Abstract
Amyloid-β (Aβ) deposition, a hallmark of Alzheimer’s disease, is known to induce free radical production and oxidative stress, leading to neuronal damage. During oxidative stress, several cell types (including astrocytes) can activate the nuclear factor erythroid 2-related factor 2 (Nrf2), a regulator of several phase II detoxifying and antioxidant genes, such as the System Xc− subunit xCT. Here, we studied (i) the effect of the Aβ fragment 25-35 (Aβ25-35) on Nrf2-dependent System Xc− expression in U373 human astroglial cells and (ii) the effect of Aβ25-35-induced astrocytic response on neuronal cell viability using an in vitro co-culture system. We found that Aβ25-35 was able to activate an antioxidant response in astrocytes, by inducing both Nrf2 activation and System Xc− up-regulation. However, this astrocytic response caused an enhanced cell mortality of co-cultured SH-SY5Y cells, taken as a neuronal model. Consistently, the specific System Xc− inhibitor sulfasalazine prevented the increase of both neuronal mortality and extracellular glutamate levels, thus indicating that the neurotoxic effect was due to an augmented release of glutamate through the transporter. The involvement of NMDA receptor activation in this pathway was also demonstrated using the specific inhibitor MK801 that completely restored neuronal viability at the control levels. The present study sheds light on the Nrf2/system Xc− pathway in the toxicity induced by Aβ25-35 and may help to better understand the involvement of astrocytes in neuronal death during Alzheimer’s disease.
1. Introduction
Alzheimer’s disease (AD) is recognized by World Health Organization as a global public health priority, being the leading cause of dementia, responsible for 50–75% of all world cases and results in the deterioration of selective cognitive performance, including memory and mental processing [1,2].
AD is characterized by deposition of amyloid-β peptide (Aβ) in senile plaques, intracellular neurofibrillary tangles consisting of hyper-phosphorylated tau, synaptic dysfunction and neuronal death. Some or all of these hallmarks are causally linked to the cognitive and behavioral deficits that denote this disease [1,3,4].
Although the molecular mechanisms leading to neuronal damage in AD have not been completely understood, it is well established that accumulation of Aβ, in soluble and/or aggregated form, is a key pathogenetic event for AD [5,6]. The predominant forms of Aβ in the human brain as well as in human cerebrospinal fluid (CSF) are the full length Aβ1-40 and Aβ1-42 peptides and shorter carboxyterminal Aβ peptides, as well as amino-terminal truncated species [7]. In senile plaques, the predominant N-terminal truncated Aβ peptides are Aβ3-40/42, Aβ11-40/42, and Aβ17-40/42 [7]. Note that many Aβ fragments (e.g., Aβ1-16, Aβ1-33, Aβ1-39) in CSF can discriminate AD patients from non-demented controls [8]. Interestingly, Aβ fragment 25-35 (Aβ25-35) is the shortest fragment that exhibits large β-sheet fibrils and retains the toxicity of the full-length peptide [9]. Aβ25-35, physiologically present in elderly people, is the more toxic region and has been found to play a relevant role in free radical-associated neurotoxicity in AD, due to its peculiar aggregation properties [9].
Most of the known genetic, medical, environmental, and lifestyle-related risk factors for AD are associated with increased oxidative stress [10,11]. Indeed, AD brain is under strong oxidative stress, manifested by increased protein and DNA oxidation, lipid peroxidation, free radical formation, nitro-tyrosine levels, and advanced glycation end products [12,13,14]. Noteworthy, we previously demonstrated that both Aβ25-35 and Aβ1-42 are able to induce oxidative stress in endothelial cells, by producing superoxide and hydroxyl radicals [15]. Moreover, Aβ can form pore in astrocytes membranes and allow the influx of calcium from the extracellular space [16]. This modulation of calcium levels can induce the activation of NADPH oxidase and the subsequent reactive oxygen species (ROS) production, thus leading to Aβ-induced oxidative stress in astrocytes [17,18]. Chronic oxidative stress conditions arise because of an imbalance between the production of pro-oxidant molecules (e.g., ROS) and antioxidant system (e.g., intracellular glutathione (GSH) production), in favor of pro-oxidant molecules [19].
During oxidative stress, a variety of cell types are able to up-regulate the activity of nuclear factor erythroid 2-related factor 2 (Nrf2), the main regulator of the antioxidant response, thus counteracting intracellular ROS accumulation and GSH depletion [20,21]. Unlike neurons, astrocytes can strongly up-regulate Nrf2-mediated gene expression, thus leading to a major resistance to oxidative injury than isolated neurons [22]. Upon changes in cellular redox state, Nrf2 migrates to the nucleus and successively binds to promoter regions, known as antioxidant responsive element (ARE), of many phase II detoxifying and antioxidant genes, such as catalase (CAT), γ-glutamyl-cysteine ligase (GCL), superoxide dismutase (SOD), heme-oxygenase-1 (HO-1), glutathione peroxidase (GPX), and System Xc− subunit xCT [23,24]. System Xc− is an amino acid antiporter and mediates the exchange of intracellular L-glutamate and extracellular L-cystine across the plasma membrane [25]. In astrocytes, L-cystine import through System Xc− is crucial to glutathione production and protection from oxidative stress.
On the other side, however, glutamate export is a further route of release through which this neurotransmitter may provoke excitotoxicity [26,27]. Thus, System Xc− has currently been related to both pathological and physiological processes in the central nervous system (CNS) [28]. Even though the induction of Nrf2-dependent gene expression has been commonly reported as a protective mechanism to withstand the effects of oxidative stress in astrocytes, the up-regulation of xCT induced by Nrf2 could be a possible source for excitotoxicity due to excessive release of glutamate [29].
Despite many clinical and experimental data suggesting astrocytes as the cell population liable for most of the neuronal death in several neurodegenerative disorders, the specific cellular mechanisms are not yet clearly defined.
Here, we studied the effect of the Aβ25-35 on the induction of astroglial antioxidant response, focusing on the activation of Nrf2 transcription factor and on System Xc− expression. We also analyzed the effect of astrocytic System Xc− upregulation on the viability of neuronal cells in a co-culture system.
2. Materials and Methods
2.1. Materials
Amyloid-β Protein fragment 25-35 (Aβ25-35), DMEM (Dulbecco’s modified Eagle’s medium), FBS (fetal bovine serum), Trypsin–EDTA 0.25% solution, gentamicin 50 mg/mL solution, sulfasalazine (SSZ; a specific inhibitor of System Xc−), MK-801 hydrogen maleate (MK-801; an NMDA receptor antagonist), and a kit for MTT assay were obtained from Sigma–Aldrich (Milan, Italy). The reagent for Bradford assay was from Bio-Rad Italia (Milan, Italy). All chemicals were of reagent or analytical grade and were used without further purification. TRIzol Reagent was from Life technologies Italia-Invitrogen, (Monza, Italy). The kit Go Taq 2-Step RT-qPCR System was obtained from Promega (Promega Italia Srl, Milan, Italy). For Western blot analysis and immunofluorescence, the following primary antibodies were used: anti-actin 1:1000 (a2066 Sigma-Aldrich; Milan, Italy), anti-Nrf2 (ab31163 Abcam), anti-Lamin A (ab26300 Abcam; Milan, Italy), anti-System Xc− (TA301518 OriGene; Bologna, Italy). For Western blot, secondary peroxidase-labeled anti-rabbit IgG antibodies were from Bio-Rad Italia (Milan, Italy). For immunofluorescence, secondary anti-Rabbit IgG Alexa Fluor 488 was obtained from Invitrogen. Hoechst 33342 (Cod. H3570) was from Thermofisher Scientific.
2.2. Cell Cultures and Treatments
U373-MG human glioblastoma astrocytoma cells and SH-SY5Y human neuroblastoma cells were purchased from ATCC (Manassas, VA, USA). Cells were grown in DMEM supplemented with 2 mM L-glutamine, 10% FBS and 40 μg/mL gentamicin at 37 °C in a humidified 5% CO2 incubator. Confluent monolayers of U373 cells were sub-cultured by conventional trypsinization. For the experiments, 2.5 × 105 or 4 × 105 cells were seeded in 35 or 60 mm tissue culture dishes, respectively, and grown up to 80% confluence for 18–24 h before treatments. Stock solution of Aβ25-35 was prepared at 2.5 mM concentration in bi-distilled water and kept frozen at −20 °C. For aggregation, Aβ25–35 was aged overnight at 25 °C before being added to the culture medium to the final desired concentration (for further details on Aβ25–35 preparation, conformation and aggregation properties see [9,30]).
2.3. SH-SY5Y Cell Differentiation
SH-SY5Y cells were seeded in a confluent monolayer in culture dishes established for the experiments. For neuronal differentiation, cells were cultured for a week in Neurobasal medium (Gibco; Milan, Italy) supplemented with 2 mM L-glutamine, 10 µM Retinoic Acid (Sigma, Milan, Italy) and 1X B-27 supplement (Gibco). The medium was changed every two days.
2.4. MTT Assay
To test neuronal viability, differentiated SH-SY5Y cells were grown alone and in co-cultures with U373 using a Transwell culture system as previously reported [29]. For each sample in co-cultures, 1.5 × 104 neuronal cells were seeded in Transwell insert and 3 × 104 astroglial cells were plated in the lower compartment of a 6-well plate and allowed to grow for 24 h.
MTT assay was performed at the end of the incubation period as indicated by manufacturer’s instructions and as reported elsewhere [29].
2.5. Quantitative Real-Time Reverse Transcription–Polymerase Chain Reaction
Total RNA was purified by using TRIzol Reagent and reverse transcribed with GoTaq 2-step RT-qPCR system. cDNA was then amplified for the following genes: System Xc− (xCT subunit; NM_014331.4), superoxide dismutase (SOD1; NM_000454.5 and SOD2; NM_000636.4), heme-oxygenase-1 (HO-1; NM_002133.3), catalase (CAT; NM_001752), glutathione peroxidase-3 (GPX3; NM_001329790.2), and glutamate-cysteine ligase (GCLC; AB262176.1). mRNA for Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; NM_002046.7) was examined as the reference cellular transcript. The sequences of primers used for RT-qPCR were reported elsewhere [29]. The SYBR-Green method was applied to calculate PCR product quantification. Reactions were performed in an Agilent Aria Mx machine (Agilent technologies) using the following program: 45 cycles of 95 °C for 15 s, 60° C for 60 s, 72 °C for 20 s. GAPDH mRNA amplification products were present at equivalent levels in all cell lysates. Values were calculated relative to the internal housekeeping gene according to the second derivative test (delta–delta Ct (2−ΔΔCT) method).
2.6. Preparation of Nuclear and Total Extracts
After treatments, the cells were mechanically detached with a scraper in cold PBS. Nuclear extracts were prepared as reported elsewhere [29]. The protein content of nuclear extracts was measured according to Bradford method [31]. The quality of fraction separation was verified by blotting nuclear and cytosolic fractions for the specific markers, lamin A and actin, respectively. Total extracts were prepared by mechanically detaching the cells with a scraper in cold PBS. Extracts were then prepared as reported in [29]. The total protein content was determined according to Bradford method [31].
2.7. Evaluation of Nrf2 Activation and System Xc− Expression by Western Blotting
To measure Nrf2 nuclear levels, equal amounts of nuclear extracts (20 μg proteins/sample) were loaded in an 8% polyacrylamide gel, subjected to electrophoresis and transferred to nitrocellulose. After incubation with 5% non-fat dry milk for 1 h, membranes were incubated at 4 °C overnight with the polyclonal anti-Nrf2 antibody (1:1000) or with a polyclonal anti-lamin A (1:1000). To analyze System Xc− expression, equal amounts of total extracts (15 μg proteins/sample) were subjected to SDS-PAGE. Electrophoresis was performed using a 10% polyacrylamide gel. Membranes were blotted with anti-xCT polyclonal antibody (1:5000) or polyclonal anti-actin antibody (1:1000).
Actin and lamin A were used as reference proteins for total and nuclear extracts, respectively. Anti-rabbit secondary antibody labeled with peroxidase was used at 1:10,000 dilution. ECL Western blotting detection reagents was used to detect immunoreactive bands that were captured by Chemi Doc TM XRS 2015 (Bio-Rad Laboratories, Hercules, CA, USA). Densitometric analysis was carried out using Image Lab software (Version 5.2.1; © Bio-Rad Laboratories).
2.8. Analysis of Glutamate Concentration in Cell Supernatants
The release of glutamate in co-culture supernatants was evaluated using Glutamate Assay kit (BioVision; Florence, Italy), as reported elsewhere [29]. The concentration of glutamate in each sample was estimated using glutamate standard curve.
2.9. Immunofluorescence Analysis
1.5 × 105 U373 cells were seeded on 6-well dishes containing poly-L-lysine-treated glass coverslip. After treatments, cells were washed in PBS and fixed with 4% paraformaldehyde for 10 min at room temperature (RT) and permeabilized in methanol for 10 min at −20 °C. Cells were then washed and incubated for 1 h at RT with a blocking solution (5% FBS, 1% BSA in PBS). Next, coverslips were incubated overnight at 4 °C with polyclonal anti-System Xc− (1:100). The secondary antibody anti-Rabbit IgG conjugated with Alexa Fluor 488 was diluted 1:500 and incubated at RT for 1 h. The nuclei were counterstained using Hoechst 33342 (Cod. H3570, Invitrogen; Milan, Italy, Thermofisher Scientific).
2.10. Statistical Analysis
Values are expressed as the mean ± standard error of the mean (SEM) of n observations. Statistical analysis was carried out by one-way ANOVA and subsequently by Bonferroni post-test. Differences are considered statistically significant at p ≤ 0.05.
3. Results and Discussion
Free radical production and oxidative stress play crucial roles in many neurodegenerative diseases, including Alzheimer’s disease [12]. In many cell types, including astrocytes, ROS can activate a protective antioxidant response through Nrf2-mediated induction of antioxidant and phase II detoxifying genes (i.e., ARE genes).
3.1. Aβ25-35 Activates Nrf2 in Astroglial Cells
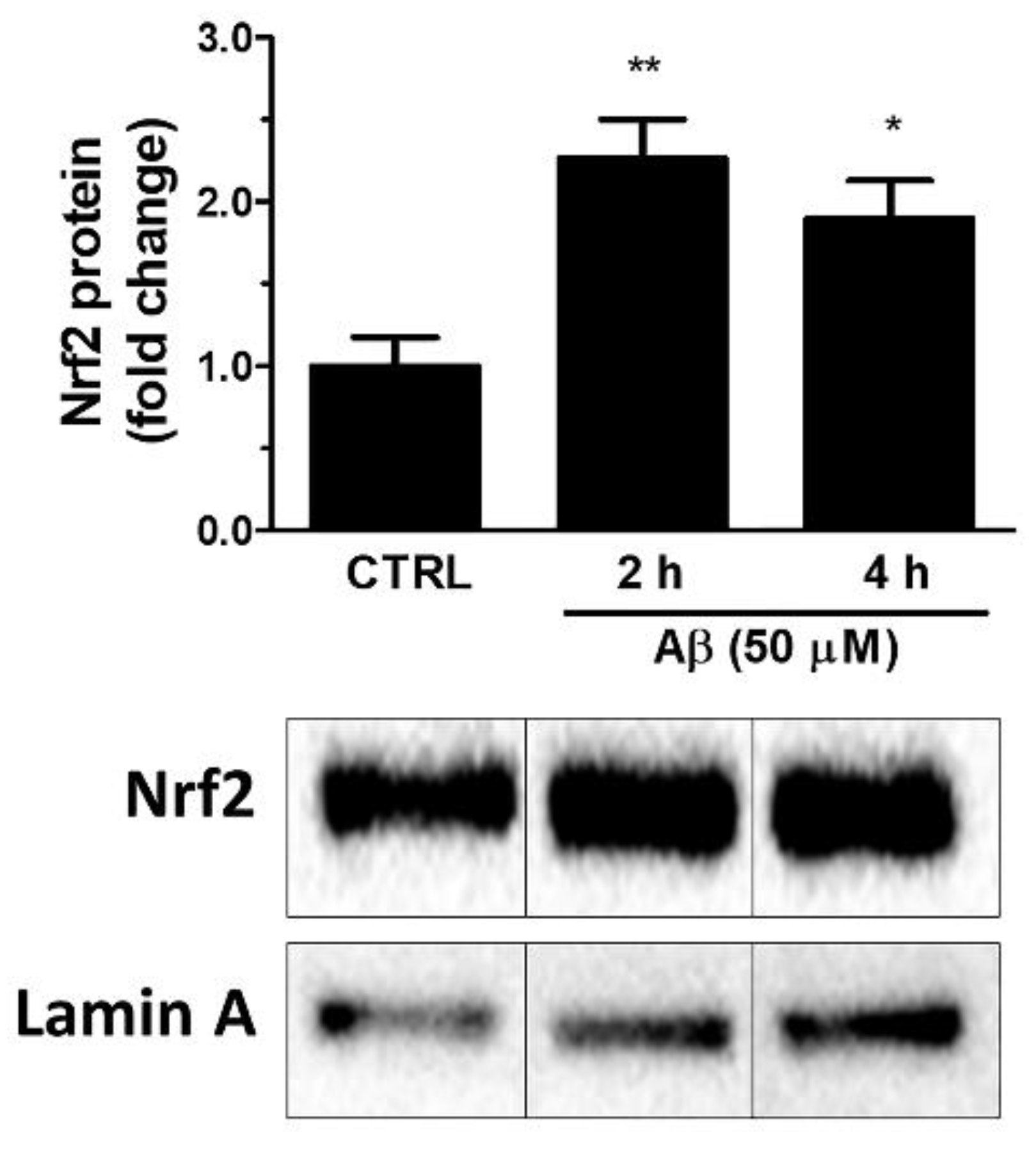
Firstly, we investigated whether Aβ25-35 could activate Nrf2 in astroglial cells. To this aim, U373 cells were treated with Aβ25-35 (50 μM) for 2, 4 and 24 h and Nrf2 levels were measured in nuclear extracts by Western blot analysis. The results shown in Figure 1 indicate that Aβ25-35 induced a 2.3-fold increase of the nuclear Nrf2 levels already at 2 h post-treatment and a 1.9-fold increase after 4 h of treatment.
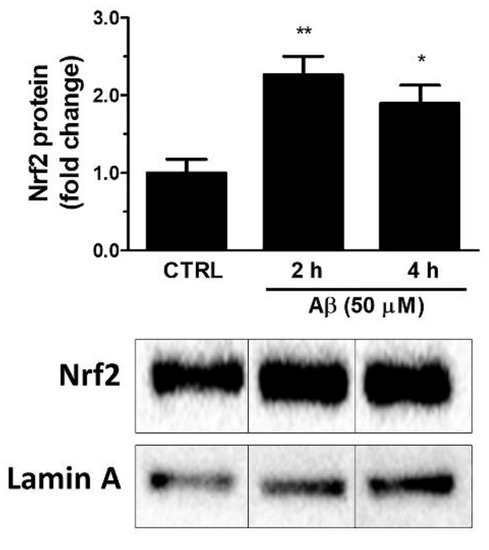
Figure 1.
Effects of Aβ25-35 on nuclear translocation of Nrf2 in U373 cells. Cells were treated with Aβ25-35 (50 µM) for 2 and 4 h. The histograms show the densitometric analysis of the Western blots for each sample. Data are calculated relative to the housekeeping gene (i.e., nuclear lamin A) content and are the means ± SEM from three separate experiments, each performed in duplicate. One-way ANOVA, followed by Bonferroni’s test, was used to define significant differences. * p ≤ 0.05 vs. CTRL; ** p ≤ 0.01 vs. CTRL.
3.2. Aβ25-35 Induces ARE Gene Expression in Astroglial Cells
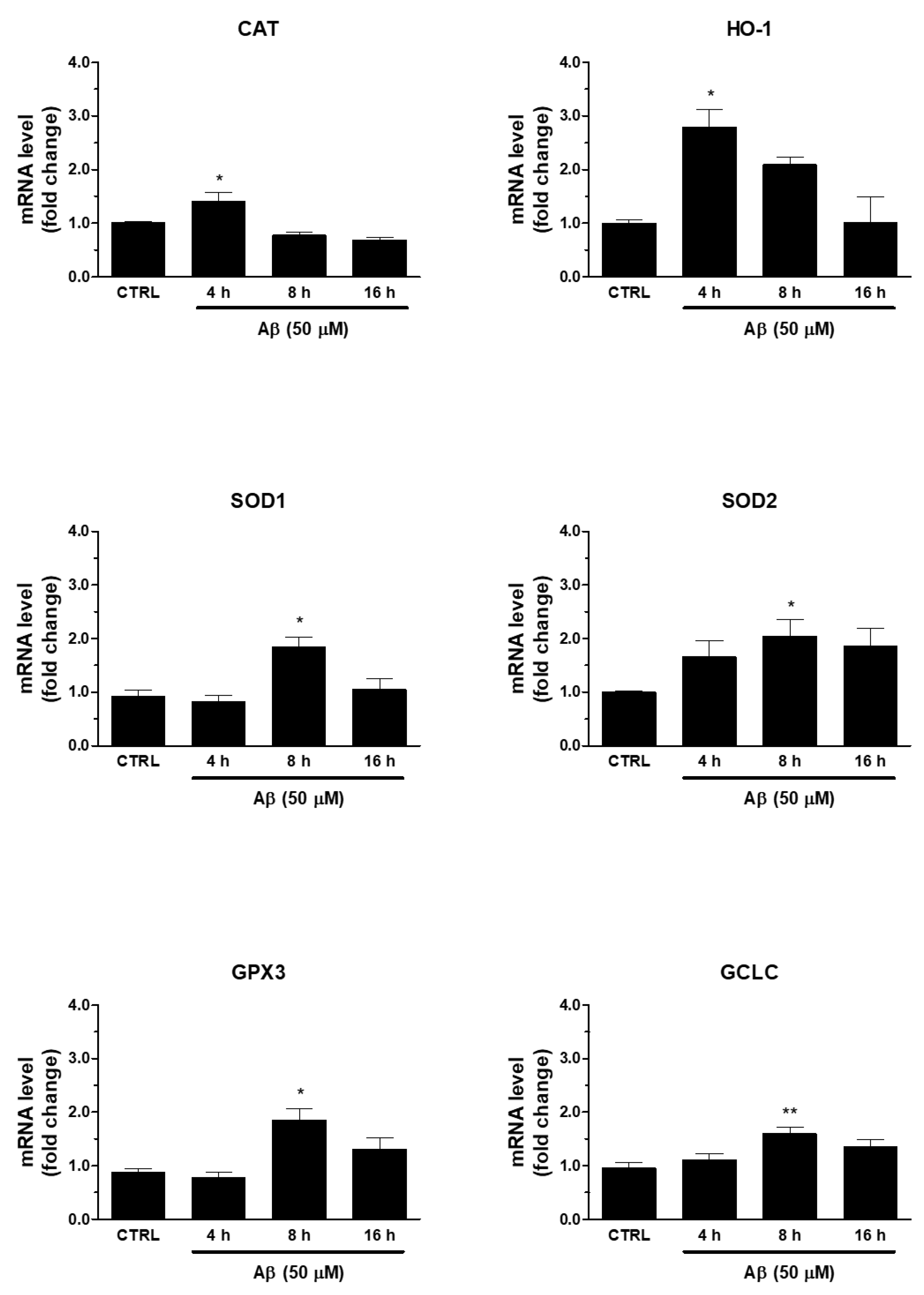
Secondly, we verified whether Aβ-induced Nrf2 was able to regulate antioxidant ARE genes. In this respect, we found that the treatment of human U373 astroglial cells with Aβ25-35 (50 μM) for 4, 8, and 16 h was able to increase the mRNA expression of enzymes involved in maintenance of redox state, such as SOD1, SOD2, CAT, HO-1, GPX3, and GCLC (Figure 2). The peak was reached at 8 h for SOD1, SOD2, GPX3, and GCLC, whereas CAT and HO-1 expression peaked at 4 h after Aβ25-35 treatment. This time frame was compatible with the earlier transcriptional activation of Nrf2.
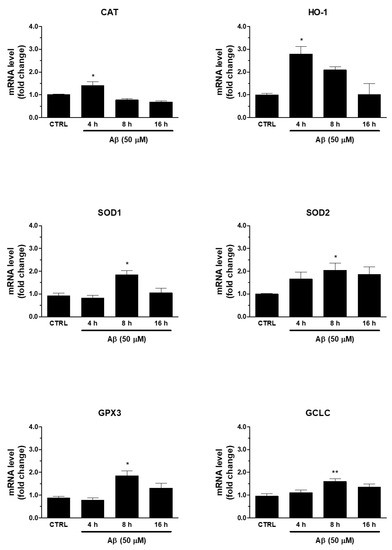
Figure 2.
Effects of Aβ25-35 on ARE-dependent gene expression in U373 cells. Cells were treated with Aβ25-35 (50 µM) for 4, 8, and 16 h. The samples were then analyzed by RT-qPCR to evaluate the mRNA expression of CAT, HO-1, SOD1, SOD2, GCLC and GPX3 genes. Data are calculated relative to GAPDH content, taken as an internal housekeeping gene. Bars are the means ± SEM from three separate experiments, each carried out in duplicate. One-way ANOVA, followed by Bonferroni’s test, was used to define significant differences. * p ≤ 0.05 vs. CTRL; ** p ≤ 0.01 vs. CTRL.
3.3. Aβ25-35 Induces System Xc− in Astroglial Cells
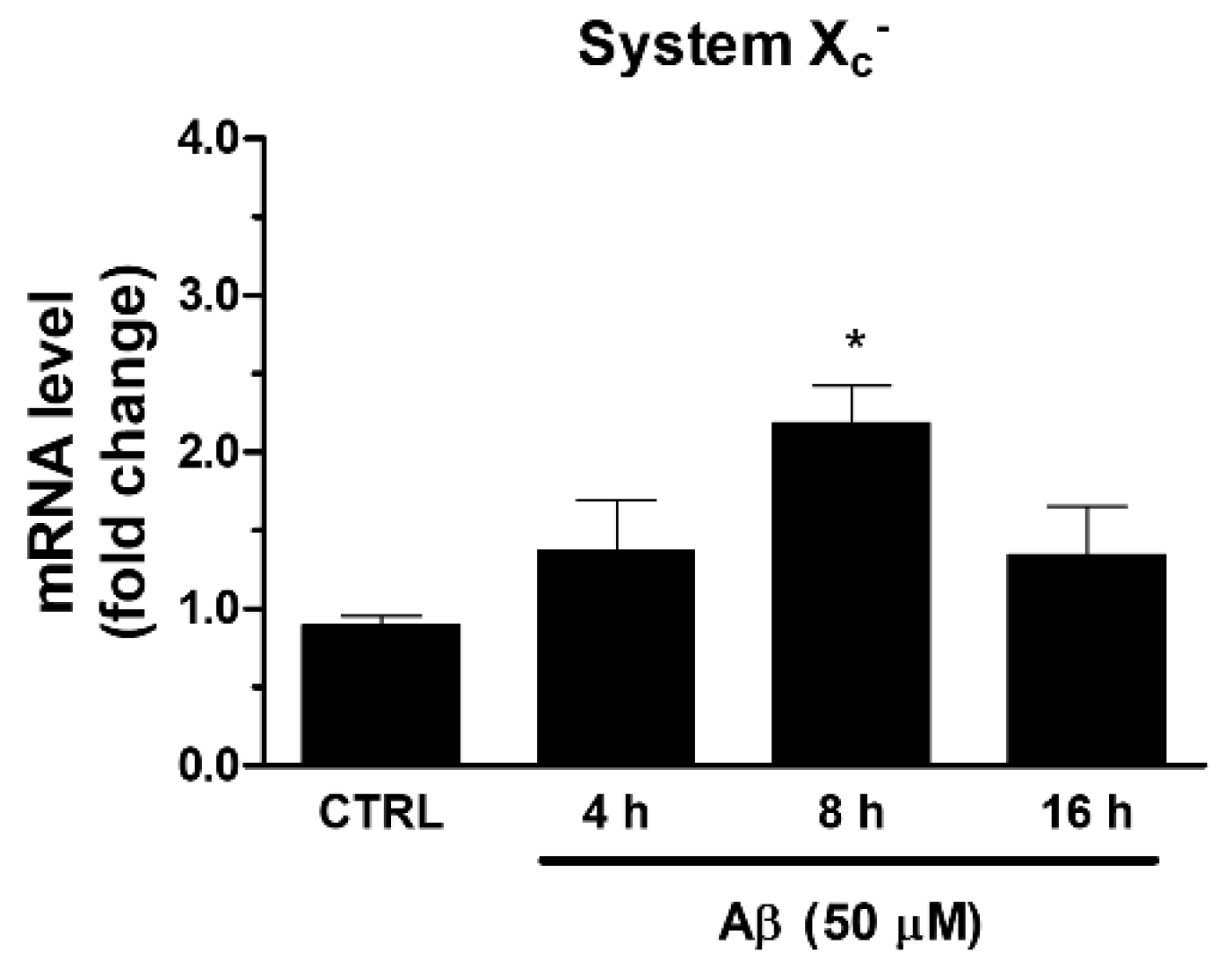
Among ARE genes, we further focused our attention on System Xc−, which is involved in the maintenance of GSH intracellular levels and the redox state. In the same experimental conditions described above, we found that Aβ25-35 was able to increase the mRNA expression of xCT, the catalytic subunit of System Xc−. As shown in Figure 3, we observed a maximum reached at 8 h post-treatment.
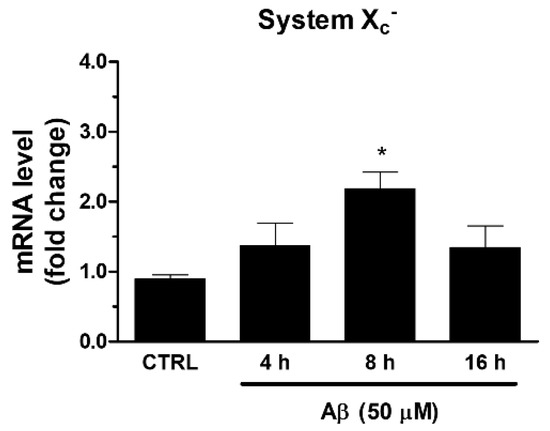
Figure 3.
Effects of Aβ25-35 on System Xc− gene expression in U373 cells. Cells were treated with Aβ25-35 (50 µM) for 4, 8, and 16 h. After incubation at 37 °C, cells were homogenized, and total RNA has been purified to evaluate mRNA content of System Xc− by RT-qPCR. Results are computed relative to GAPDH content, taken as a housekeeping gene. Bars are the means ± SEM from three separate experiments, each performed in duplicate. One-way ANOVA, followed by Bonferroni’s test, was used to define significant differences. * p ≤ 0.05 vs. CTRL.
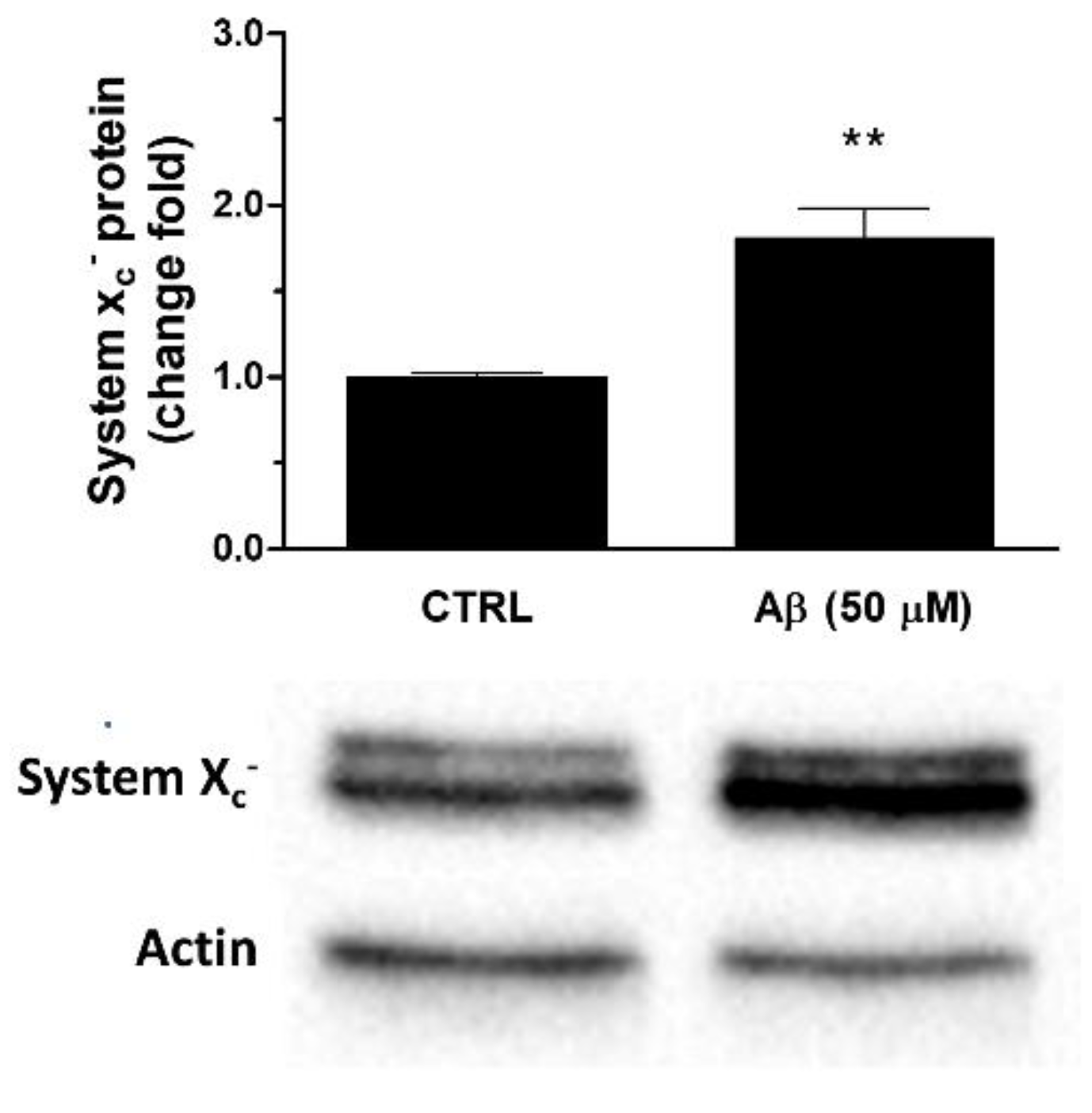
Consistently, we observed that Aβ25-35 was able to up-regulate System Xc− also at protein level. In particular, a 24 h treatment of U373 cells with Aβ25-35 (50 μM) caused a two-fold increase of System Xc− protein levels in whole cell extracts when compared to controls, as verified by Western blot analyses (see Figure 4).
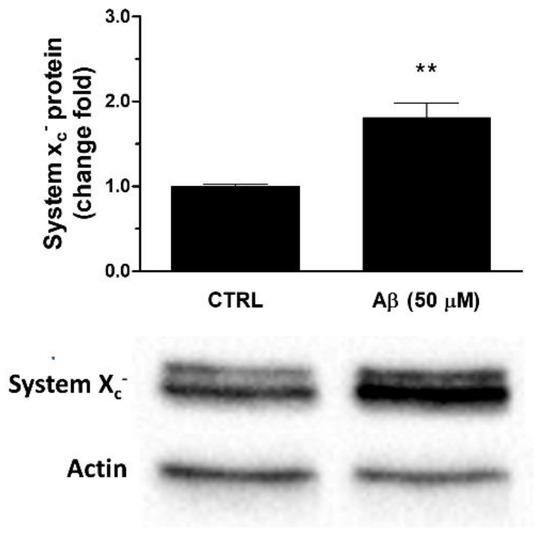
Figure 4.
Effects of Aβ25-35 on System Xc− protein expression in U373 cells. Cells were treated with Aβ25-35 (50 µM) for 24 h. The graph shows the densitometric analysis of the western blots for each sample. Data are computed relative to the internal housekeeping gene (actin) and are the means ± SEM from three separate experiments, each carried out in duplicate. One-way ANOVA, followed by Bonferroni’s test, was used to define significant differences. ** p ≤ 0.01 vs. CTRL.
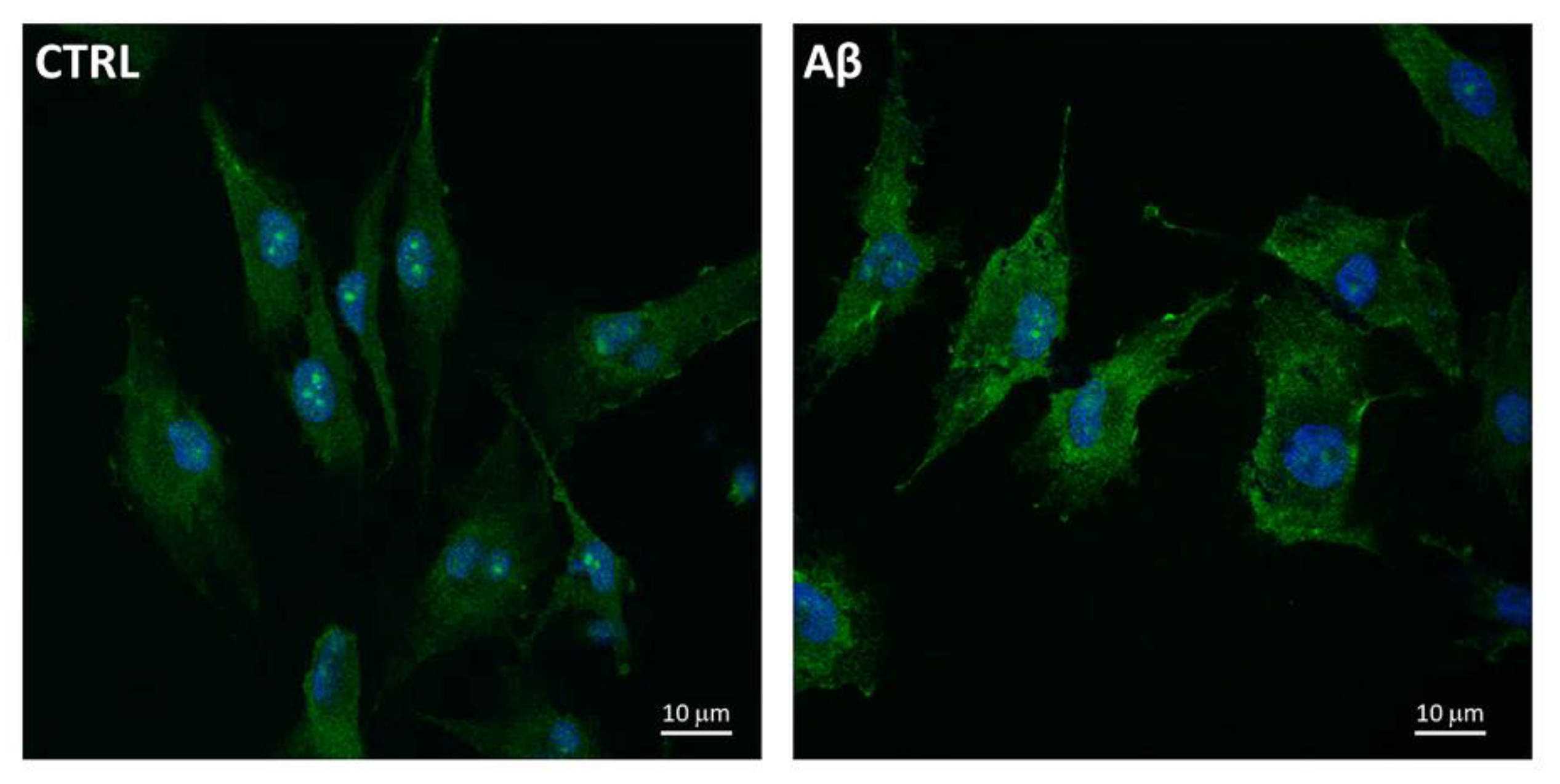
Furthermore, we used the confocal microscopy to also evaluate the expression and localization of System Xc− in U373 cells treated with Aβ25-35 (50 μM) for 24 h. Figure 5 shows an increased levels of System Xc−, the latter being mainly localized on plasma membrane of treated cells compared to controls at 24 h post-treatment.
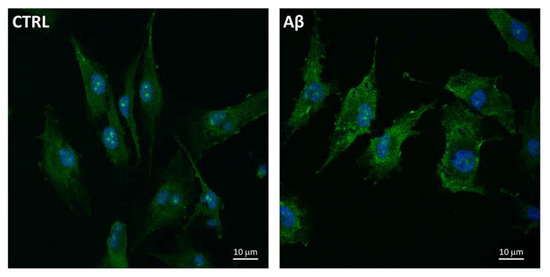
Figure 5.
System Xc− localization in Aβ25-35-treated U373. Cells were treated with Aβ25-35 (50 µM) for 24 h and subjected to immunofluorescence staining using anti-System Xc− 1:100 (OriGene, green) antibodies as reported in Materials and Methods. Nuclei (blue) are stained with Hoechst 33342.
3.4. Aβ25-35 Induces Glutamate Release through System Xc−
The above results would seem to be in agreement with the concept that astrocytes play an important role in providing antioxidant support to neighboring neurons. In fact, post-mitotic neurons are thought to survive for many decades despite their relatively low intrinsic antioxidant defenses [22,32,33,34]. Given the role of System Xc− in providing the cell with cystine, its augmented expression and/or activity increases intracellular levels of cysteine. That is the rate-limiting substrate for the synthesis of GSH, thereby endowing astrocytes with an effective antioxidant response.
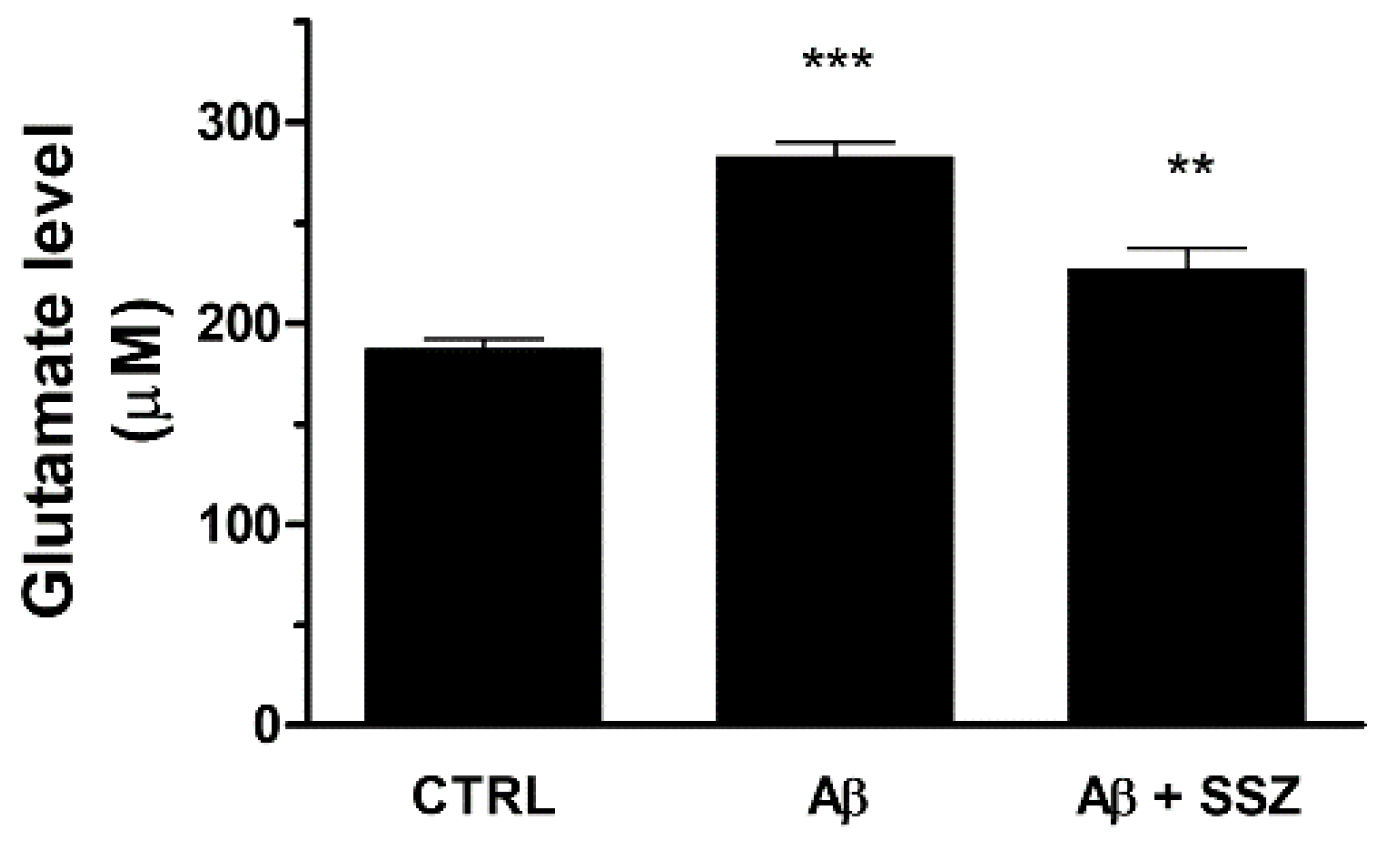
Nevertheless, the up-regulation of System Xc− can increase extracellular glutamate release and potentially cause excitotoxicity. To verify whether the treatment of astrocytes with Aβ25-35 can enhance the release of glutamate in the extracellular space, we quantified the levels of glutamate in the supernatant of U373 co-cultured with differentiated SH-SY5Y cells in the presence of Aβ25-35 (50 μM) for 24 h. As shown in Figure 6, Aβ25-35-treated cells released about 50% more glutamate with respect to untreated cells. As a control, we found that glutamate was also released from mono-cultured U373 cells treated with Aβ25-35 for 24 h, thus proving its astroglial origin (data not shown).
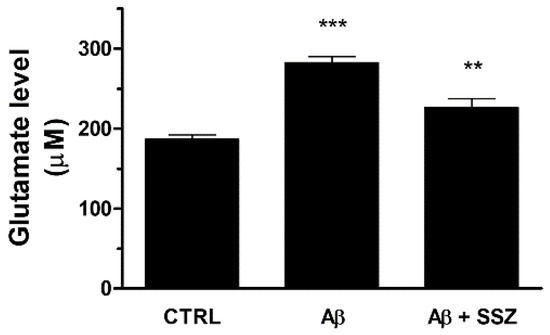
Figure 6.
Effects of Aβ25-35 and System Xc− activity on release of extracellular glutamate in differentiated SH-SY5Y/U373 co-cultures. Cells were treated for 24 h and the glutamate assay was performed as specified in the Materials and Methods. The graph shows the extracellular release of glutamate (µM). Data are calculated relative to a glutamate standard curve and are the means ± SEM from three separate experiments, each carried out in duplicate. One-way ANOVA, followed by Bonferroni’s test, was used to define significant differences. *** p ≤ 0.001 between CTRL and Aβ25-35; ** p < 0.01 between Aβ25-35 and Aβ25-35 + SSZ.
To confirm that Aβ25-35-elicited glutamate release occurred through System Xc− activation, we analyzed the levels of glutamate in the supernatants of co-cultures in the presence of sulfasalazine (SSZ; 300 μM), a specific inhibitor of System Xc−. We observed that SSZ treatment prevented Aβ25-35-induced glutamate release, reducing its levels in the extracellular space. These data clearly demonstrate that in astroglial cells the treatment with Aβ25-35 increases the release of glutamate by eliciting System Xc− up-regulation (see Figure 6).
3.5. Aβ25-35 Affects Viability of Co-Cultured SH-SY5Y Cells via System Xc− and NMDA Receptor
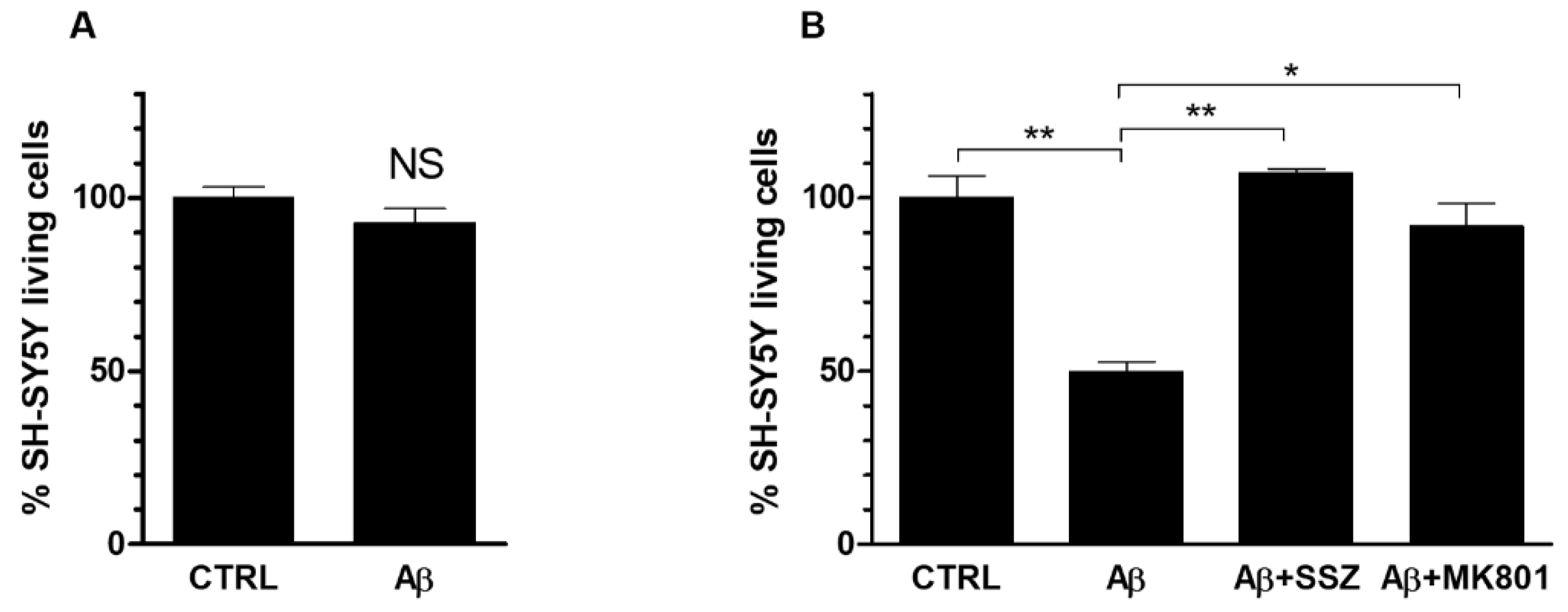
To verify whether the System Xc−-mediated increase of glutamate release by astroglial cells caused neurotoxicity, we investigated the viability of neurons cocultured with astrocytes in the presence of Aβ25-35. First of all, we verified that a 24 h treatment with Aβ25-35 (50 μM) of differentiated SH-SY5Y cells, cultured alone, did not affected cell viability (Figure 7a). Afterwards, we evaluated the viability of neuronal cells grown in co-cultures with astrocytes, in the presence of Aβ25-35. As shown in Figure 7b, a treatment with Aβ25-35 for 24 h caused a significant reduction of 50% less viability of differentiated neuronal SH-SY5Y cells co-cultured with U373 cells in comparison to untreated cells. To verify whether Aβ25-35-induced neurotoxicity was due to an enhancement of glutamate export through System Xc−, we carried out the MTT assay in co-cultures treated with Aβ25-35 for 24 h in the presence of SSZ (300 μM). Our results indicate that SSZ prevented neurotoxicity in SH-SY5Y cells co-cultured with U373 cells, thus restoring neuronal viability at the control level (see Figure 7b). These data suggest that Aβ25-35-induced neurotoxic effect is mediated by increased glutamate release due to System Xc− up-regulation in astroglial cells.
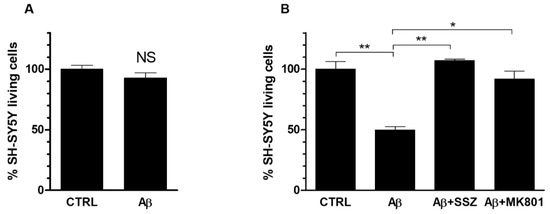
Figure 7.
Role of System Xc− and NMDA receptor activation on the viability of Aβ-treated SH-SY5Y differentiated cells. (A) SH-SY5Y cells were grown in mono-cultures and treated for 24 h with Aβ25-35 (50 μM). (B) SH-SY5Y cells were co-cultured with U373 cells and treated for 24 h with Aβ25-35 (50 μM) alone or in the presence of either SSZ (300 μM) or MK801 (10 μM). MTT cell viability assay was carried out as specified in Materials and Methods. The histograms show the percentage of living cells, and the rate of reduction was calculated by setting the control (CTRL) equal to 100%. Values are the means ± SEM from three separate experiments, each performed in duplicate. One-way ANOVA, followed by Bonferroni’s test, was used to determine significant differences. NS not significant; ** p ≤ 0.01 between CTRL and Aβ25-35; ** p ≤ 0.01 between Aβ25-35 and Aβ25-35 + SSZ; * p ≤ 0.05 between Aβ25-35 and Aβ25-35 + MK801.
The release of glutamate via System Xc− from both microglia and astrocytes has been found to increase excitotoxicity of cortical neurons [35,36,37,38,39,40]. Noteworthy, changes in glutamate transport have been demonstrated in a mouse model for Alzheimer’s disease. Enhanced cortical expression of VGLUT3 and xCT along with a strong trend towards increased cortical extracellular glutamate levels have been reported elsewhere [41]. In neurons, excitotoxicity occurs via glutamate-induced overactivation of NMDA receptor and subsequent perturbed cellular calcium homeostasis and mitochondrial alterations [42,43].
To assess whether the reduced neuronal viability, as triggered by extracellular glutamate release through System Xc−, was effectively due to the activation of NMDA receptor, we performed MTT assay on SH-SY5Y (grown in co-cultures with U373 cells) treated for 24 h with Aβ25-35 in the presence of MK801 (10 μM), an NMDA receptor antagonist. As shown in Figure 7b, MK801 prevented neuronal toxicity restoring the percentage of neuronal living cells at the control level. These results clearly indicate that Aβ25-35-induced neurotoxicity is mediated by the activation of NMDA receptor elicited by System Xc−-dependent glutamate release.
Note that during neuroinflammation, activated astrocytes and microglia have been reported to release and maintain high concentrations of extracellular glutamate [40]. Moreover, excitotoxic glutamate release and high levels of System Xc− expression were observed in microglia, due to a prolonged need for oxidative protection [44]. Interestingly, neurons co-cultured with astrocytes were observed to be more susceptible than neurons alone to hypoxic cell death after treatment with IL-1β, an effect being mediated by enhanced glutamate efflux from astrocytes through System Xc− [36]. Very recently, we have reported that HIV-1 Tat protein was able to induce neurotoxicity by eliciting Nrf2-mediated System Xc− activation [29]. Previously, we reported that HIV-1 Tat can induce neuro-toxicity by eliciting the spermine oxidase-dependent ROS generation through NMDA receptor stimulation in SH-SY5Y cells, which in turn leads to GSH depletion and oxidative stress [45]. Finally, we have recently demonstrated that System Xc− participated in the increase of glutamate excitotoxicity in the neocortex of a mouse model (Dach-SMOX), displaying a chronic oxidative stress [46].
Although Aβ25-35 may have some limits in representing the whole Aβ peptide and is a scarce version in vivo, it is particularly worthy of attention in light of its great oxidative stress generation capacity and extreme toxicity in neuronal cells and synaptosomes [9]. Altogether, our data show how inflammatory pathways and oxidative stress may converge to an intersection point, represented by activation of System Xc−, thereby suggesting a possible explanation for the mechanism involved in excitotoxicity induced by Aβ25-35. It should be pointed out, however, that Aβ25-35 is just one of the fragments and this does not exclude the possibility that other parts of Aβ, than the 25-35 fragment, can be involved in the induction of Nrf2/SystemXc− pathway.
4. Conclusions
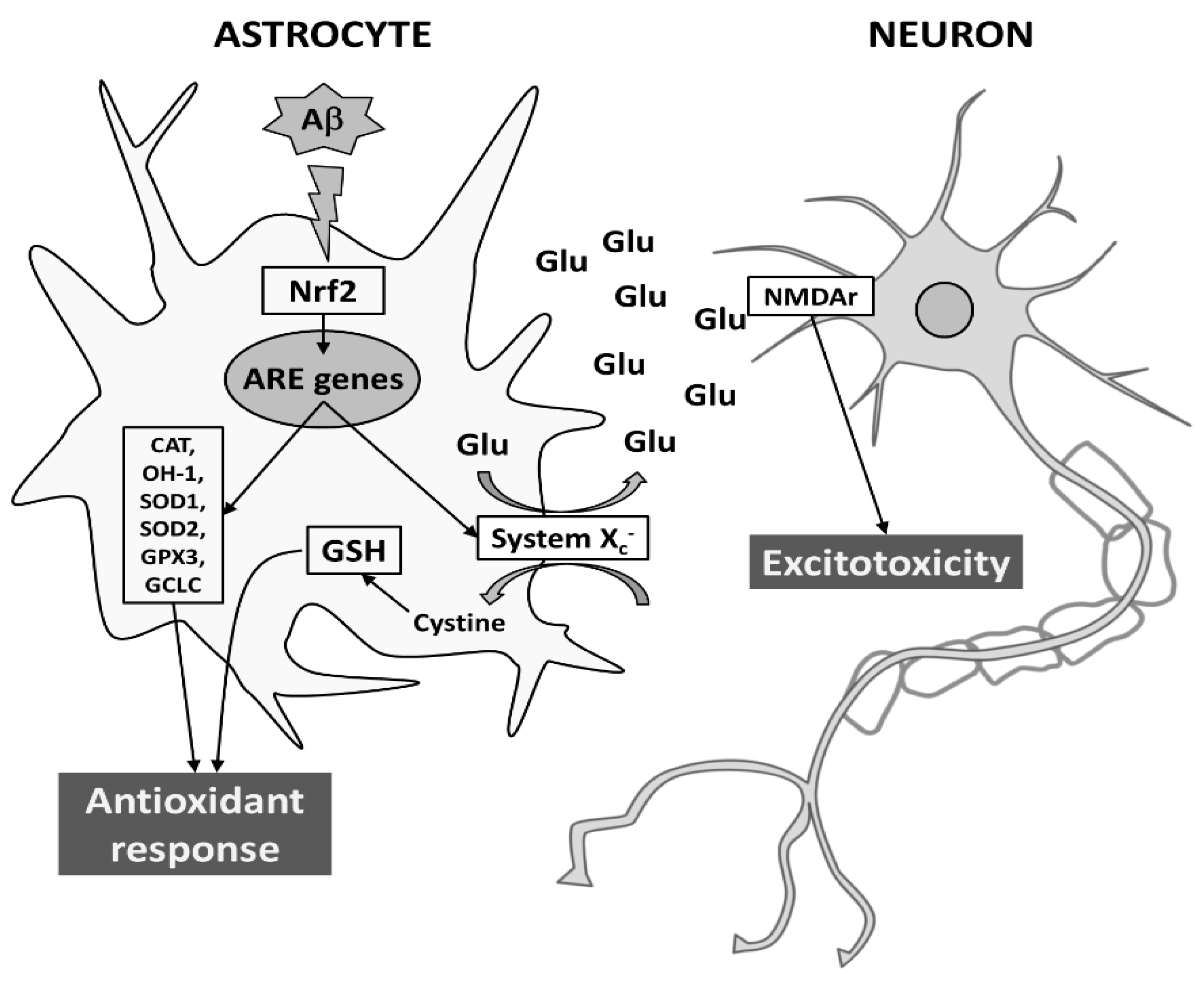
Given the role played by astrocytes in maintaining the homeostasis of extracellular space in the brain, their response may affect neuronal function and provide antioxidant support to neighboring neurons both in normal and pathological conditions. Here, we show that Aβ25-35 is able to trigger an antioxidant response in astrocytes, by inducing both Nrf2 and ARE-driven genes, including System Xc−. However, the induction of System Xc− in astrocytes seems to be also responsible for mortality of neuronal cells, due to sustained glutamate release and neuronal NMDA receptor activation. Although further studies are needed, it is tempting to speculate that neurodegeneration can be exacerbated by converting oxidative stress to excitotoxicity via System Xc− (for a schematic model see Figure 8).
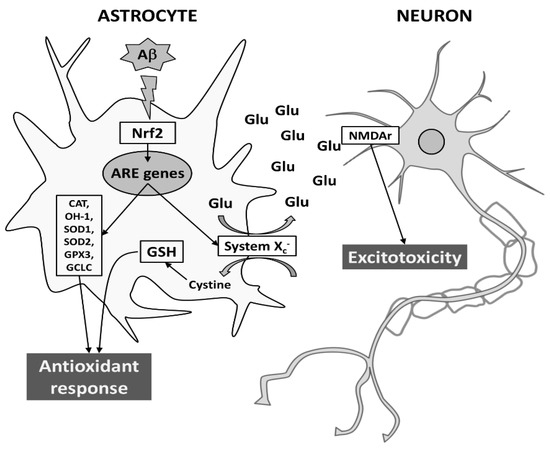
Figure 8.
Proposed model for Aβ-dependent neurodegeneration during AD. In astrocytes, Aβ25-35 elicits an antioxidant response by transcriptionally inducing Nrf2-driven ARE genes, such as CAT, HO-1, SOD1, SOD2, GPX3, GCLC, and System Xc−. While the L-cystine import through System Xc− is crucial to protection from oxidative stress (e.g., GSH production), the export of glutamate may cause neurodegeneration through the activation of NMDAr on neuronal cells. For more details see text.Abbreviations: Aβ, amyloid-β; ARE, antioxidant responsive element; CAT, catalase; GCLC, glutamate-cysteine ligase; GPX3, glutathione peroxidase; GSH, reduced glutathione; HO-1, heme-oxygenase-1; NMDAr, N-methyl-D-aspartate receptor; Nrf2, nuclear factor erythroid 2-related factor 2; SOD, superoxide dismutase.
In conclusion, the present study highlights the importance of the Nrf2/System Xc− pathway for a better understanding of the role of astrocytes as a cell population responsible for the death of neurons in AD.
Author Contributions
Conceptualization, T.P.; methodology, V.D. and T.P.; software, M.C.; validation, V.D. and T.P.; formal analysis, V.D., M.C. and T.P.; investigation, V.D.; data curation, V.D. and T.P.; writing—original draft preparation, V.D. and T.P.; writing—review and editing, M.C. and T.P.; supervision and project administration, T.P.; funding acquisition, M.C. and T.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR (ARTICOLO 1, COMMI 314–337 LEGGE 232/2016), grant of Excellence Department of Sciences (ROMA TRE University).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
We thank Roberta Mastrantonio for excellent technical assistance in the use of confocal microscopy as well as Andrea Sperati and Marta Russo for their invaluable support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.; Lee, B.Y.; Hane, F.T. Recent progress in Alzheimer’s disease research, part 2: Genetics and epidemiology. J. Alzheimers Dis. 2017, 57, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S. Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Trambauer, J.; Fukumori, A.; Steiner, H. Pathogenic Abeta generation in familial Alzheimer’s disease: Novel mechanistic Insights and therapeutic implications. Curr. Opin. Neurobiol. 2020, 61, 73–81. [Google Scholar] [CrossRef]
- Chen, X.; Ji, B.; Hao, X.; Li, X.; Eisele, F.; Nystrom, T.; Petranovic, D. FMN Reduces amyloid-beta toxicity in yeast by regulating redox status and cellular metabolism. Nat. Commun. 2020, 11, 867. [Google Scholar] [CrossRef]
- Li, Y.; Lu, J.; Cao, X.; Zhao, H.; Gao, L.; Xia, P.; Pei, G. A Newly Synthesized rhamnoside derivative alleviates Alzheimer’s Amyloid-beta-induced oxidative stress, mitochondrial dysfunction, and cell senescence through upregulating SIRT3. Oxid. Med. Cell Longev. 2020, 2020, 7698560. [Google Scholar] [CrossRef] [Green Version]
- Wiltfang, J.; Esselmann, H.; Bibl, M.; Smirnov, A.; Otto, M.; Paul, S.; Schmidt, B.; Klafki, H.-W.; Maler, M.; Dyrks, T.; et al. Highly Conserved and disease-specific patterns of carboxyterminally truncated Aβ peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation: Carboxyterminally truncated Aβ peptides. J. Neurochem. 2002, 81, 481–496. [Google Scholar] [CrossRef]
- Portelius, E.; Zetterberg, H.; Andreasson, U.; Brinkmalm, G.; Andreasen, N.; Wallin, A.; Westman-Brinkmalm, A.; Blennow, K. An Alzheimer’s disease-specific β-amyloid fragment signature in cerebrospinal fluid. Neurosci. Lett. 2006, 409, 215–219. [Google Scholar] [CrossRef]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25-35. CPPS 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular mechanisms and genetics of oxidative stress in Alzheimer’s disease. J. Alzheimers Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-beta peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, R.C.; Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M.; et al. Amyloid β peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive ENOS/HSP90 interaction and disruption of agonist-mediated akt activation. J. Neuroinflamm. 2015, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Drews, A.; Flint, J.; Shivji, N.; Jonsson, P.; Wirthensohn, D.; De, G.E.; Vincke, C.; Muyldermans, S.; Dobson, C.; Klenerman, D. Individual aggregates of amyloid beta induce temporary calcium influx through the cell membrane of neuronal cells. Sci. Rep. 2016, 6, 31910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox. Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; He, J.; Zhang, H.; Yao, L.; Li, H. Oleanolic acid alleviates oxidative stress in Alzheimer’s disease by regulating Stanniocalcin-1 and uncoupling Protein-2 signalling. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Liddell, J.R. Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system Xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox. Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking outside the cleft to understand synaptic activity: Contribution of the cystine-glutamate antiporter (system Xc−) to normal and pathological glutamatergic signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrantonio, R.; D’Ezio, V.; Colasanti, M.; Persichini, T. Nrf2-mediated system Xc− activation in astroglial cells is involved in HIV-1 tat-induced neurotoxicity. Mol. Neurobiol. 2019, 56, 3796–3806. [Google Scholar] [CrossRef]
- Millucci, L.; Raggiaschi, R.; Franceschini, D.; Terstappen, G.; Santucci, A. Rapid aggregation and assembly in aqueous solution of Aβ (25-35) peptide. J. Biosci. 2009, 34, 293–303. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Chandran, S.; Hardingham, G.E. Human stem cell-derived astrocytes and their application to studying N Rf2-mediated neuroprotective pathways and therapeutics in neurodegeneration. Br. J. Clin. Pharm. 2013, 75, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, J.A. The Nrf2–ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Jin, B.; Ondrejcak, T.; Rowan, M.J. Opposite in vivo effects of agents that stimulate or inhibit the glutamate/cysteine exchanger system Xc- on the inhibition of hippocampal LTP by ass. Hippocampus 2016, 26, 1655–1665. [Google Scholar] [CrossRef]
- Fogal, B.; Li, J.; Lobner, D.; McCullough, L.D.; Hewett, S.J. System Xc activity and astrocytes are necessary for interleukin-1 -mediated hypoxic neuronal injury. J. Neurosci. 2007, 27, 10094–10105. [Google Scholar] [CrossRef] [Green Version]
- Jackman, N.A.; Uliasz, T.F.; Hewett, J.A.; Hewett, S.J. Regulation of system Xc−activity and expression in astrocytes by interleukin-1β: Implications for hypoxic neuronal injury. Glia 2010, 58, 1806–1815. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Albano, R.; Lobner, D. FGF-2 induces neuronal death through upregulation of system Xc−. Brain Res. 2014, 1547, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S. System Xc− and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J. Neurosci. 2006, 26, 3345–3356. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [Green Version]
- Schallier, A.; Smolders, I.; Van, D.D.; Loyens, E.; De Deyn, P.P.; Michotte, A.; Michotte, Y.; Massie, A. Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer’s disease. J. Alzheimers Dis. 2011, 24, 287–300. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [PubMed]
- Gupta, K.; Hardingham, G.E.; Chandran, S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013, 543, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piani, D.; Fontana, A. Involvement of the cystine transport system Xc− in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J. Immunol. 1994, 152, 3578–3585. [Google Scholar] [PubMed]
- Capone, C.; Cervelli, M.; Angelucci, E.; Colasanti, M.; Macone, A.; Mariottini, P.; Persichini, T. A Role for Spermine Oxidase as a Mediator of Reactive Oxygen Species Production in HIV-Tat-Induced Neuronal Toxicity. Free Radic. Biol. Med. 2013, 63, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Pietropaoli, S.; Leonetti, A.; Cervetto, C.; Venturini, A.; Mastrantonio, R.; Baroli, G.; Persichini, T.; Colasanti, M.; Maura, G.; Marcoli, M.; et al. Glutamate Excitotoxicity Linked to Spermine Oxidase Overexpression. Mol. Neurobiol 2018, 55, 7259–7270. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).