
Inhibition of Peroxiredoxin 6 PLA2 Activity Decreases Oxidative Stress and the Severity of Acute Lung Injury in the Mouse Cecal Ligation and Puncture Model

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. PIP-2
2.3. Generation of CLP and Experimental Plan
2.4. Bacterial Culture
2.5. Biochemical Assays
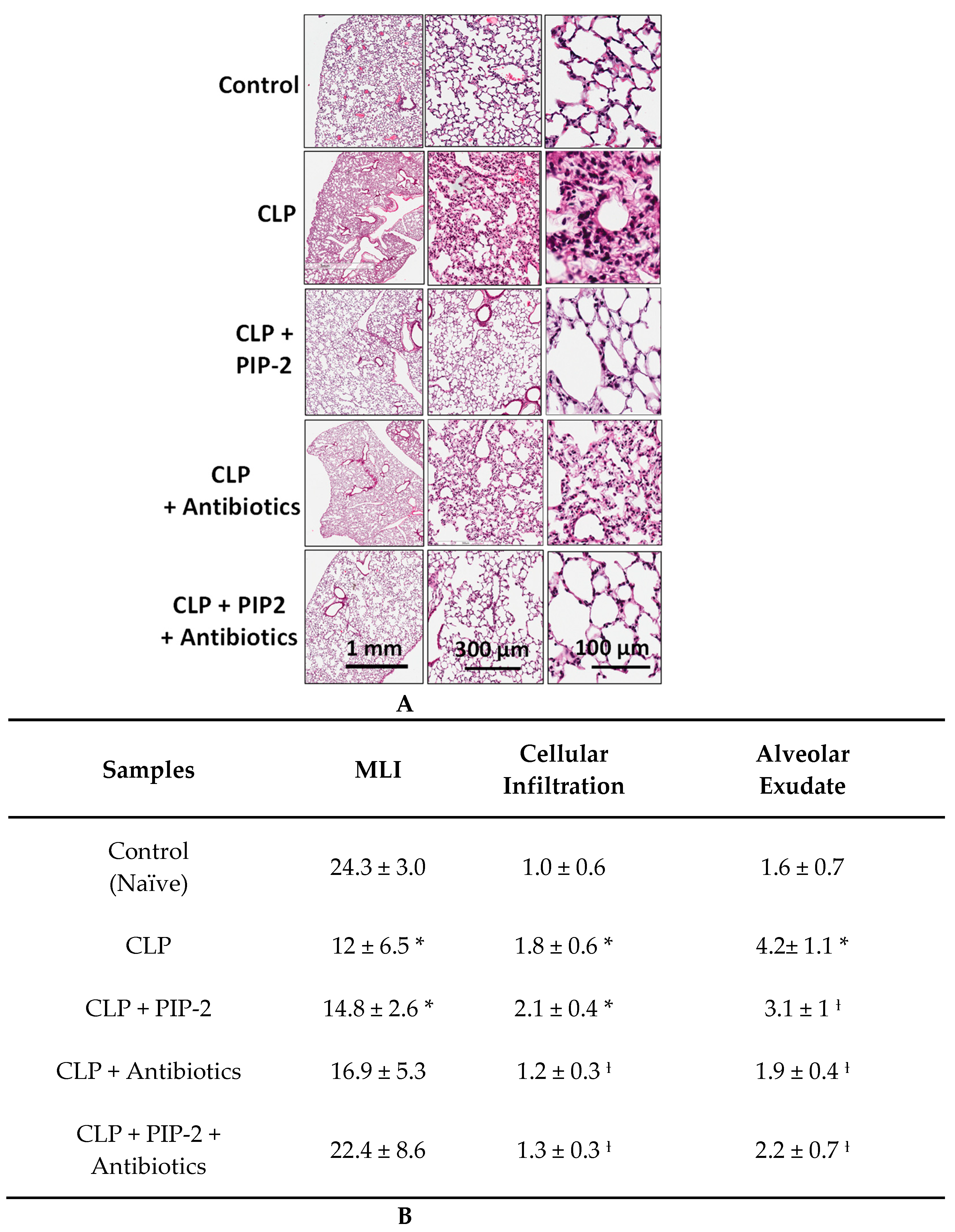
2.6. Lung Histology
2.7. Statistical Analysis
3. Results
3.1. Tissue Uptake of PIP-2
3.2. Mortality
3.3. Bacterial Culture
3.4. Lung Histology
3.5. Lung MPO Activity and W/D
3.6. Lung Cytokines
3.7. Lung Oxidative Stress
4. Discussion
5. Conclusions
6. Patent Pending
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol. Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Downey, G.P. Neutrophil activation and acute lung injury. Curr. Opin. Crit. Care 2001, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.F.; Ward, P.A. Role of oxidants in lung injury during sepsis. Antioxid. Redox Signal. 2007, 9, 1991–2002. [Google Scholar] [CrossRef]
- Aldridge, A.J. Role of the neutrophil in septic shock and the adult respiratory distress syndrome. Eur. J. Surg. 2002, 168, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Chabot, F.; Mitchell, J.A.; Gutteridge, J.M.; Evans, T.W. Reactive oxygen species in acute lung injury. Eur. Respir. J. 1998, 11, 745–757. [Google Scholar]
- Sato, K.; Kadiiska, M.B.; Ghio, A.J.; Corbett, J.; Fann, Y.C.; Holland, S.M.; Thurman, R.G.; Mason, R.P. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: A model for ARDS. FASEB J. 2002, 16, 1713–1720. [Google Scholar] [CrossRef]
- Davidson, B.A.; Vethanayagam, R.R.; Grimm, M.J.; Mullan, B.A.; Raghavendran, K.; Blackwell, T.S.; Freeman, M.L.; Ayyasamy, V.; Singh, K.K.; Sporn, M.B.; et al. NADPH oxidase and Nrf2 regulate gastric aspiration-induced inflammation and acute lung injury. J. Immunol. 2013, 190, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Dodia, C.; Chatterjee, S.; Zagorski, J.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Jain, M.; Fisher, A.B. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. J. Pharmacol. Exp. Ther. 2013, 345, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B.; Zhang, G. NADPH and NADPH Oxidase. In Encyclopedia of Respiratory Medicine, Laurent, G.J., Shapiro, S.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 3, pp. 77–83. [Google Scholar]
- Kreck, M.L.; Freeman, J.L.; Abo, A.; Lambeth, J.D. Membrane association of Rac is required for high activity of the respiratory burst oxidase. Biochemistry 1996, 35, 15683–15692. [Google Scholar] [CrossRef]
- Sarfstein, R.; Gorzalczany, Y.; Mizrahi, A.; Berdichevsky, Y.; Molshanski-Mor, S.; Weinbaum, C.; Hirshberg, M.; Dagher, M.C.; Pick, E. Dual role of Rac in the assembly of NADPH oxidase, tethering to the membrane and activation of p67phox: A study based on mutagenesis of p67phox-Rac1 chimeras. J. Biol. Chem. 2004, 279, 16007–16016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Sulciner, D.J.; Gutkind, J.S.; Irani, K.; Goldschmidt-Clermont, P.J.; Finkel, T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem. J. 1996, 318 Pt 2, 379–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Wang, W.; Suzuki, Y.; Tanigaki, T.; Rank, D.R.; Raffin, T.A. Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. Am. J. Respir. Crit. Care Med. 1994, 150, 1449–1452. [Google Scholar] [CrossRef]
- Dodd-o, J.M.; Welsh, L.E.; Salazar, J.D.; Walinsky, P.L.; Peck, E.A.; Shake, J.G.; Caparrelli, D.J.; Ziegelstein, R.C.; Zweier, J.L.; Baumgartner, W.A.; et al. Effect of NADPH oxidase inhibition on cardiopulmonary bypass-induced lung injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H927–H936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, L.; Lu, S.; Mittwede, P.N.; Clemmer, J.S.; Hester, R.L. Inhibition of NADPH oxidase prevents acute lung injury in obese rats following severe trauma. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H684–H689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd-O,, J.M.; Pearse, D.B. Effect of the NADPH oxidase inhibitor apocynin on ischemia-reperfusion lung injury. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H303–H312. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Zhu, G.; Yuan, D.; Yao, W.; Chi, X.; Hei, Z. Propofol alleviates acute lung injury following orthotopic autologous liver transplantation in rats via inhibition of the NADPH oxidase pathway. Mol. Med. Rep. 2015, 11, 2348–2354. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.P.; Standiford, T.J.; Rahman, A.; Newstead, M.; Holland, S.M.; Dinauer, M.C.; Liu, Q.H.; Malik, A.B. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: Studies in p47phox-/- and gp91phox-/- mice. J. Immunol. 2002, 168, 3974–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, E.; Greineder, C.F.; Dodia, C.; Han, J.; Mesaros, C.; Shuvaev, V.; Blair, I.; Fisher, A.B.; Muzykantov, V. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Control. Release 2012, 163, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmageed, M.E.; El-Awady, M.S.; Suddek, G.M. Apocynin ameliorates endotoxin-induced acute lung injury in rats. Int. Immunopharmacol. 2016, 30, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Kokkinaki, D.; Valenti, M.C.; Kim, G.J.; Barca, E.; Tomar, D.; Hoffman, N.E.; Subramanyam, P.; Colecraft, H.M.; Hirano, M.; et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S. Emerging drugs for acute lung injury. Expert Opin. Emerg. Drugs 2015, 20, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; McAuley, D.F.; Ware, L.B. Clinical trials in acute respiratory distress syndrome: Challenges and opportunities. Lancet Respir. Med. 2017, 5, 524–534. [Google Scholar] [CrossRef]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Medina, J.P.; Tao, J.Q.; Patel, P.; Bannitz-Fernandes, R.; Dodia, C.; Sorokina, E.M.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. Genetic inactivation of the phospholipase A2 activity of peroxiredoxin 6 in mice protects against LPS-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L656–L668. [Google Scholar] [CrossRef]
- Ambruso, D.R.; Ellison, M.A.; Thurman, G.W.; Leto, T.L. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim. Biophys. Acta 2011, 1823, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Chatterjee, S. A Peptide Inhibitor of Peroxiredoxin 6 Phospholipase A2 Activity Significantly Protects against Lung Injury in a Mouse Model of Ventilator Induced Lung Injury (VILI). Antioxidants 2021, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50 (Suppl.), S237–S242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 2006, 1761, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Maridonneau-Parini, I.; Tauber, A.I. Activation of NADPH-oxidase by arachidonic acid involves phospholipase A2 in intact human neutrophils but not in the cell-free system. Biochem. Biophys. Res. Commun. 1986, 138, 1099–1105. [Google Scholar] [CrossRef]
- Henderson, L.M.; Chappell, J.B.; Jones, O.T. Superoxide generation is inhibited by phospholipase A2 inhibitors. Role for phospholipase A2 in the activation of the NADPH oxidase. Biochem. J. 1989, 264, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benipal, B.; Feinstein, S.I.; Chatterjee, S.; Dodia, C.; Fisher, A.B. Inhibition of the phospholipase A2 activity of peroxiredoxin 6 prevents lung damage with exposure to hyperoxia. Redox Biol. 2015, 4, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism-based inhibitor of NADPH oxidase (type 2). Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L635–L644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanshan, Y.; Beibei, J.; Li, T.; Minna, G.; Shipeng, L.; Li, P.; Yong, Z. Phospholipase A2 of Peroxiredoxin 6 Plays a Critical Role in Cerebral Ischemia/Reperfusion Inflammatory Injury. Front. Cell. Neurosci. 2017, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Chander, A. Inhibition of lung calcium-independent phospholipase A2 by surfactant protein A. Am. J. Physiol. 1994, 267, L335–L341. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Manevich, Y.; Baldwin, J.L.; Dodia, C.; Yu, K.; Feinstein, S.I.; Fisher, A.B. Interaction of surfactant protein A with peroxiredoxin 6 regulates phospholipase A2 activity. J. Biol. Chem. 2006, 281, 7515–7525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaiah, S.Y.; Dodia, C.; Sorokina, E.M.; Li, H.; Feinstein, S.I.; Fisher, A.B. Binding sites for interaction of peroxiredoxin 6 with surfactant protein A. Biochim. Biophys. Acta 2016, 1864, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I. Identification of Small Peptides that Inhibit NADPH Oxidase (Nox2) Activation. Antioxidants 2018, 7, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B.; Dodia, C.; Chatterjee, S.; Feinstein, S.I. A Peptide Inhibitor of NADPH Oxidase (NOX2) Activation Markedly Decreases Mouse Lung Injury and Mortality Following Administration of Lipopolysaccharide (LPS). Int. J. Mol. Sci. 2019, 20, 2395. [Google Scholar] [CrossRef] [Green Version]
- Ware, L.B. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care. Med. 2006, 27, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I.; Ho, Y.S. Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J. Lipid Res. 2005, 46, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Feinstein, S.I.; Wang, Y.; Dodia, C.; Fisher, D.; Yu, K.; Ho, Y.S.; Fisher, A.B. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free. Radic. Biol. Med. 2010, 49, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Pourfathi, M.; Cereda, M.; Chatterjee, S.; Xin, Y.; Kadlecek, S.; Duncan, I.; Hamedani, H.; Siddiqui, S.; Profka, H.; Ehrich, J.; et al. Lung Metabolism and Inflammation during Mechanical Ventilation; An Imaging Approach. Sci. Rep. 2018, 8, 3525. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Habertheuer, A.; Xin, Y.; Pourfathi, M.; Tao, J.Q.; Hamedani, H.; Kadlecek, S.; Duncan, I.; Vallabhajosyula, P.; Naji, A.; et al. Detection of lung transplant rejection in a rat model using hyperpolarized [1-(13) C] pyruvate-based metabolic imaging. NMR Biomed. 2019, 32, e4107. [Google Scholar] [CrossRef]
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellner, M.; Noonepalle, S.; Lu, Q.; Srivastava, A.; Zemskov, E.; Black, S.M. ROS Signaling in the Pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). Adv. Exp. Med. Biol. 2017, 967, 105–137. [Google Scholar] [CrossRef]
- Helmstadter, J.; Keppeler, K.; Aust, F.; Kuster, L.; Frenis, K.; Filippou, K.; Vujacic-Mirski, K.; Tsohataridis, S.; Kalinovic, S.; Kroller-Schon, S.; et al. GLP-1 Analog Liraglutide.Improves Vascular Function in Polymicrobial Sepsis by Reduction of Oxidative Stress and Inflammation. Antioxidants 2021, 10, 1175. [Google Scholar] [CrossRef] [PubMed]
- Ortega, E.; de Pablo, M.A.; Gallego, A.M.; Gaforio, J.J.; Alvarez, C.; Ruiz-Bravo, A.; de Cienfuegos, G.A. Modification of acquired immunity in mice by imipenem/cilastatin. J. Antimicrob. Chemother. 1999, 44, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; An, X.; Hu, X.; Bi, J.; Tong, L.; Yang, D.; Song, Y.; Bai, C. Peroxiredoxin 6 knockout aggravates cecal ligation and puncture-induced acute lung injury. Int. Immunopharmacol. 2019, 68, 252–258. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Manevich, Y.; Shuvaeva, T.; Dodia, C.; Kazi, A.; Feinstein, S.I.; Fisher, A.B. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A(2) activities. Arch. Biochem. Biophys. 2009, 485, 139–149. [Google Scholar] [CrossRef] [Green Version]
- To, E.E.; Luong, R.; Diao, J.; JJ, O.L.; Brooks, D.A.; Vlahos, R.; Selemidis, S. Novel endosomal NOX2 oxidase inhibitor ameliorates pandemic influenza A virus-induced lung inflammation in mice. Respirology 2019, 24, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Yajjala, V.K.; Bauer, C.; Talmon, G.A.; Fischer, K.J.; Kielian, T.; Metzger, D.W. Nox2-derived oxidative stress results in inefficacy of antibiotics against post-influenza S. aureus pneumonia. J. Exp. Med. 2016, 213, 1851–1864. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| PLA2 Activity nmol/h/mg Protein | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mouse Organs | Lung | Kidney | Heart | Liver | ||||
| 2 h | 24 h | 2 h | 24 h | 2 h | 24 h | 2 h | 24 h | |
| Liposomes | 8.21 ± 0.28 | ND | 6.61 ± 0.27 | ND | 4.75 ± 0.39 | ND | 5.32 ± 0.33 | ND |
| Liposomes + PIP-2 (2 µg)/g | 1.49 ± 0.06 | 1.51 ± 0.09 | 1.25 ± 0.06 | 1.28 ± 0.04 | 0.95 ± 0.02 | 0.96 ± 0.06 | 1.26 ± 0.02 | 1.27 ± 0.09 |
| Liposomes + PIP-2 (20 µg)/g | 1.44 ± 0.06 | ND | 1.09 ± 0.02 | ND | 0.85 ± 0.03 | ND | 1.22 ± 0.04 | ND |
| Number of Mice | Mice Dead by 24 h | % Mortality | |
|---|---|---|---|
| CLP | 15 | 1 | 7 |
| CLP + Antibiotics | 13 | 1 | 8 |
| CLP + PIP-2 (2 µg/g) | 10 | 0 | 0 |
| CLP + PIP – 2 (2µ/g) + Antibiotics | 11 | 1 | 9 |
| CLP + PIP – 2 (10 µg/g) | 6 | 6 | 100 |
| CLP + PIP – 2 (10 µg/g) + Antibiotics | 5 | 0 | 0 |
| Colonies /mL of Lung Homogenate X 10−3 | Colonies /mL of Peritoneal Fluid X 10−3 | |||
|---|---|---|---|---|
| 6 h | 24 h | 6 h | 24 h | |
| Control | 0.32 ± 0.10 | 0.38 ± 0.80 | ||
| CLP | 0.06 ± 0.05 | 68.4 ± 21.6 * | 10.5 ± 0.2 | 1522 ± 440 * |
| CLP + PIP-2 (2 µg/g) | 0.09 ± 0.04 | 133 ± 16.5 ƚ | 15.8 ± 0.16 | 3894 ± 536 ƚ |
| CLP + Antibiotics | 0 | 0.45 ± 0.14 | 0 | 0.30 ± 0.14 |
| CLP + PIP – 2 (2 µg/g) + Antibiotics | 0 | 0.42 ± 0.16 | 0 | 0.32 ± 0.12 |
| CLP + PIP – 2 (10 µg/g) + Antibiotics | 0 | 0.37 ± 0.09 | 0 | 0.34 + 0.11 |
| MPO Activity nmol/mg Protein | Wet/Dry Weight | |||
|---|---|---|---|---|
| 6 h | 24 h | 6 h | 24 h | |
| Control | 4.94 ± 3.8 | 4.90 ± 0.14 | ||
| CLP | 14.02 ± 7.3 | 68.7 ±9.8 * | 5.13 ± 0.26 | 7.32 ± 0.51 * |
| CLP + PIP-2 (2 mg/g) | 0.66 ± 0.24 | 23.3 ± 6.6 * ƚ | 5.07 ± 0.23 | 6.24 ± 0.42 * ƚ |
| CLP + Antibiotics | 0.29 ± 0.14 | 8.95 ± 4.1 ƚ | 5.00 ± 0.22 | 7.52 ± 0.16 * |
| CLP + PIP - 2(2 µg/g) + Antibiotics | 0.04 ± 0.15 | 2.40 ± 6.5 ƚ | 5.01 ± 0.18 | 6.27 ± 0.47 * ƚ |
| CLP + PIP - 2(10 µg/g) + Antibiotics | 0.26 ± 0.18 | 2.94 ± 1.6 ƚ | 5.02 ± 0.14 | 6.23 ± 0.21 * ƚ |
| TBARS pmol/mg Prot. | 8-Isoprostanes pmol/mg Prot. | Protein Carbonyls nmol/mg Prot. | ||||
|---|---|---|---|---|---|---|
| 6 h | 24 h | 6 h | 24 h | 6 h | 24 h | |
| Control | 69.3 ± 2.6 | 32.3 ± 1.6 | 5.25 ± 0.40 | |||
| CLP | 73.4 ± 5.9 | 243 ± 12 * | 32.9 ± 1.4 | 121 ± 5.8 * ƚ | 5.26 ± 0.02 | 16.1 ± 1.26 * |
| CLP + PIP-2 (2 µg/g) | 72.8 ± 2.2 | 105 ± 5.5 * ƚ | 31.8 ± 4.8 | 59.6 ± 5.3 * ƚ | 5.31 ± 0.74 | 7.67 ± 0.23 * ƚ |
| CLP + Antibiotics | 71.9 ± 2.8 | 231 ± 20 * | 32.1 ± 1.0 | 111 ± 9.4 * | 5.08 ± 0.33 | 15.8 ± 0.89 * |
| CLP + PIP – 2 (2 µg/g) Antibiotics | 74.7 ± 5.6 | 108 ± 13 * ƚ | 33.7 ± 2.0 | 53.4 ± 6.3 * ƚ | 5.19 ± 0.33 | 7.74 ± 0.28 * ƚ |
| CLP + PIP – 2 (10 µg/g) Antibiotics | 74 .5 ± 7.2 | 111 ± 2.6 * ƚ | 32.7 ± 1.8 | 56.6 ± 6.5 * ƚ | 4.96 ± 0.33 | 7.60 ± 0.15 * ƚ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, A.B.; Dodia, C.; Tao, J.-Q.; Feinstein, S.I.; Chatterjee, S. Inhibition of Peroxiredoxin 6 PLA2 Activity Decreases Oxidative Stress and the Severity of Acute Lung Injury in the Mouse Cecal Ligation and Puncture Model. Antioxidants 2021, 10, 1676. https://doi.org/10.3390/antiox10111676
Fisher AB, Dodia C, Tao J-Q, Feinstein SI, Chatterjee S. Inhibition of Peroxiredoxin 6 PLA2 Activity Decreases Oxidative Stress and the Severity of Acute Lung Injury in the Mouse Cecal Ligation and Puncture Model. Antioxidants. 2021; 10(11):1676. https://doi.org/10.3390/antiox10111676
Chicago/Turabian StyleFisher, Aron B., Chandra Dodia, Jian-Qin Tao, Sheldon I. Feinstein, and Shampa Chatterjee. 2021. "Inhibition of Peroxiredoxin 6 PLA2 Activity Decreases Oxidative Stress and the Severity of Acute Lung Injury in the Mouse Cecal Ligation and Puncture Model" Antioxidants 10, no. 11: 1676. https://doi.org/10.3390/antiox10111676