
Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology



Abstract
:
1. H2O2 Reduction by Organoselenides
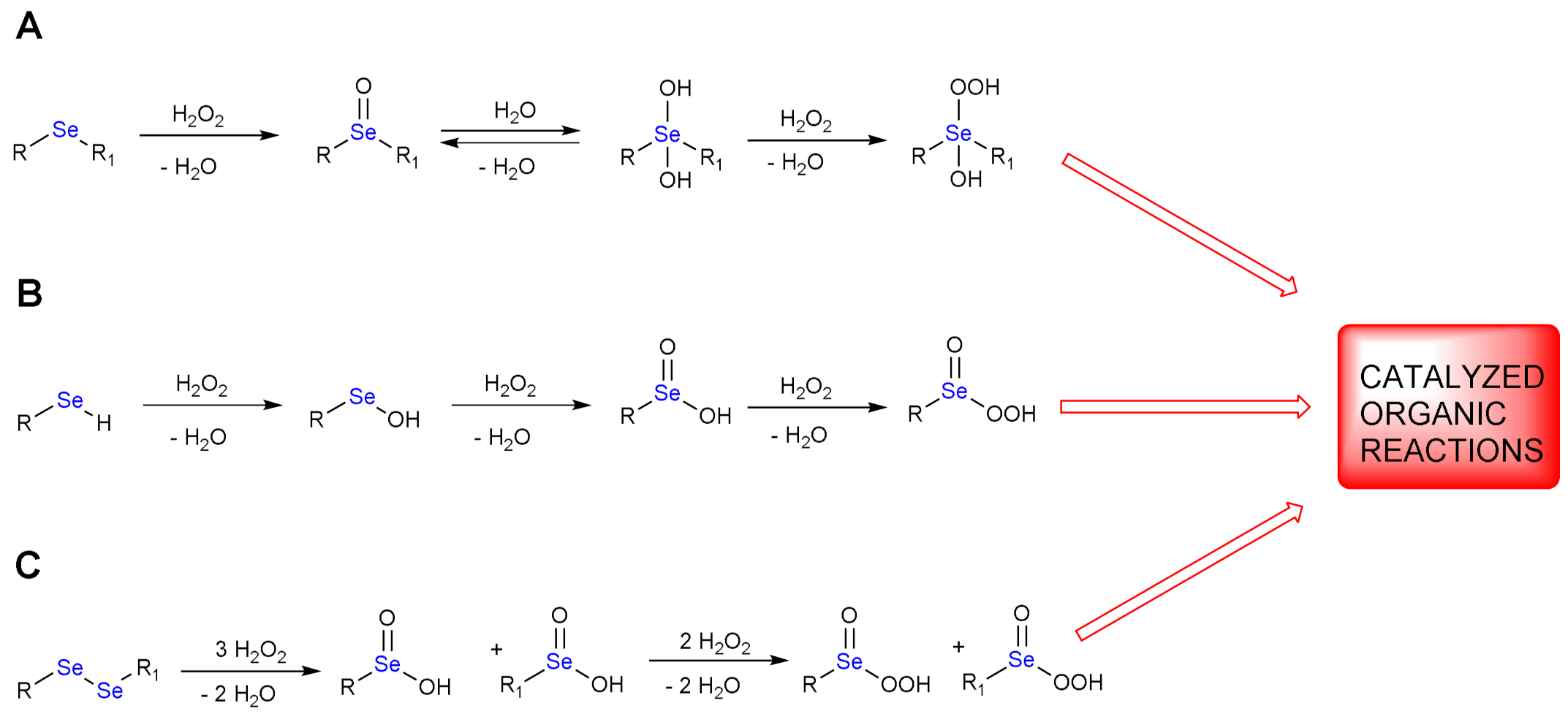
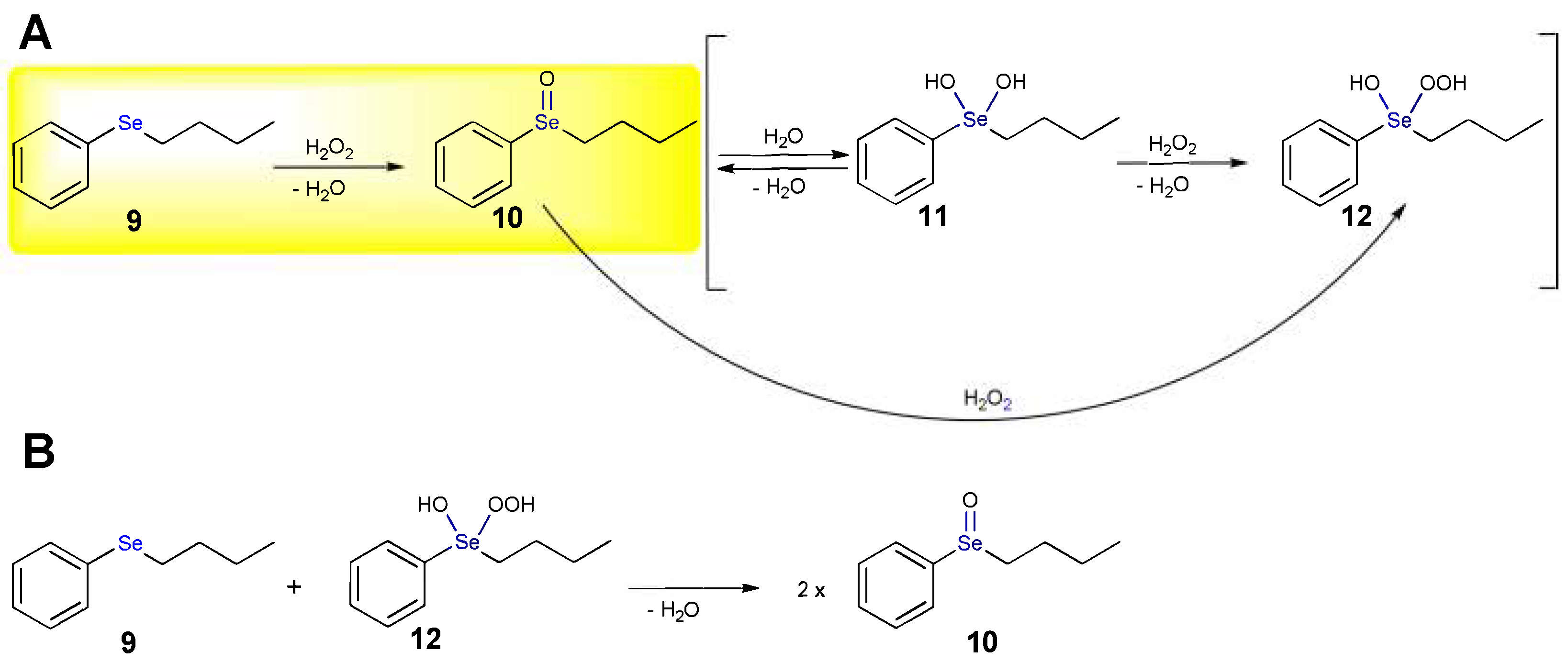
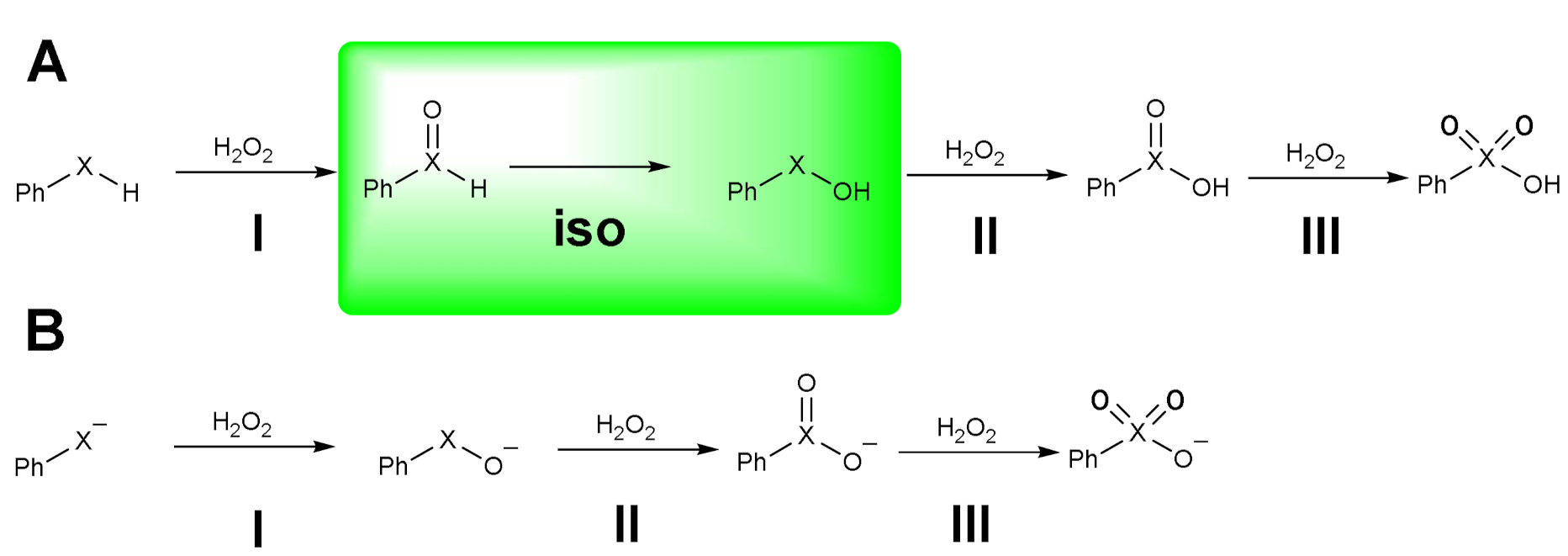
1.1. Oxidation of n-butyl Phenyl Selenide by H2O2
1.2. Oxidation of Phenylselenol by H2O2
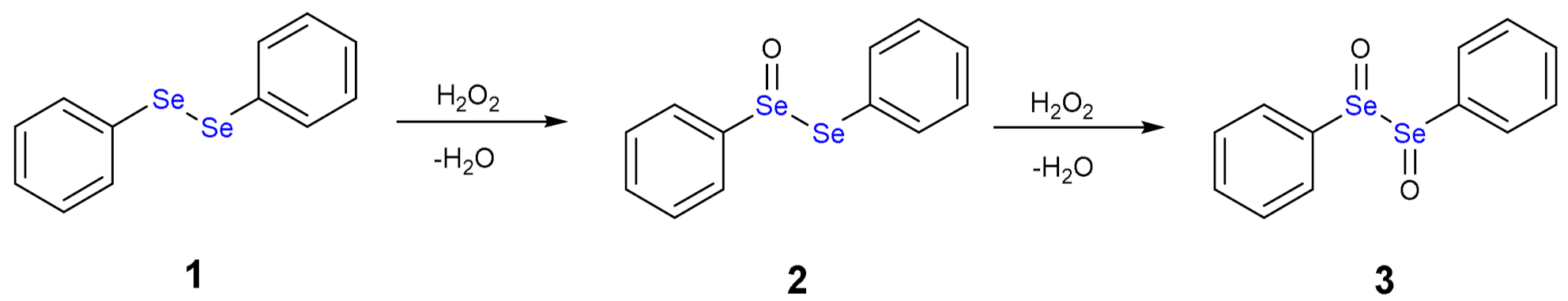
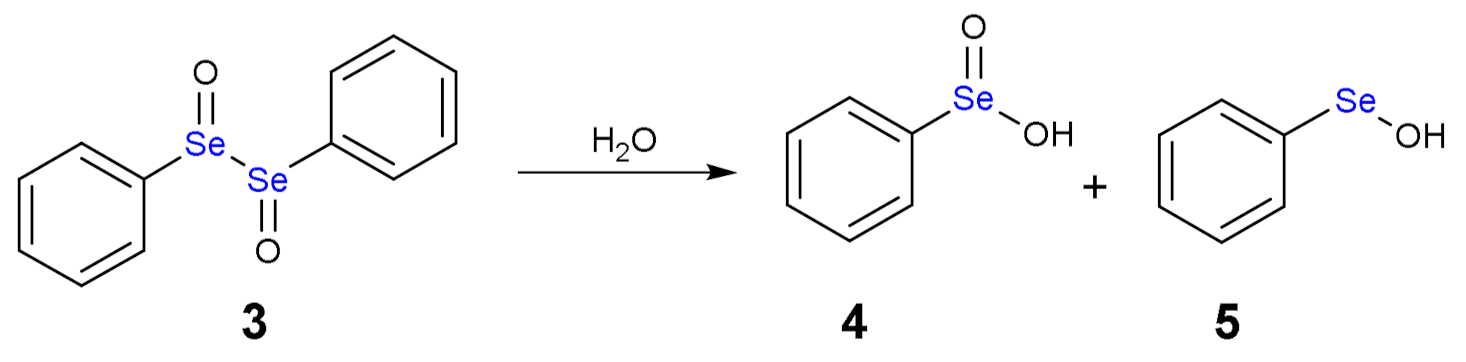
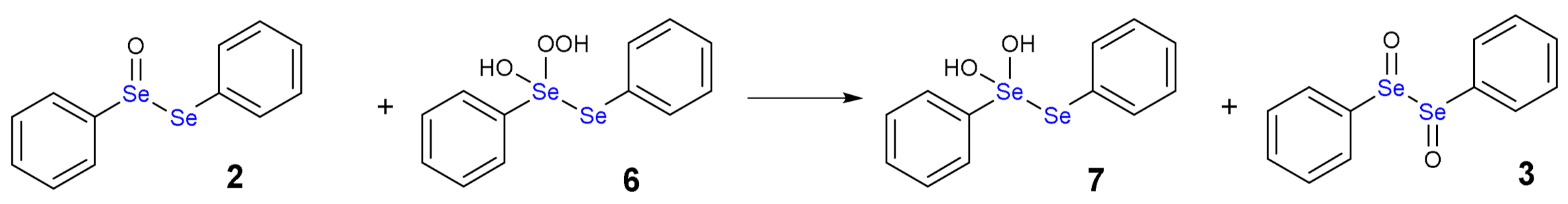
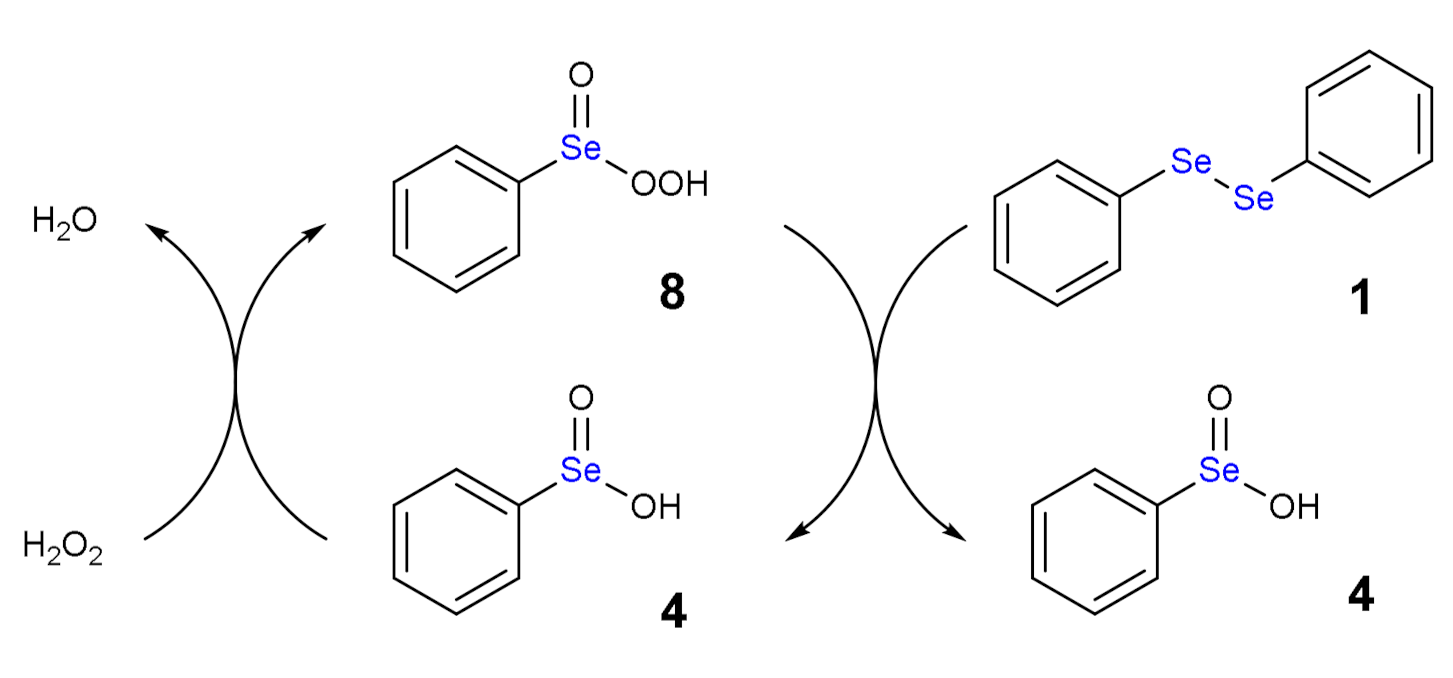
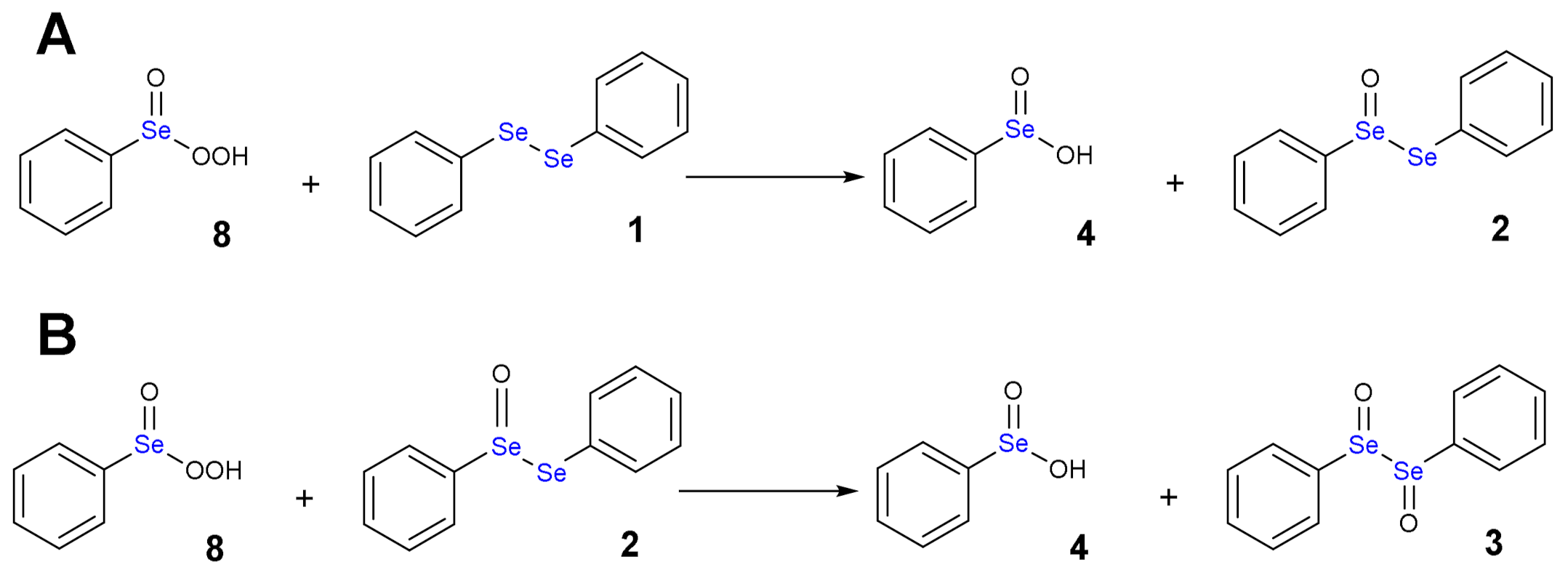
1.3. Oxidation of Diphenyl Diselenide by H2O2
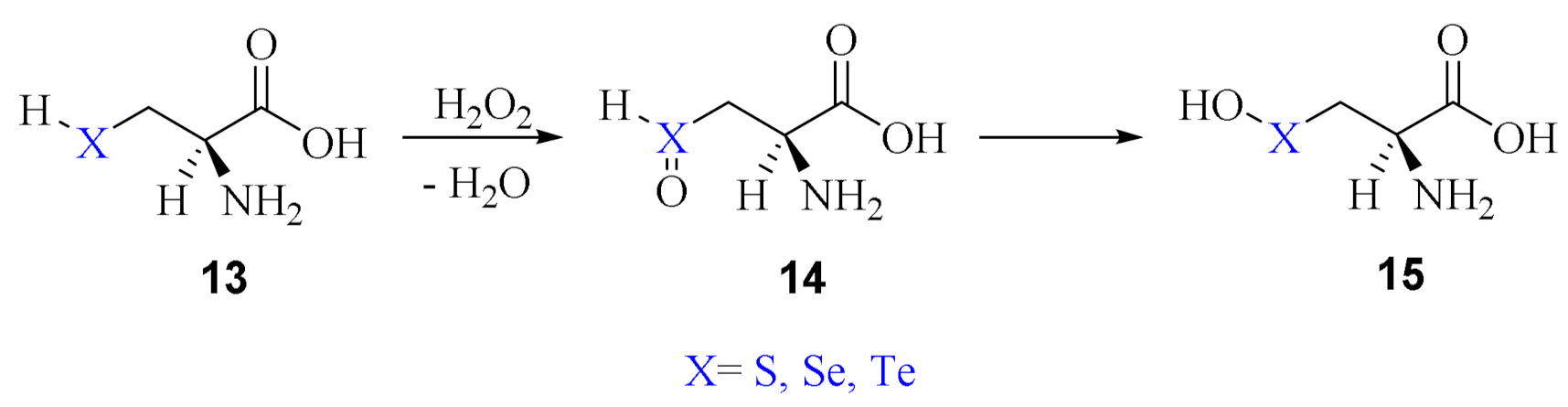
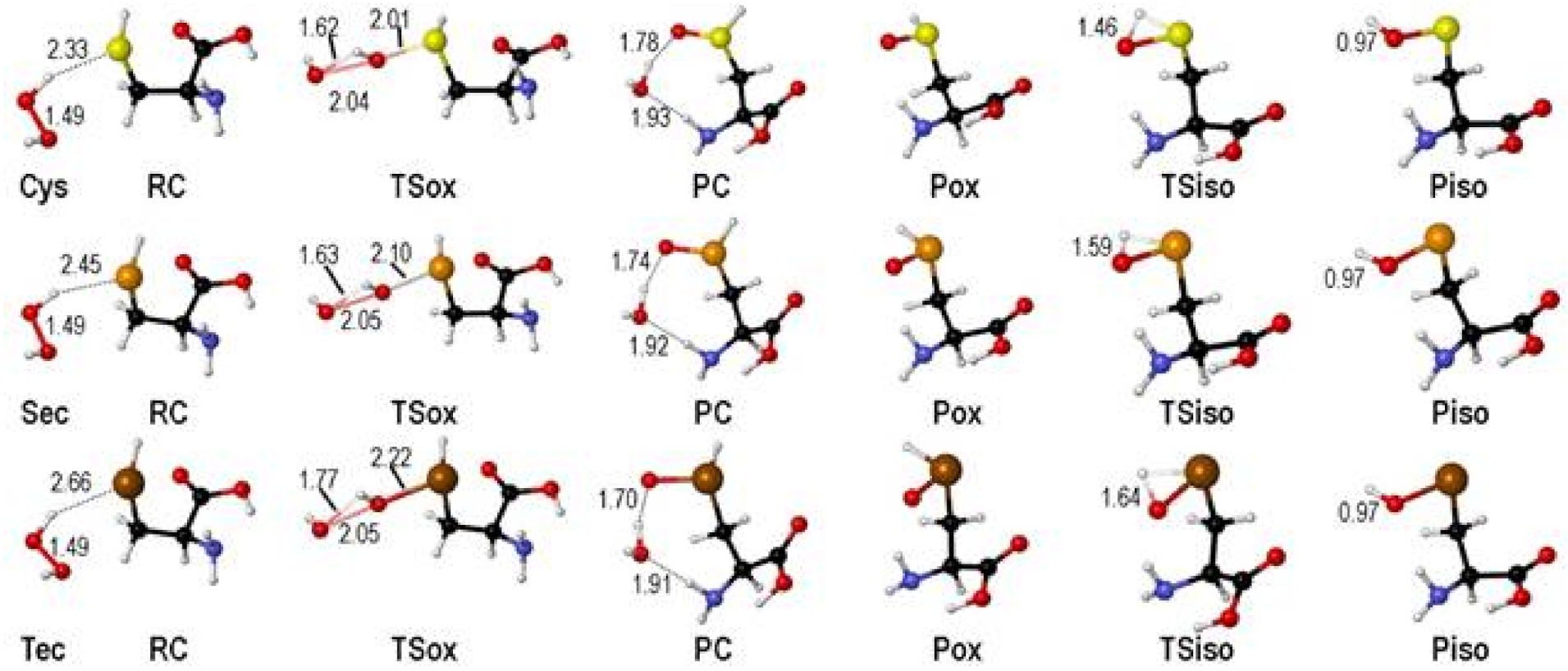
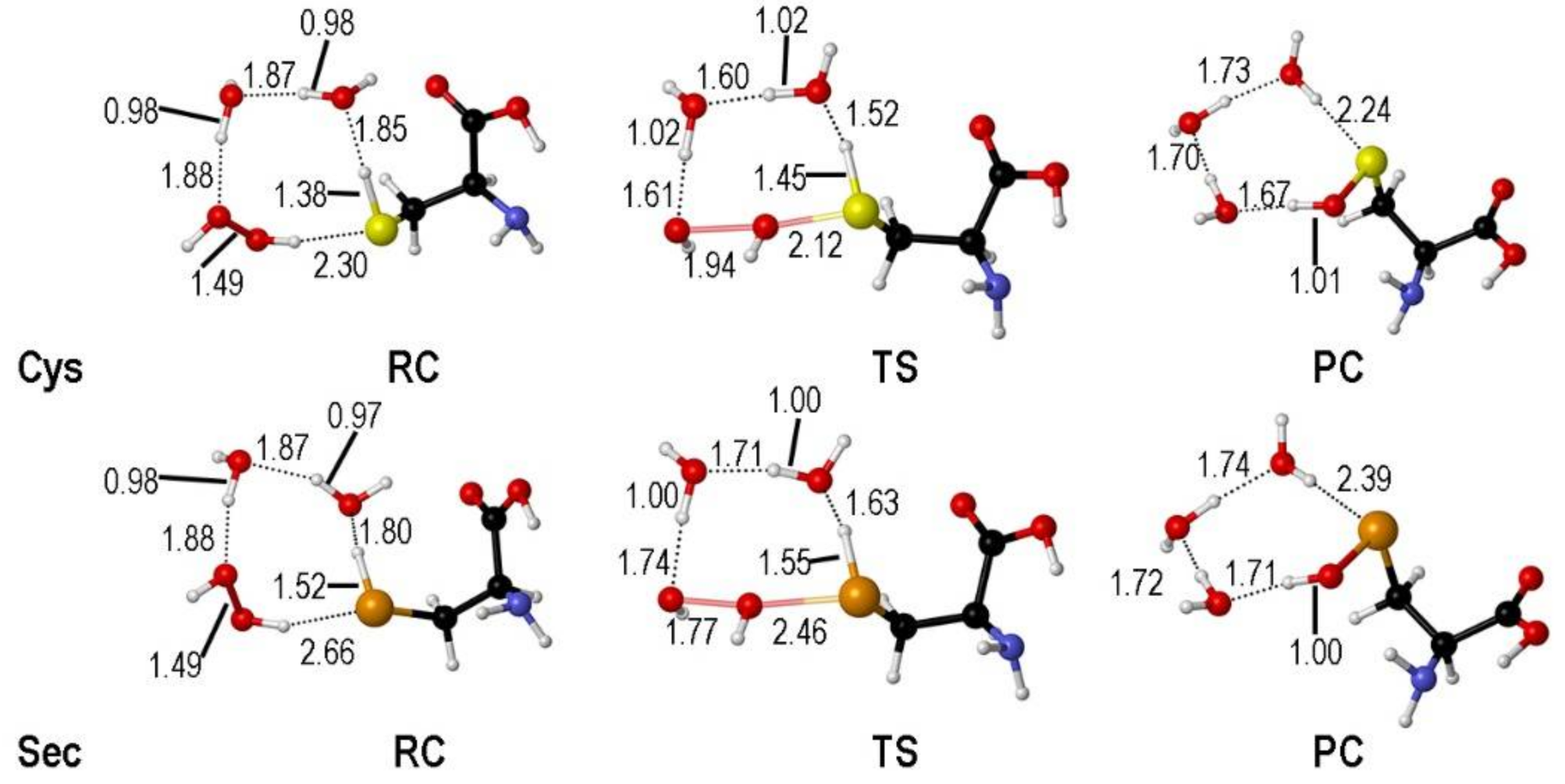
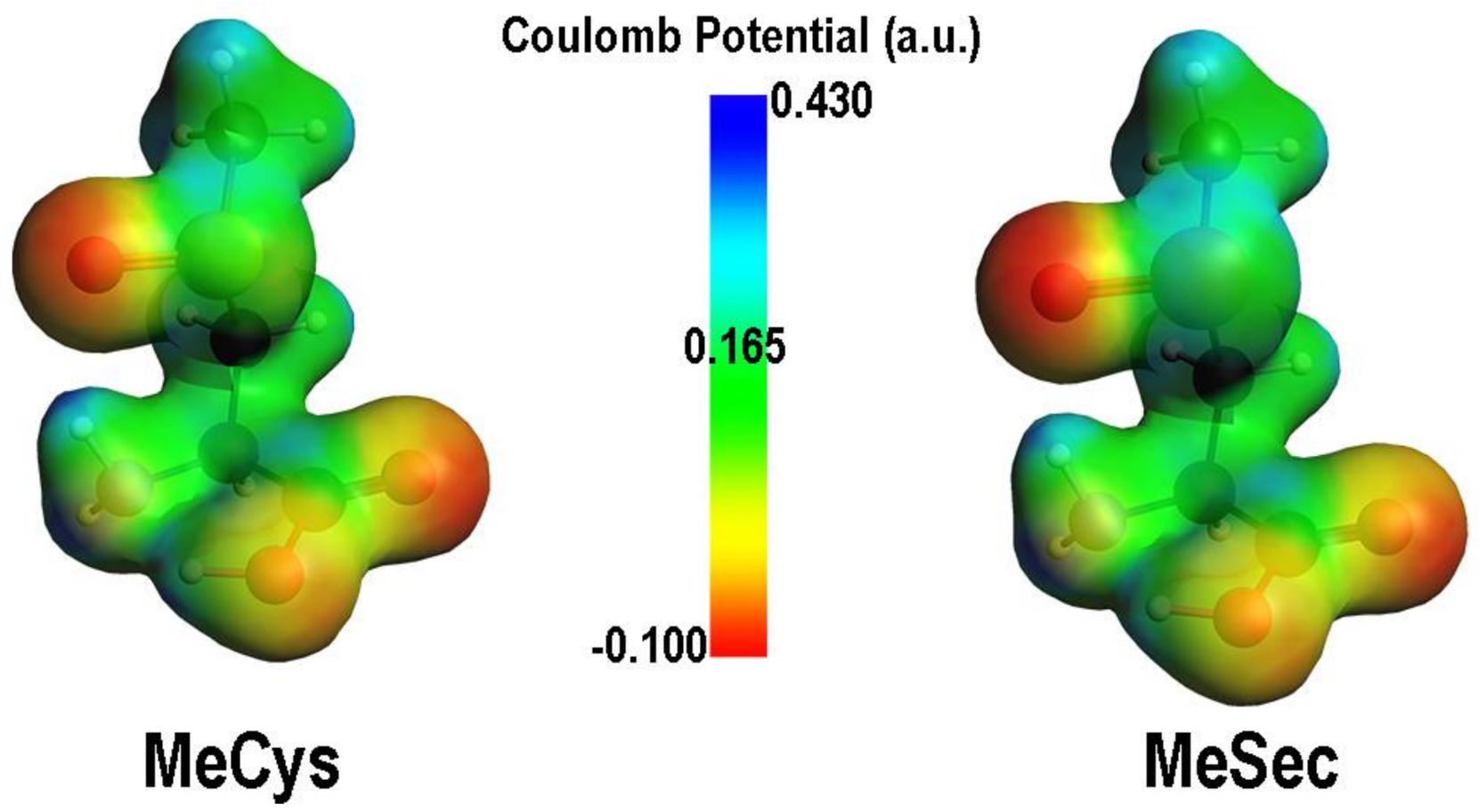
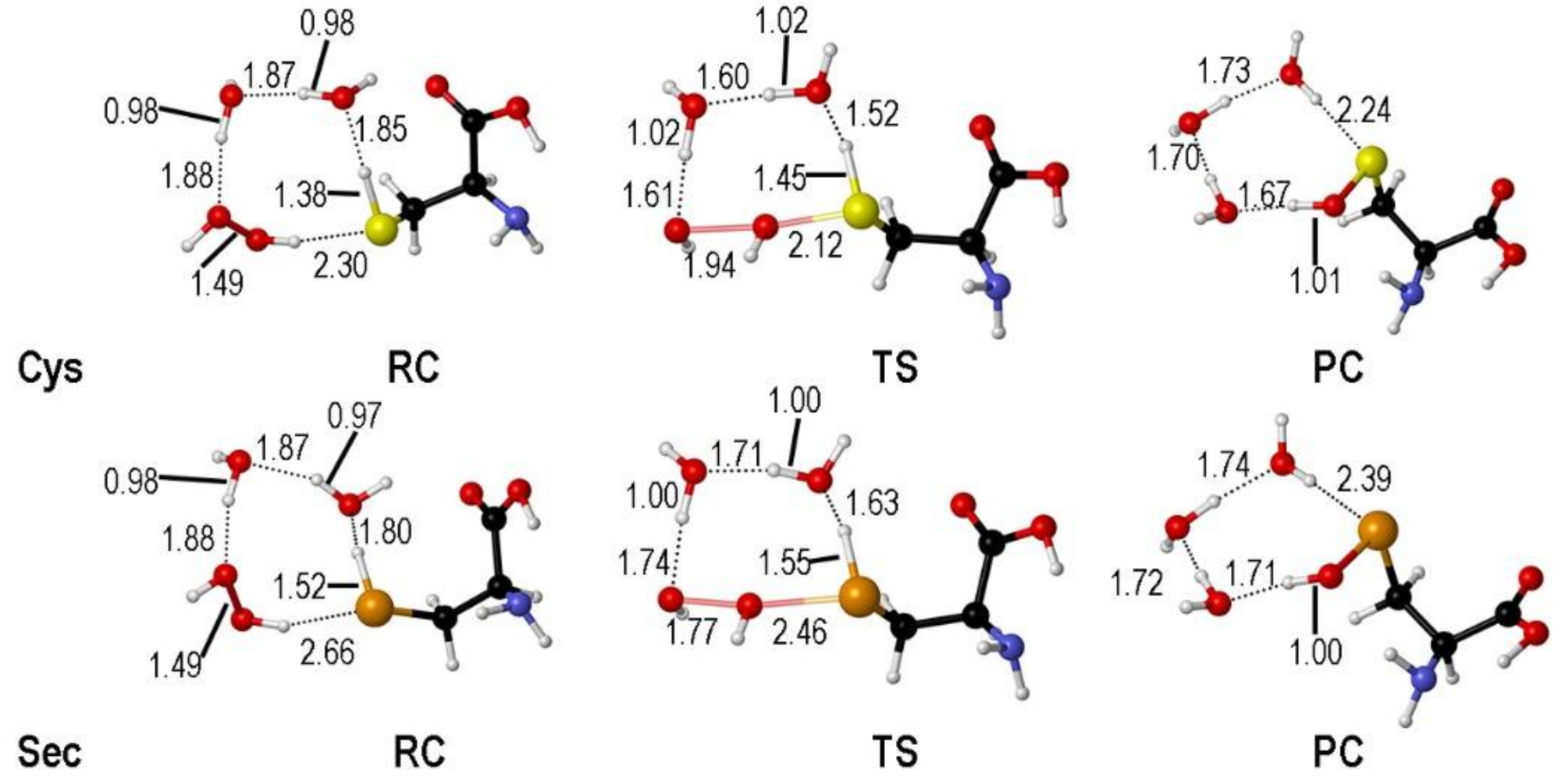
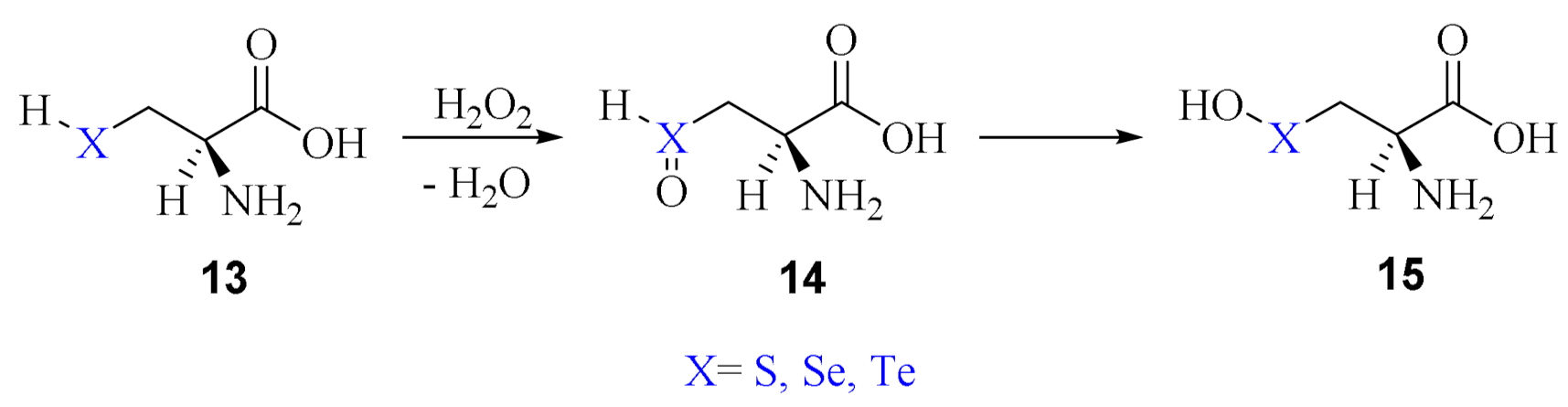
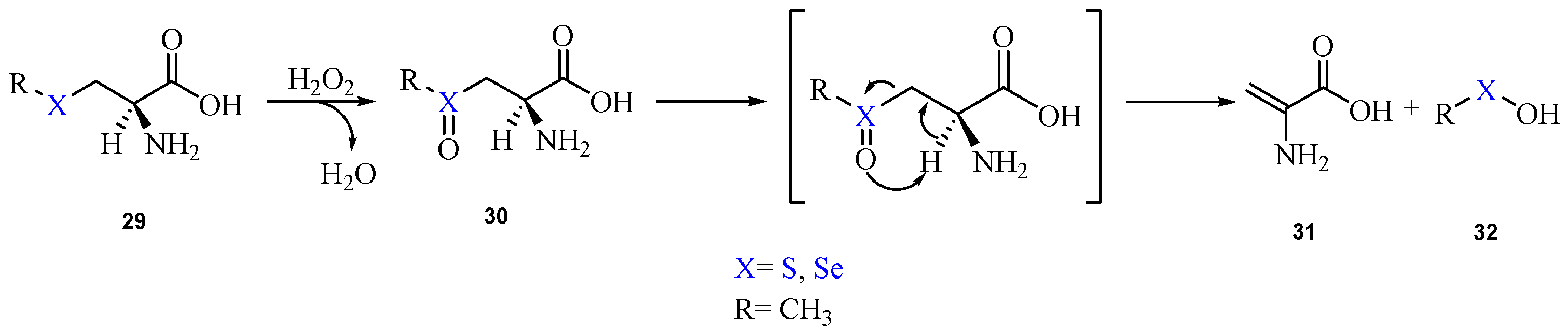
2. Oxidation of Cysteine and Selenocysteine by H2O2
3. The Reduction of Hydroperoxides in Selenoproteins
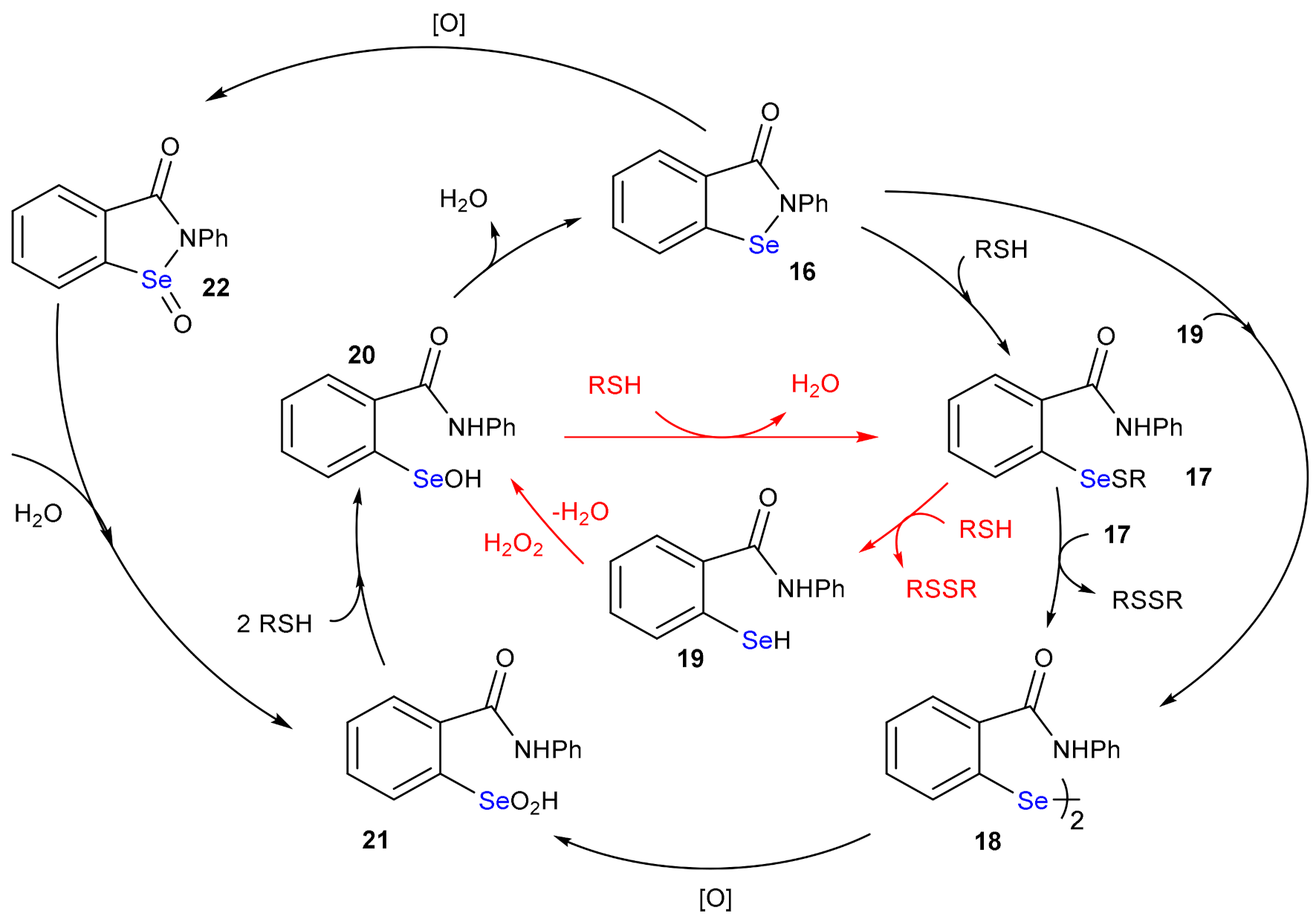
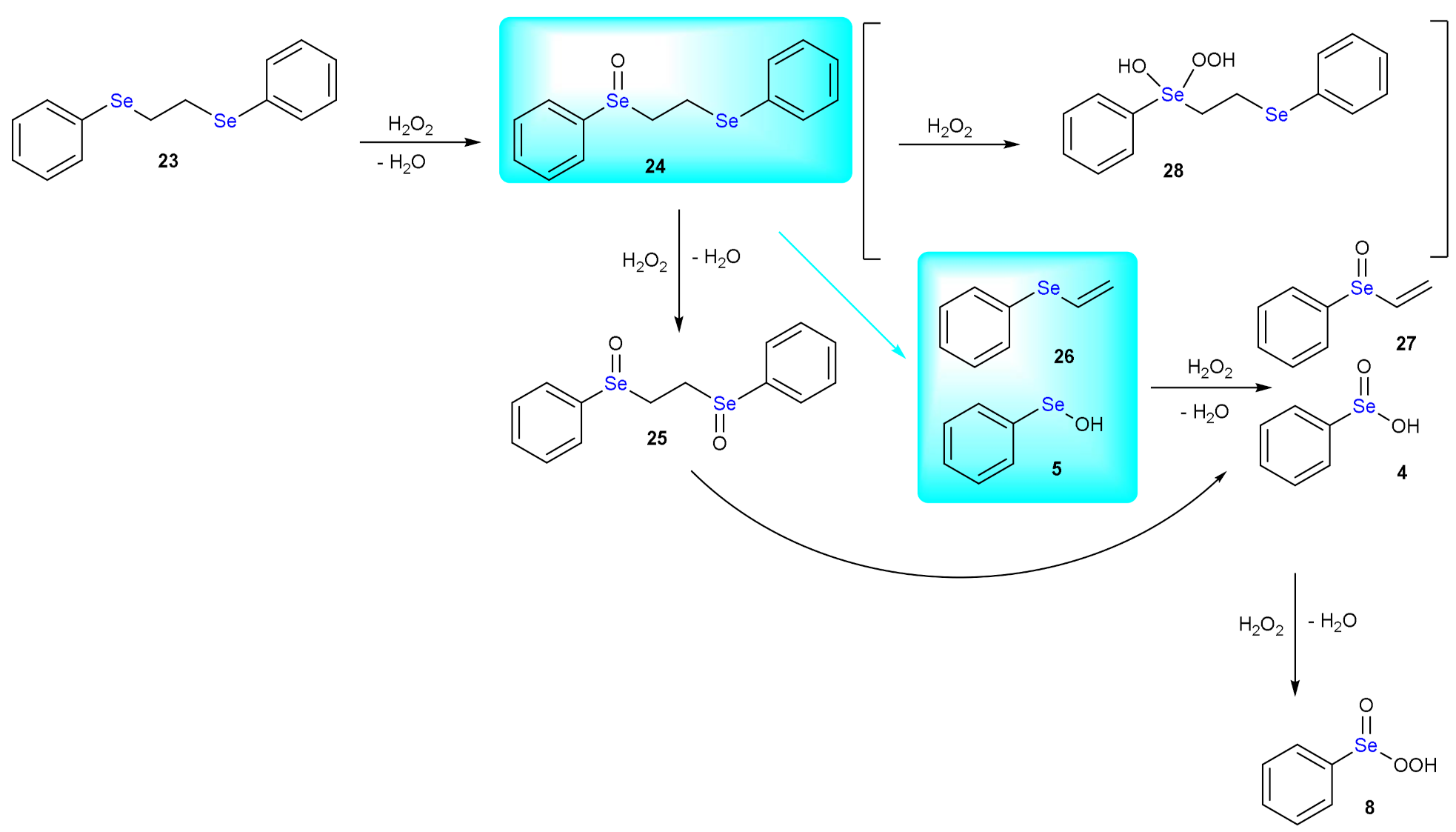
4. The Reduction of Hydroperoxides by GPx Mimics: The Case of Ebselen
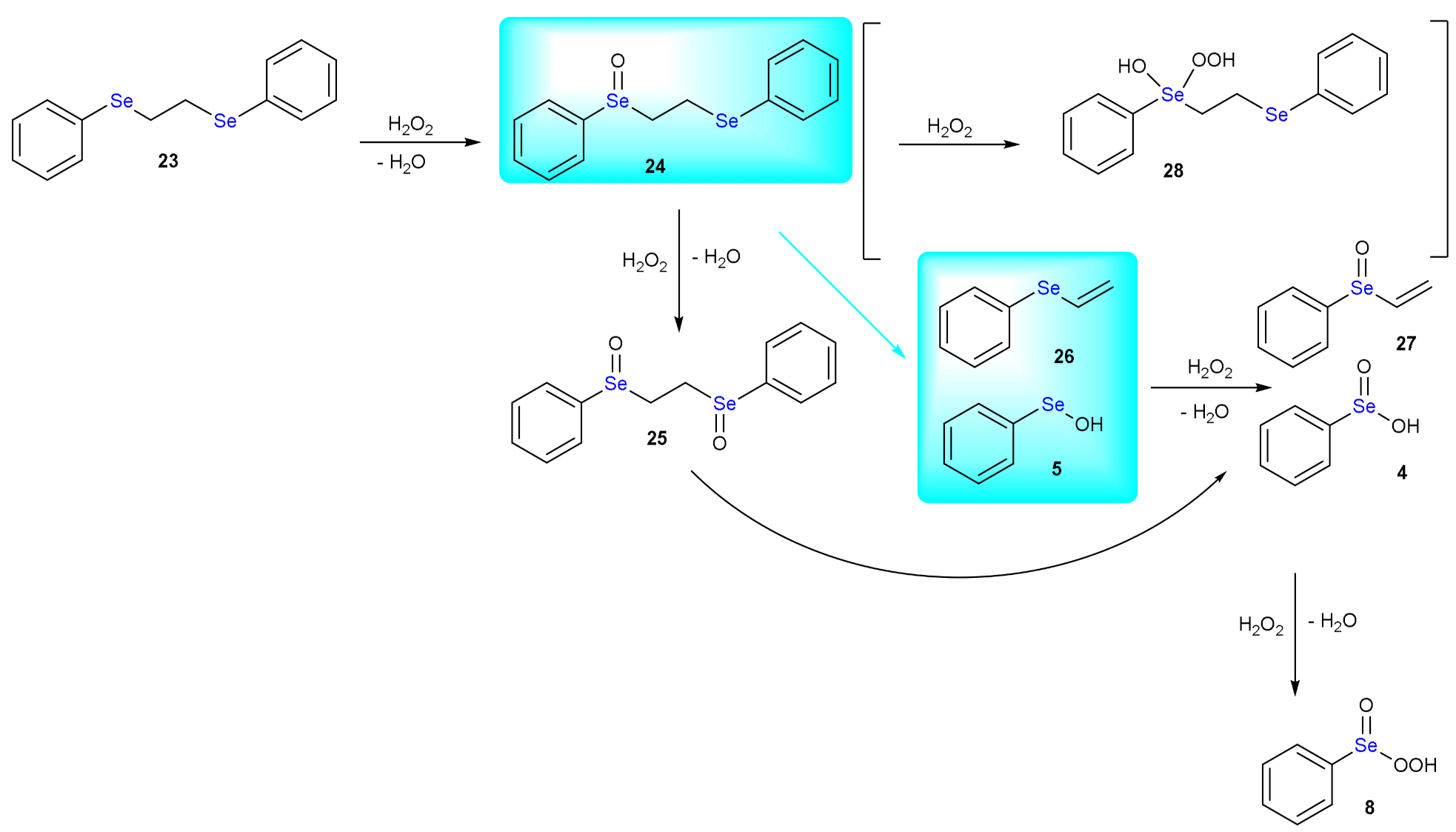
5. Deselenylation Paths
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BP86 | GGA functional with Becke exchange and Perdew correlation |
| B3PW91 | Hybrid functional with Becke exchange and Perdew Wang correlation |
| COSMO | Conductor-like Screening Model for solvation |
| Cys | Cysteine |
| DFT | Density Functional Theory |
| DHA | Dehydroalanine |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GGA | Generalized Gradient Approximation |
| GPx | Glutathione Peroxidase |
| GTO | Gaussian Type Orbital |
| IMPase | Inositol MonoPhosphatase |
| OLYP | GGA functional with OPTX exchange and LYP correlation |
| OPBE | GGA functional with OPTX exchange and PBEc correlation |
| D3(BJ) | Dispersion correction (D3) by Grimme with damping function |
| HOMO | Highest Occupied Molecular Orbital |
| LUMO | Lowest Unoccupied Molecular Orbital |
| MeCys | Methyl Cysteine |
| MEP | Molecular Electrostatic Potential |
| MPn | Møller Plesset perturbative method of order n |
| MeSec | Methyl Selenocysteine |
| NMR | Nuclear Magnetic Resonance |
| PC | Product Complex |
| PCM | Polarizable Continuum Model |
| PES | Potential Energy Surface |
| QCISD(T) | Quadratic Configuration Interaction with Single Double (Triple) excitations |
| QM | Quantum Mechanics |
| QZ4P | uncontracted set of Slater-type orbitals of quadruple-ζ quality, augmented with four sets of polarization functions per atom |
| RC | Reactant Complex |
| SAPE | Solvent Assisted Proton Exchange |
| Sec | Selenocysteine |
| S-GPx | Sulfur mutant of glutathione peroxidase |
| SMD | Solvation Model based on Density |
| STO | Slater Type Orbital |
| Tec | Tellurocysteine |
| Te-GPx | Tellurium mutant of glutathione peroxidase |
| TS | Transition State |
| TZP | uncontracted set of Slater-type orbitals of triple-ζ quality, augmented with one set of polarization functions per atom |
| TZ2P | uncontracted set of Slater-type orbitals of triple-ζ quality, augmented with two sets of polarization functions per atom |
| XC | Exchange- Correlation |
| ZORA | Zeroth Order Regular Approximation |
References
- Jain, V.K. An overview of organoselenium chemistry: From fundamentals to synthesis. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; Royal Socienty of Chemistry: London, UK, 2017. [Google Scholar]
- Singh, F.V.; Wirth, T. Selenium reagents as catalysts. Catal. Sci. Techol. 2019, 9, 1073–1091. [Google Scholar] [CrossRef]
- Back, T.G. Oxidations catalyzed by seleninic acids and anhydrides, their precursors and congeners. Curr. Green Chem. 2016, 3, 76–91. [Google Scholar] [CrossRef]
- Miochowski, J.; Wojtowicz-Miochowska, H. Developments in synthetic applications of selenium (IV) oxide and organoselenium compounds as oxygen donors and oxygen-transfer agents. Molecules 2015, 6, 10205–10243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Torres, M.; Arends, I.W.C.E.; Mayoral, J.A.; Pires, E.; Jimenez-Oses, G. A highly efficient, green and recoverable catalytic system for the epoxidation of fatty esters and biodiesel with H2O2. Appl. Catal. A Gen. 2012, 425–426, 91–96. [Google Scholar] [CrossRef]
- Cerra, B.; Mangiavacchi, F.; Santi, C.; Lozza, A.M.; Gioiello, A. Selective continuous flow synthesis of hydroxy lactones from alkenoic acids. React. Chem. Eng. 2017, 2, 467–471. [Google Scholar] [CrossRef]
- Wang, T.; Jing, X.; Chen, C.; Yu, L. Organoselenium-catalyzed oxidative C=C bond cleavage: A relatively green oxidation of alkenes into carbonyl compounds with hydrogen peroxide. J. Org. Chem. 2017, 82, 9342–9349. [Google Scholar] [CrossRef]
- Sancineto, L.; Mangiavacchi, F.; Tidei, C.; Bagnoli, L.; Marini, F.; Gioiello, A.; Scianowski, J.; Santi, C. Selenium-catalyzed oxacyclization of alkenoic acids and alkenols. Asian J. Org. Chem. 2017, 6, 988–992. [Google Scholar] [CrossRef]
- Freudendahl, D.M.; Santoro, S.; Shahzad, S.A.; Santi, C.; Wirth, T. Green chemistry with selenium reagents: Development of efficient catalytic reactions. Angew. Chem. Int. Ed. 2009, 48, 8409–8411. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Azeredo, J.B.; Nascimento, V.; Sancineto, L.; Braga, A.L.; Santi, C. The green side of the moon: Ecofriendly aspects of organoselenium chemistry. RSC Adv. 2014, 4, 31521–31535. [Google Scholar] [CrossRef]
- Sands, K.N.; Mendoza Rengifo, E.; George, G.N.; Pickering, I.J.; Gelfand, B.S.; Back, T.G. The unexpected role of Se (VI) species in epoxidations with benzeneseleninic acid and hydrogen peroxide. Angew. Chem. 2020, 132, 4313–4317. [Google Scholar] [CrossRef]
- Kice, J.L.; Chiou, S.; Weclas, L. Mechanism of the oxidation of o-nitro- and o-benzoylbenzeneselenenic acids by peracids, hydroperoxides, and hydrogen peroxide. J. Org. Chem. 1985, 50, 2508–2516. [Google Scholar] [CrossRef]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B.T. Organoselenium and organotellurium compounds: Toxicology and pharmacology. Chem. Rev. 2004, 104, 6255–6286. [Google Scholar] [CrossRef]
- Masuda, R.; Kimura, R.; Karasaki, T.; Sase, S.; Goto, K. Modeling the catalytic cycle of glutathione peroxidase by nuclear magnetic resonance spectroscopic analysis of selenocysteine selenenic acids. J. Am. Chem. Soc. 2021, 143, 6345–6350. [Google Scholar] [CrossRef]
- Sase, S.; Kimura, R.; Masuda, R.; Goto, K. Model study on trapping of protein selenenic acids by utilizing a stable synthetic congener. New J. Chem. 2019, 43, 6830–6833. [Google Scholar] [CrossRef]
- Nascimento, V.; Alberto, E.E.; Tondo, D.W.; Dambrowski, D.; Detty, M.R.; Nome, F.; Braga, A.L. GPx-Like activity of selenides and selenoxides: Experimental evidence for the involvement of hydroxy perhydroxy selenane as the active species. J. Am. Chem. Soc. 2012, 134, 138–141. [Google Scholar] [CrossRef]
- Ribaudo, G.; Bellanda, M.; Menegazzo, I.; Wolters, L.P.; Bortoli, M.; Ferrer-Sueta, G.; Zagotto, G.; Orian, L. Mechanistic insight into the oxidation of organic phenylselenides by H2O2. Chem. Eur. J. 2017, 23, 2405–2422. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Trofast, J. The discovery of selenium. Berzelius Soc. Publ. 2016, 43–67. [Google Scholar]
- Heverly-Coulson, G.S.; Boyd, R.J. Systematic study of the performance of density functional theory methods for prediction of energies and geometries of organoselenium compounds. J. Phys. Chem. A 2011, 115, 4827–4831. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, F.; Wolters, L.P.; Fonseca Guerra, C.; Orian, L. Insights on selenium and tellurium diaryldichalcogenides: A benchmark DFT study. J. Comput. Chem. 2016, 37, 1672–1680. [Google Scholar] [CrossRef]
- Wolter, L.P.; Orian, L. Peroxidase activity of organic selenides: Mechanistic insights from quantum chemistry. Curr. Org. Chem. 2015, 20, 189–197. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Ribaudo, G.; Orian, L. Organodiselenides: Organic catalysis and drug design learning from glutathione peroxidase. Curr. Org. Chem. 2019, 23, 1381–1402. [Google Scholar] [CrossRef] [Green Version]
- Bortoli, M.; Wolters, L.P.; Orian, L.; Bickelhaupt, F.M. Addition-elimination or nucleophilic substitution? Understanding the energy profiles for the reaction of chalcogenolates with dichalcogenides. J. Chem. Theory Comput. 2016, 12, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Bortoli, M.; Bruschi, M.; Swart, M.; Orian, L. Sequential oxidations of phenylchalcogenides by H2O2: Insights into the redox behavior of selenium via DFT analysis. New J. Chem. 2020, 44, 6724–6731. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why nature chose selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Rocher, C.; Lalanne, J.-L.; Chaudiere, J. Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur. J. Biochem. 1992, 205, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Aumann, K.D.; Brigelius-Flohé, R.; Ursini, F.; van den Heuvel, J.; McCarthy, J.; Flohé, L. Probing the presumed catalytic triad of selenium-containing peroxidases by mutational analysis of phospholipid hydroperoxide glutathione peroxidase (PHGPx). Biol. Chem. Hoppe. Seyler. 1995, 376, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Loschen, G.; Günzler, W.A.; Eichele, E. Glutathione peroxidase, V. The kinetic mechanism. Hoppe. Seylers. Z. Physiol. Chem. 1972, 353, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, M.; Gregolin, C.; Ursini, F. Oxygen radicals in biological systems part B: Oxygen radical and antioxidants. In Methods in Enzymology; Packer, L., Glazer, A.N., Eds.; Academic Press: New York, NY, USA, 1990; Volume 186. [Google Scholar]
- Lacourciere, G.M.; Stadtman, T.C. Catalytic properties of selenophosphate synthetases: Comparison of the selenocysteine-containing enzyme from Haemophilus influenzae with the corresponding cysteine-containing enzyme from Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Kortemme, T.; Creighton, T.E. Ionisation of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 1995, 253, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Cardey, B.; Enescu, M. A computational study of thiolate and selenolate oxidation by hydrogen peroxide. ChemPhysChem 2005, 6, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Cardey, B.; Enescu, M. Selenocysteine versus cysteine reactivity: A theoretical study of their oxidation by hydrogen peroxide. J. Phys. Chem. A 2007, 111, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Madabeni, A.; Nogara, P.A.; Bortoli, M.; Rocha, J.B.T.; Orian, L. Effect of methylmercury binding on the peroxide-reducing potential of cysteine and selenocysteine. Inorg. Chem. 2021, 60, 4646–4656. [Google Scholar] [CrossRef]
- Bortoli, M.; Torsello, M.; Bickelhaupt, F.M.; Orian, L. Role of the chalcogen (S, Se, Te) in the oxidation mechanism of the glutathione peroxidase active site. ChemPhysChem 2017, 18, 2990–2998. [Google Scholar] [CrossRef]
- Antony, S.; Bayse, C.A. Modeling the mechanism of the glutathione peroxidase mimic ebselen. Inorg. Chem. 2011, 50, 12075–12084. [Google Scholar] [CrossRef]
- Bayse, C.A. Transition states for cysteine redox processes modeled by DFT and solvent-assisted proton exchange. Org. Biomol. Chem. 2011, 9, 4748–4751. [Google Scholar] [CrossRef]
- Bayse, C.A.; Ortwine, K.N. Modeling the glutathione peroxidase-like activity of a cyclic seleninate by DFT and solvent-assisted proton exchange. Eur. J. Inorg. Chem. 2013, 2013, 3680–3688. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.W.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flohé, L. Looking back at the early stages of redox biology. Antioxidants 2020, 9, 1254. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef] [Green Version]
- Glutathione; Flohé, L. (Ed.) CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Ursini, F.; Heim, S.; Kiess, M.; Maiorino, M.; Roveri, A.; Wissing, J.; Flohé, L. Dual function of the selenoprotein PHGPx during sperm maturation. Science 1999, 285, 1393–1396. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, M.; Roveri, A.; Ursini, F. GPx4. From prevention of lipid peroxidation to spermatogenesis and back. In Glutathione; Flohé, L., Ed.; CRC Press: Boca Raton, FL, USA, 2019; pp. 111–127. [Google Scholar]
- Söhling, B.; Parther, T.; Rücknagel, K.P.; Wagner, M.; Andreesen, J.R. A selenocysteine-containing peroxiredoxin from the strictly anaerobic organism Eubacterium acidaminophilum. Biol. Chem. 2001, 382, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Orian, L.; Mauri, P.; Roveri, A.; Toppo, S.; Benazzi, L.; Bosello-Travain, V.; De Palma, A.; Maiorino, M.; Miotto, G.; Zaccarin, M.; et al. Selenocysteine oxidation in glutathione peroxidase catalysis: An MS-supported quantum mechanics study. Free Radic. Biol. Med. 2015, 87, 1–14. [Google Scholar] [CrossRef]
- Saiki, T.; Goto, K.; Okazaki, R. Isolation and crystal strcuture of a stable selenenic acid. Angew. Chem. Int. Ed. Engl. 1997, 36, 2223–2224. [Google Scholar] [CrossRef]
- Peroxiredoxin Systems Structures and Functions; Flohé, L.; Harris, J.R. (Eds.) Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Dubbs, J.M.; Mongkolsuk, S. Peroxiredoxins in bacterial antioxidant defense. In Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2007; Volume 44, pp. 143–193. [Google Scholar]
- Mauri, P.; Benazzi, L.; Flohé, L.; Maiorino, M.; Pietta, P.G.; Pilawa, S.; Roveri, A.; Ursini, F. Versatility of selenium catalysis in PHGPx unraveled by LC/ESI-MS/MS. Biol. Chem. 2003, 384, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Bosello-Travain, V.; Cozza, G.; Miotto, G.; Roveri, A.; Toppo, S.; Zaccarin, M.; Ursini, F. Understanding mammalian glutathione peroxidase 7 in the light of its homologs. Free Radic. Biol. Med. 2015, 83, 352–360. [Google Scholar] [CrossRef]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Ramming, T.; Appenzeller-Herzog, C. Destroy and exploit: Catalyzed removal of hydroperoxides from the endoplasmic reticulum. Int. J. Cell Biol. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Maiorino, M.; Roveri, A.; Ursini, F.; Brigelius-Flohé, R.; Flohé, L. Selenium and male reproduction. In Selenium: Its Molecular Biology and Role in Human Health, 2nd ed.; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Günzler, W.A.; Steffens, G.J.; Grossmann, A.; Kim, S.M.A.; Otting, F.; Wendel, A.; Flohé, L. The amino-acid sequence of bovine glutathione peroxidase. Hoppe. Seylers. Z. Physiol. Chem. 1984, 365, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Orian, L. Proton transfer and SN2 reactions as steps of fast selenol and thiol oxidation in proteins: A model molecular study based on GPx. Chempluschem 2021, 86, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, D.; Nauser, T.; Koppenol, W.H. Selenium and sulfur in exchange reactions: A comparative study. J. Org. Chem. 2010, 75, 6696–6699. [Google Scholar] [CrossRef] [PubMed]
- Metanis, N.; Keinan, E.; Dawson, P.E. Synthetic seleno-glutaredoxin 3 analogues are highly reducing oxidoreductases with enhanced catalytic efficiency. J. Am. Chem. Soc. 2006, 128, 16684–16691. [Google Scholar] [CrossRef] [Green Version]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Maiorino, M.; Ursini, F.; Orian, L. A dual attack on the peroxide bond. The common principle of peroxidatic cysteine or selenocysteine residues. Redox Biol. 2020, 34, 101540. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, T.; Budde, H.; Flohé, L.; Menge, U.; Singh, M.; Trujillo, M.; Radi, R. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2004, 423, 182–191. [Google Scholar] [CrossRef]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Pedre, B.; Young, D.; Charlier, D.; Mourenza, Á.; Rosado, L.A.; Marcos-Pascual, L.; Wahni, K.; Martens, E.; de la Rubia, A.G.; Belousov, V.V.; et al. Structural snapshots of OxyR reveal the peroxidatic mechanism of H2O2 sensing. Proc. Natl. Acad. Sci. USA 2018, 115, 11623–11632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santi, C.; Tidei, C.; Scalera, C.; Piroddi, M.; Galli, F. Selenium containing compounds: From poison to drug candidates: A review on the GPx-like activity. Curr. Chem. Biol. 2013, 7, 25–36. [Google Scholar] [CrossRef]
- Pacula, A.J.; Mangiavacchi, F.; Sancineto, L.; Lenardão, E.J.; Scianowski, J.; Santi, C. An update on “Selenium containing compounds: From poison to drug candidates: A review on the GPx-like activity”. Curr. Chem. Biol. 2015, 9, 97–112. [Google Scholar] [CrossRef]
- Orian, L.; Toppo, S. Organochalcogen peroxidase mimetics as potential drugs: A long story of a promise still unfulfilled. Free Radic. Biol. Med. 2014, 66, 65–74. [Google Scholar] [CrossRef]
- Obieziurska-Fabisiak, M.; Pacula, A.J.; Capoccia, L.; Drogosz-Stachowicz, J.; Janecka, A.; Santi, C.; Scianowski, J. Phenylselanyl group incorporation for ‘Glutathione peroxidase-like’ activity modulation. Molecules 2020, 25, 3354. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Mugesh, G. Functional mimics of glutathione peroxidase: Bioinspired synthetic antioxidants. Acc. Chem. Res. 2010, 43, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Cadenas, E.; Graf, P.; Sies, H. A novel biologically active seleno-organic compound-1. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem. Pharmacol. 1984, 33, 3235–3239. [Google Scholar] [CrossRef]
- Sands, K.N.; Back, T.G. Key steps and intermediates in the catalytic mechanism for the reduction of peroxides by the antioxidant ebselen. Tetrahedron 2018, 38, 4959–4967. [Google Scholar] [CrossRef]
- Bachrach, S.M.; Hayes, J.M.; Dao, T.; Mynar, J.L. Density functional theory gas- and solution-phase study of nucleophilic substitution at di- and trisulfides. Theor. Chem. Acc. 2002, 107, 266–271. [Google Scholar] [CrossRef]
- Hayes, J.M.; Bachrach, S.M. Effect of micro and bulk solvation on the mechanism of nucleophilic substitution at sulfur in disulfides. J. Phys. Chem. A 2003, 107, 7952–7961. [Google Scholar] [CrossRef]
- Mulhearn, D.C.; Bachrach, S.M. Selective nucleophilic attack of trisulfides. A ab initio study. J. Am. Chem. Soc. 1996, 118, 9415–9421. [Google Scholar] [CrossRef]
- Bachrach, S.M.; Walker, C.J.; Lee, F.; Royce, S. Effect of ring strain on nucleophilic substitution at selenium: A computational study of cyclic diselenides and selenenyl sulfides. J. Org. Chem. 2007, 72, 5174–5182. [Google Scholar] [CrossRef]
- Antoniadou, I.; Kouskou, M.; Arsiwala, T.; Singh, N.; Vasudevan, S.R.; Fowler, T.; Cadirci, E.; Churchill, G.C.; Sharp, T. Ebselen has lithium-like effects on central 5-HT 2A receptor function. Br. J. Pharmacol. 2018, 175, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Fenn, G.D.; Waller-Evans, H.; Atack, J.R.; Bax, B.D. Crystallization and structure of ebselen bound to Cys141 of human inositol monophosphatase. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76, 469–476. [Google Scholar] [CrossRef]
- Yu, Y.; Jin, Y.; Zhou, J.; Ruan, H.; Zhao, H.; Lu, S.; Zhang, Y.; Li, D.; Ji, X.; Ruan, B.H. Ebselen: Mechanisms of glutamate dehydrogenase and glutaminase enzyme inhibition. ACS Chem. Biol. 2017, 12, 3003–3011. [Google Scholar] [CrossRef]
- Larabee, J.L.; Hocker, J.R.; Hanas, J.S. Mechanisms of inhibition of zinc-finger transcription factors by selenium compounds ebselen and selenite. J. Inorg. Biochem. 2009, 103, 419–426. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020, 156, 107–112. [Google Scholar] [CrossRef]
- Madabeni, A.; Nogara, P.A.; Omage, F.B.; Rocha, J.B.T.; Orian, L. Mechanistic insight into SARS-CoV-2 Mpro inhibition by organoselenides: The ebselen case study. Appl. Sci. 2021, 11, 6291. [Google Scholar] [CrossRef]
- Nogara, P.A.; Omage, F.B.; Bolzan, G.R.; Delgado, C.P.; Aschner, M.; Orian, L.; Rocha, J.B.T. In silico studies on the interaction between Mpro and PLpro from SARS-CoV-2 and ebselen, its metabolites and derivatives. Mol. Inform. 2021, 40, 2100028. [Google Scholar] [CrossRef] [PubMed]
- Saytzeff, A.; Grabowsky, N. Eigenschaften der normalen Sulfobutylsäure und ihrer Salze. Justus Liebigs Ann. Chem. 1875, 175, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Emerson, D.W.; Craig, A.P.; Potts, I.W. The pyrolysis of unsymmetrical dialkyl sulfoxides. Rates of alkene formation and composition of the gaseous products. J. Org. Chem. 1967, 32, 102–105. [Google Scholar] [CrossRef]
- Ribaudo, G.; Bortoli, M.; Ongaro, A.; Oselladore, E.; Gianoncelli, A.; Zagotto, G.; Orian, L. Fluoxetine scaffold to design tandem molecular antioxidants and green catalysts. RSC Adv. 2020, 10, 18583. [Google Scholar] [CrossRef]
- Nogara, P.A.; Madabeni, A.; Bortoli, M.; Teixeira Rocha, J.B.; Orian, L. Methylmercury can facilitate the formation of dehydroalanine in selenoenzymes: Insight from DFT molecular modeling. Chem. Res. Toxicol. 2021, 34, 1655–1663. [Google Scholar] [CrossRef]
- Ma, S.; Caprioli, R.M.; Hill, K.E.; Burk, R.F. Loss of selenium from selenoproteins: Conversion of selenocysteine to dehydroalanine in vitro. J. Am. Soc. Mass Spectrom. 2003, 14, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Palacios, Ò.; Lobinski, R. Investigation of the stability of selenoproteins during storage of human serum by size-exclusion LC-ICP-MS. Talanta 2007, 71, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin ii by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]






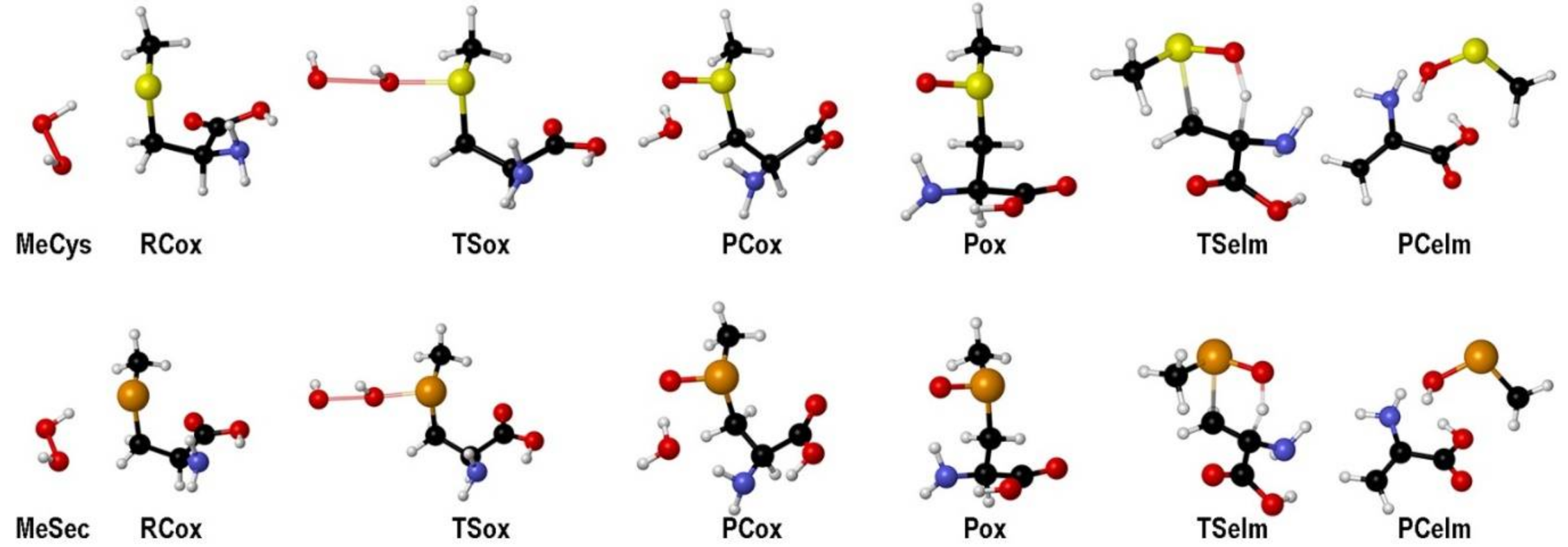

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
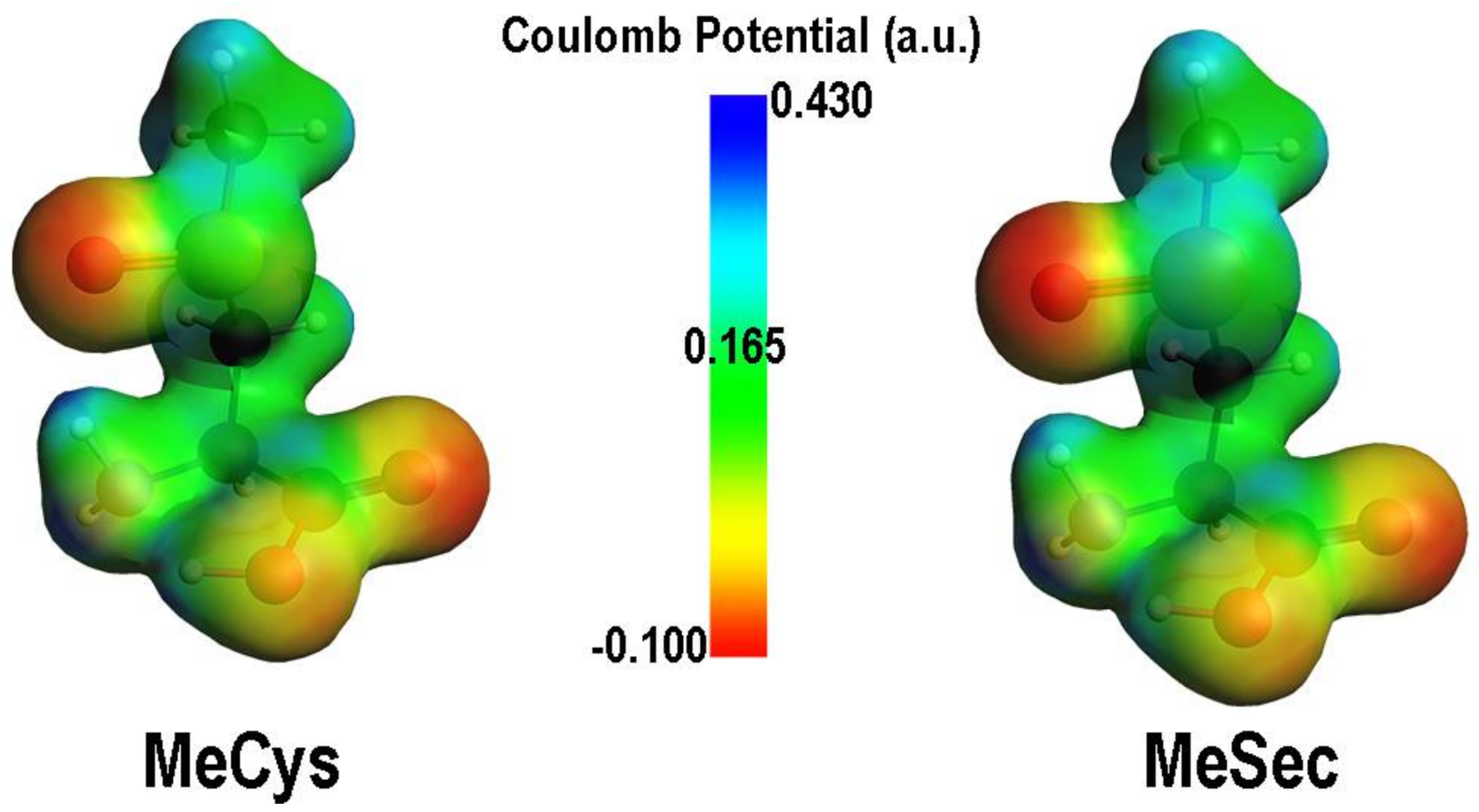{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
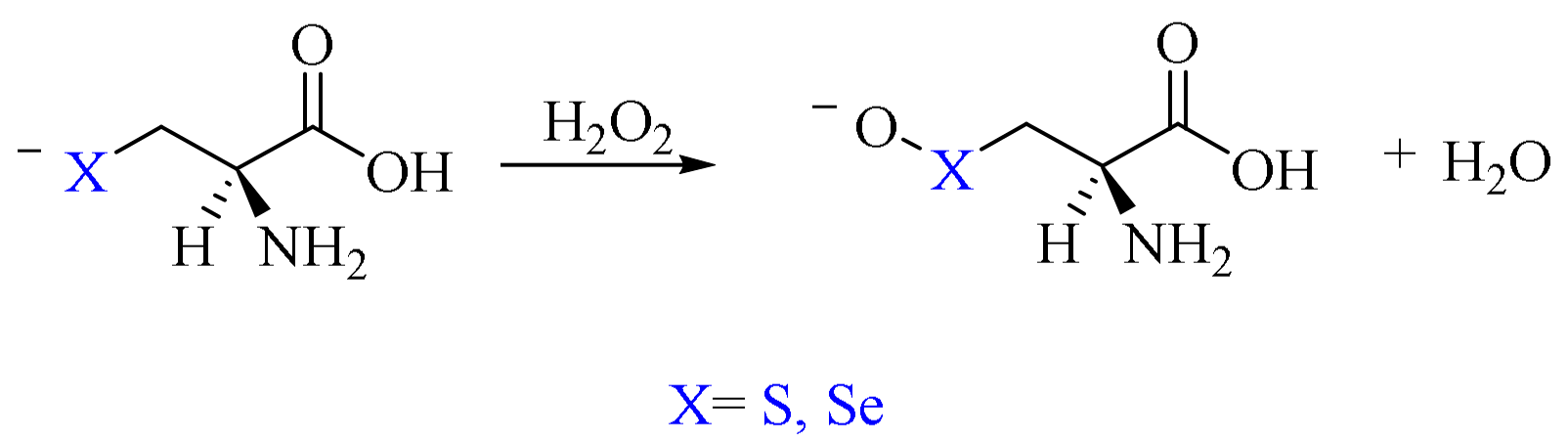{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elementary Steps | S | Se | Te |
|---|---|---|---|
| I | 27.1 | 24.0 | 16.5 |
| iso | 38.8 | 31.0 | 31.5 |
| II | 19.3 | 16.8 | 9.4 |
| III | 26.2 | 32.3 | 29.8 |
| Elementary Steps | S | Se | Te |
|---|---|---|---|
| I | 12.3 (7.4) | 10.2 (5.5) | 8.2 (3.7) |
| II | 11.2 (8.4) | 12.0 (9.4) | 12.0 (8.9) |
| III | 11.2 (10.0) | 16.5 (16.2) | 16.8 (16.4) |
| RCOX | TSOX | PCOX | POX | ΔE#OX a | TSiso | Piso | ΔE#iso b | |
|---|---|---|---|---|---|---|---|---|
| Cys | −6.01 | 17.84 | −47.88 | −38.84 | 23.85 | 39.80 | −49.83 | 39.80 |
| Sec | −6.02 | 14.06 | −40.04 | −29.64 | 20.08 | 30.77 | −55.42 | 30.77 |
| Tec | −6.04 | 6.25 | −48.51 | −37.72 | 12.29 | 32.09 | −65.75 | 32.09 |
| Cys− | −19.86 | −13.01 | −65.14 | −50.17 | 6.85 | - | - | - |
| Sec− | −18.70 | −13.51 | −59.92 | −44.08 | 5.19 | - | - | - |
| RCOX | TSOX | PCOX | POX | ΔE#OX a | |
|---|---|---|---|---|---|
| Cys | −22.92 | −12.65 | −80.81 | −49.83 | 10.27 |
| Sec | −22.43 | −15.67 | −84.88 | −55.42 | 6.77 |
| RCox | TSox | ΔG‡ox | PCox | Pox | TSelm | ΔG‡elm | PCelm | Pelm | ΔGelm | |
|---|---|---|---|---|---|---|---|---|---|---|
| MeCys | 2.65 | 10.24 | 7.59 | −47.48 | −49.25 | −30.94 | 18.31 | −42.52 | −48.17 | 1.08 |
| MeSec | 3.80 | 6.37 | 2.57 | −41.24 | −41.87 | −31.64 | 10.23 | −45.99 | −51.58 | −9.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orian, L.; Flohé, L. Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology. Antioxidants 2021, 10, 1560. https://doi.org/10.3390/antiox10101560
Orian L, Flohé L. Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology. Antioxidants. 2021; 10(10):1560. https://doi.org/10.3390/antiox10101560
Chicago/Turabian StyleOrian, Laura, and Leopold Flohé. 2021. "Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology" Antioxidants 10, no. 10: 1560. https://doi.org/10.3390/antiox10101560
APA StyleOrian, L., & Flohé, L. (2021). Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology. Antioxidants, 10(10), 1560. https://doi.org/10.3390/antiox10101560