction of Glutathione (GSH)
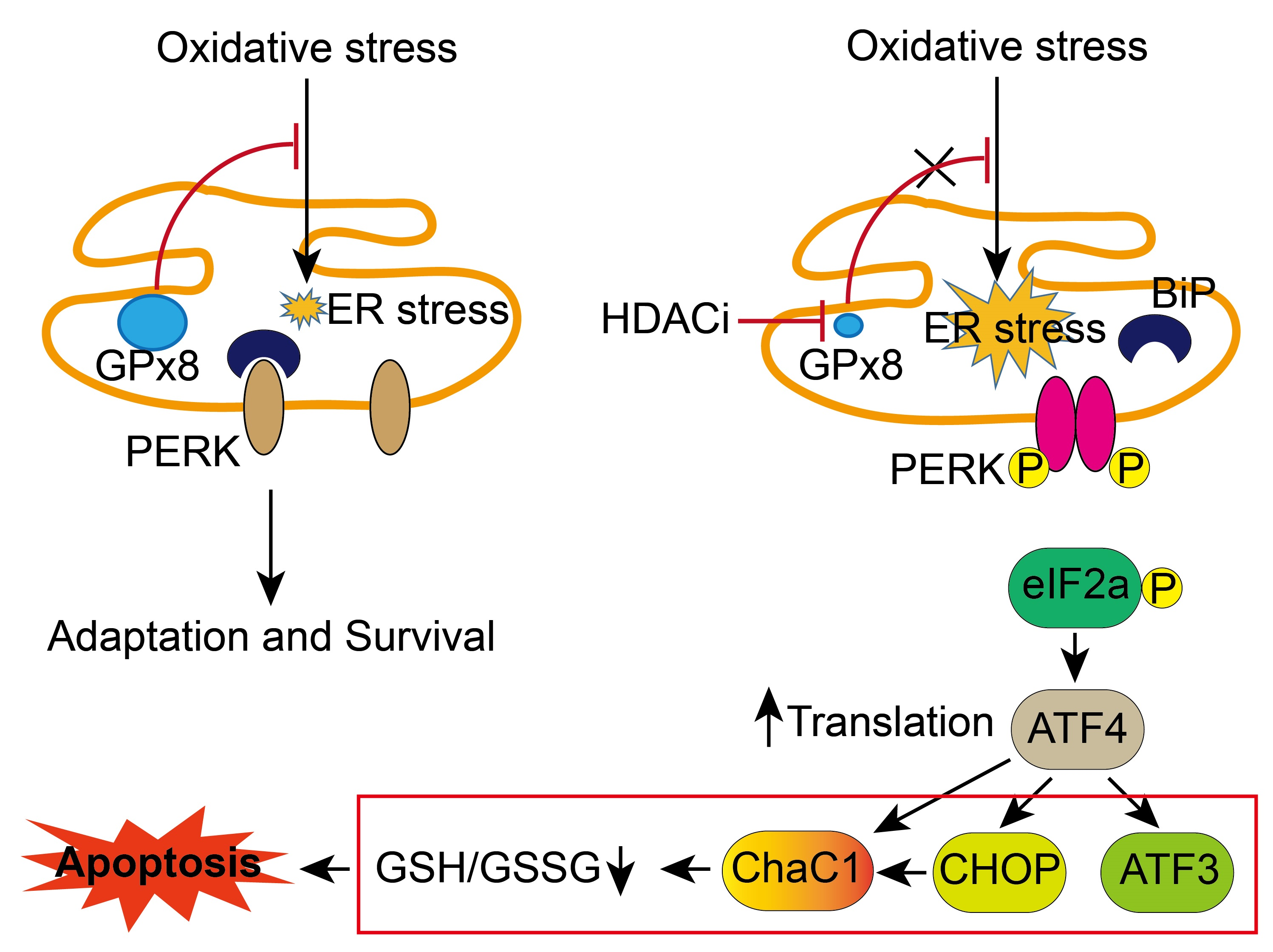
3.6. GPX8 Overexpression Restored Oxidative Stress and HDACi-Induced ER Stress and Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef]
- Grandhi, M.S.; Kim, A.K.; Ronnekleiv-Kelly, S.M.; Kamel, I.R.; Ghasebeh, M.A.; Pawlik, T.M. Hepatocellular carcinoma: From diagnosis to treatment. Surg. Oncol. 2016, 25, 74–85. [Google Scholar] [CrossRef]
- Bruix, J.; Llovet, J.M. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology 2002, 35, 519–524. [Google Scholar] [CrossRef]
- Eatrides, J.; Wang, E.; Kothari, N.; Kim, R. Role of Systemic Therapy and Future Directions for Hepatocellular Carcinoma. Cancer Control. 2017, 24, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Li, D.; Sedano, S.; Allen, R.; Gong, J.; Cho, M.; Sharma, S. Current Treatment Landscape for Advanced Hepatocellular Carcinoma: Patient Outcomes and the Impact on Quality of Life. Cancers 2019, 11, 841. [Google Scholar] [CrossRef] [Green Version]
- Cabral, L.K.D.; Tiribelli, C.; Sukowati, C.H.C. Sorafenib Resistance in Hepatocellular Carcinoma: The Relevance of Genetic Heterogeneity. Cancers 2020, 12, 1576. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Goey, A.K.; Sissung, T.M.; Peer, C.J.; Figg, W.D. Pharmacogenomics and histone deacetylase inhibitors. Pharmacogenomics 2016, 17, 1807–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells In Vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Worns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Tak, W.Y.; Ryoo, B.Y.; Lim, H.Y.; Kim, D.Y.; Okusaka, T.; Ikeda, M.; Hidaka, H.; Yeon, J.E.; Mizukoshi, E.; Morimoto, M.; et al. Phase I/II study of first-line combination therapy with sorafenib plus resminostat, an oral HDAC inhibitor, versus sorafenib monotherapy for advanced hepatocellular carcinoma in east Asian patients. Investig. New Drugs 2018, 36, 1072–1084. [Google Scholar] [CrossRef]
- Tapadar, S.; Fathi, S.; Wu, B.; Sun, C.Q.; Raji, I.; Moore, S.G.; Arnold, R.S.; Gaul, D.A.; Petros, J.A.; Oyelere, A.K. Liver-Targeting Class I Selective Histone Deacetylase Inhibitors Potently Suppress Hepatocellular Tumor Growth as Standalone Agents. Cancers 2020, 12, 3095. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, A.M.; Brown, J.D.; Taylor, S.R.; Rand, J.D.; Morgan, B.A.; Veal, E.A. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 2012, 45, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toppo, S.; Vanin, S.; Bosello, V.; Tosatto, S.C. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid. Redox Signal. 2008, 10, 1501–1514. [Google Scholar] [CrossRef]
- Chen, Y.I.; Wei, P.C.; Hsu, J.L.; Su, F.Y.; Lee, W.H. NPGPx (GPx7): A novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. Am. J. Transl. Res. 2016, 8, 1626–1640. [Google Scholar]
- Lee, H.A.; Chu, K.B.; Moon, E.K.; Kim, S.S.; Quan, F.S. Sensitization to oxidative stress and G2/M cell cycle arrest by histone deacetylase inhibition in hepatocellular carcinoma cells. Free Radic. Biol. Med. 2020, 147, 129–138. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Magne, L.; Blanc, E.; Legrand, B.; Lucas, D.; Barouki, R.; Rouach, H.; Garlatti, M. ATF4 and the integrated stress response are induced by ethanol and cytochrome P450 2E1 in human hepatocytes. J. Hepatol. 2011, 54, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, X.; Sun, J.; Lei, Q.; Wang, Q.; Pan, D.; Ding, M.; Ding, Y. Reactive oxygen species mediate anlotinib-induced apoptosis via activation of endoplasmic reticulum stress in pancreatic cancer. Cell Death Dis. 2020, 11, 766. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [Green Version]
- Oka, O.B.V.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Bba Mol. Cell Res. 2013, 1833, 2425–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.C.; Ruddock, L.W. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide during Disulfide Bond Formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Kanemura, S.; Sofia, E.F.; Hirai, N.; Okumura, M.; Kadokura, H.; Inaba, K. Characterization of the endoplasmic reticulum-resident peroxidases GPx7 and GPx8 shows the higher oxidative activity of GPx7 and its linkage to oxidative protein folding. J. Biol. Chem. 2020, 295, 12772–12785. [Google Scholar] [CrossRef]
- Subramanian, K.; Rauniyar, N.; Lavallee-Adam, M.; Yates, J.R., 3rd; Balch, W.E. Quantitative Analysis of the Proteome Response to the Histone Deacetylase Inhibitor (HDACi) Vorinostat in Niemann-Pick Type C1 disease. Mol. Cell. Proteom. 2017, 16, 1938–1957. [Google Scholar] [CrossRef] [Green Version]
- Mazzio, E.A.; Soliman, K.F.A. Whole-transcriptomic Profile of SK-MEL-3 Melanoma Cells Treated with the Histone Deacetylase Inhibitor: Trichostatin A. Cancer Genom. Proteom. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Khatib, A.; Solaimuthu, B.; Ben Yosef, M.; Abu Rmaileh, A.; Tanna, M.; Oren, G.; Schlesinger Frisch, M.; Axelrod, J.H.; Lichtenstein, M.; Shaul, Y.D. The glutathione peroxidase 8 (GPX8)/IL-6/STAT3 axis is essential in maintaining an aggressive breast cancer phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 21420–21431. [Google Scholar] [CrossRef]
- Chueh, A.C.; Tse, J.W.; Togel, L.; Mariadason, J.M. Mechanisms of Histone Deacetylase Inhibitor-Regulated Gene Expression in Cancer Cells. Antioxid. Redox Signal. 2015, 23, 66–84. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Suzuki, R.; Ohguchi, H.; Yoshida, Y.; Lu, D.; Cottini, F.; Jakubikova, J.; Bianchi, G.; Harada, T.; Gorgun, G.; et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia 2015, 29, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. The unfolded protein response: A stress signaling pathway critical for health and disease. Neurology 2006, 66, S102–S109. [Google Scholar] [CrossRef] [PubMed]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Hayner, J.N.; Shan, J.X.; Kilberg, M.S. Regulation of the ATF3 gene by a single promoter in response to amino acid availability and endoplasmic reticulum stress in human primary hepatocytes and hepatoma cells. Bba Gene Regul. Mech. 2018, 1861, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.H.; Du, Z.X.; Li, N.; Li, C.; Zhang, Q.; Liu, B.Q.; Guan, Y.F.; Wang, H.Q. Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Bba Mol. Cell Res. 2012, 1823, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Mungrue, I.N.; Pagnon, J.; Kohannim, O.; Gargalovic, P.S.; Lusis, A.J. CHAC1/MGC4504 Is a Novel Proapoptotic Component of the Unfolded Protein Response, Downstream of the ATF4-ATF3-CHOP Cascade. J. Immunol. 2009, 182, 466–476. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Gebert, M.; Janaszak-Jasiecka, A.; Cabaj, A.; Kroliczewski, J.; Bartoszewska, S.; Sobolewska, A.; Crossman, D.K.; Ochocka, R.; Kamysz, W.; et al. Genome-wide mRNA profiling identifies RCAN1 and GADD45A as regulators of the transitional switch from survival to apoptosis during ER stress. FEBS J. 2020, 287, 2923–2947. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-A.; Chu, K.-B.; Moon, E.-K.; Quan, F.-S. Glutathione Peroxidase 8 Suppression by Histone Deacetylase Inhibitors Enhances Endoplasmic Reticulum Stress and Cell Death by Oxidative Stress in Hepatocellular Carcinoma Cells. Antioxidants 2021, 10, 1503. https://doi.org/10.3390/antiox10101503
Lee H-A, Chu K-B, Moon E-K, Quan F-S. Glutathione Peroxidase 8 Suppression by Histone Deacetylase Inhibitors Enhances Endoplasmic Reticulum Stress and Cell Death by Oxidative Stress in Hepatocellular Carcinoma Cells. Antioxidants. 2021; 10(10):1503. https://doi.org/10.3390/antiox10101503
Chicago/Turabian StyleLee, Hae-Ahm, Ki-Back Chu, Eun-Kyung Moon, and Fu-Shi Quan. 2021. "Glutathione Peroxidase 8 Suppression by Histone Deacetylase Inhibitors Enhances Endoplasmic Reticulum Stress and Cell Death by Oxidative Stress in Hepatocellular Carcinoma Cells" Antioxidants 10, no. 10: 1503. https://doi.org/10.3390/antiox10101503
APA StyleLee, H.-A., Chu, K.-B., Moon, E.-K., & Quan, F.-S. (2021). Glutathione Peroxidase 8 Suppression by Histone Deacetylase Inhibitors Enhances Endoplasmic Reticulum Stress and Cell Death by Oxidative Stress in Hepatocellular Carcinoma Cells. Antioxidants, 10(10), 1503. https://doi.org/10.3390/antiox10101503