
Thunbergia laurifolia Leaf Extract Inhibits Glutamate-Induced Neurotoxicity and Cell Death through Mitophagy Signaling

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Material and Extraction
2.3. Qualitative Bioactive Compounds of TLE by LC–MS Analysis
2.4. Cell Culture
2.5. Cell Viability Assay
2.6. Cytotoxicity Assay
2.7. Intracellular ROS Assay
2.8. Mitochondrial Membrane Potential Staining (TMRE) Assay
2.9. Western Blot Analysis
2.10. Immunofluorescent Colocalization Analysis
2.11. Real-Time PCR Analysis
2.12. Molecular Docking
2.13. Lipinski’s Rule of Five Parameters and ADMET Property Analysis
2.14. Statistical Analysis
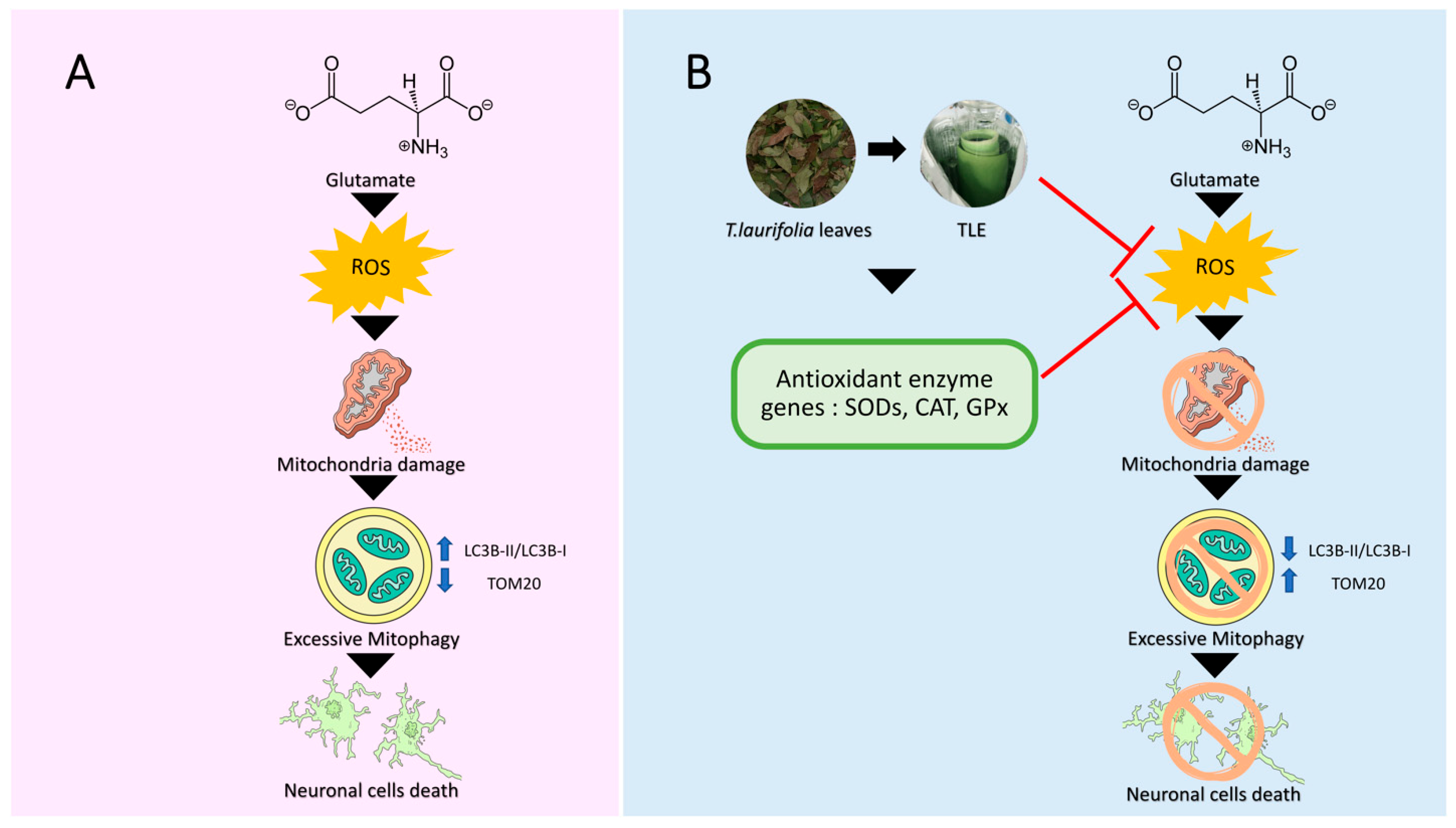
3. Results
3.1. Characterization of Bioactive Compounds from TLE
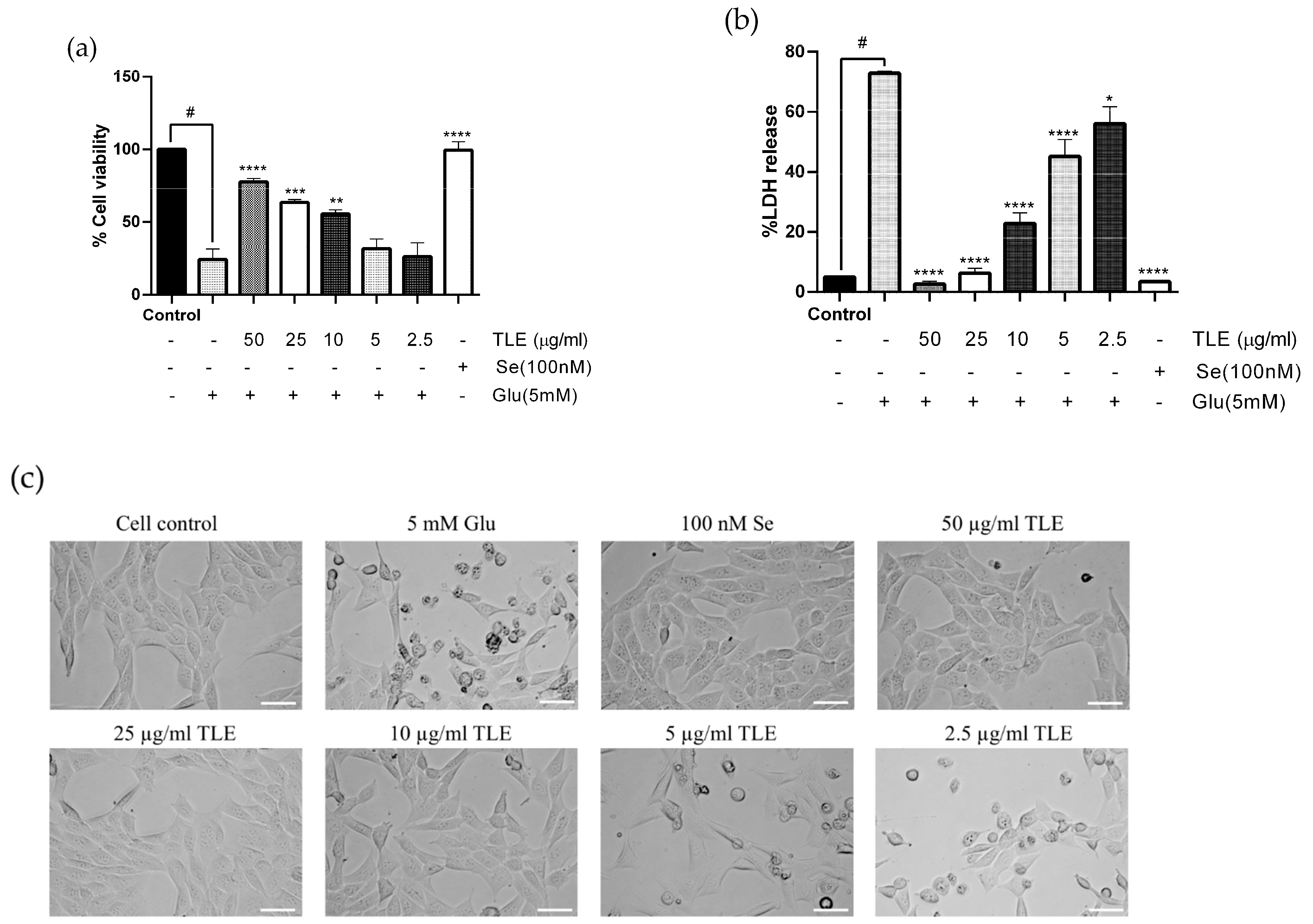
3.2. TLE Attenuates Glutamate-Induced Toxicity in HT-22 Cells
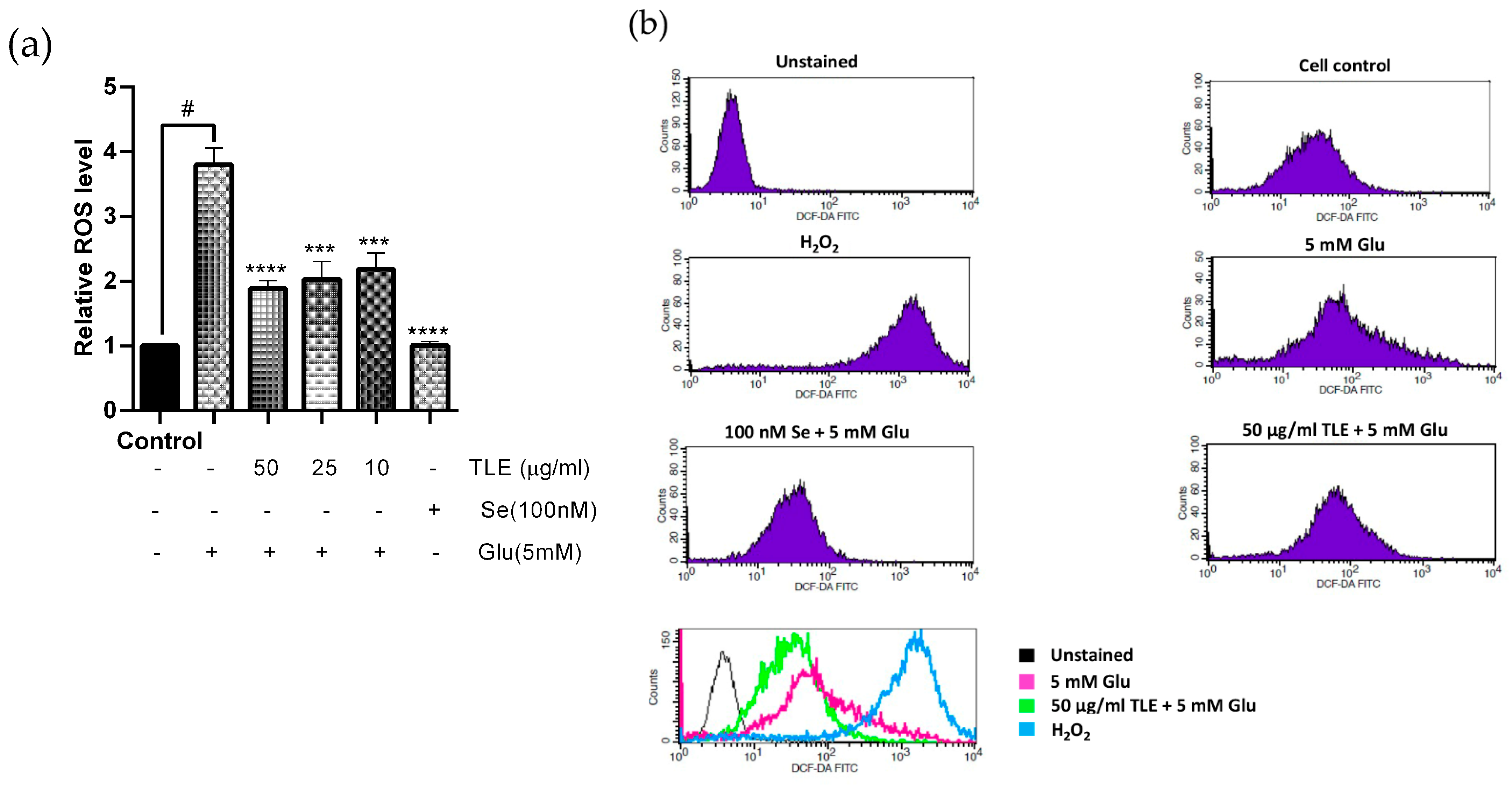
3.3. TLE Inhibits Glutamate-Induced Intracellular ROS Generation
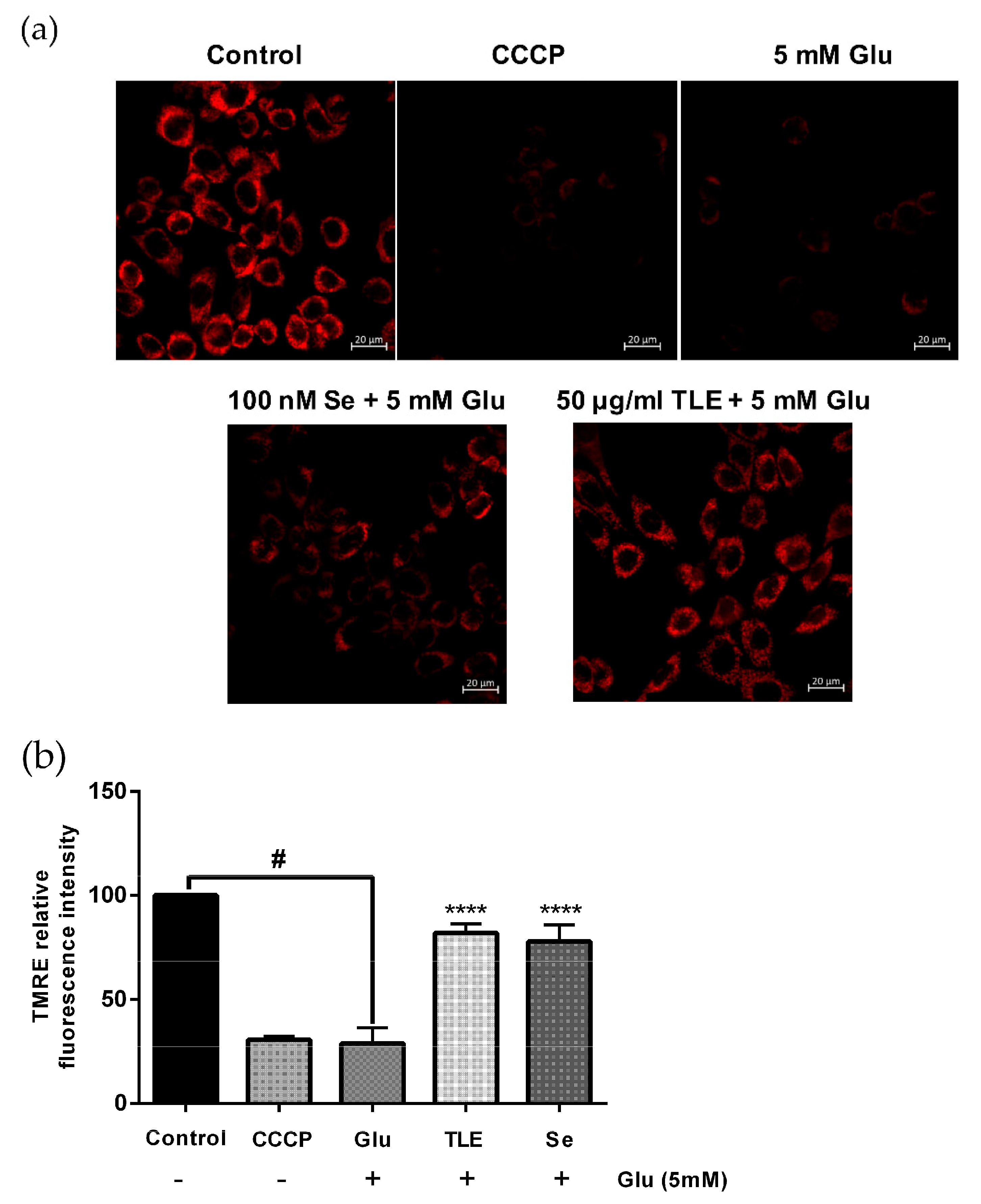
3.4. TLE Sustains the Membrane Potential of Mitochondria
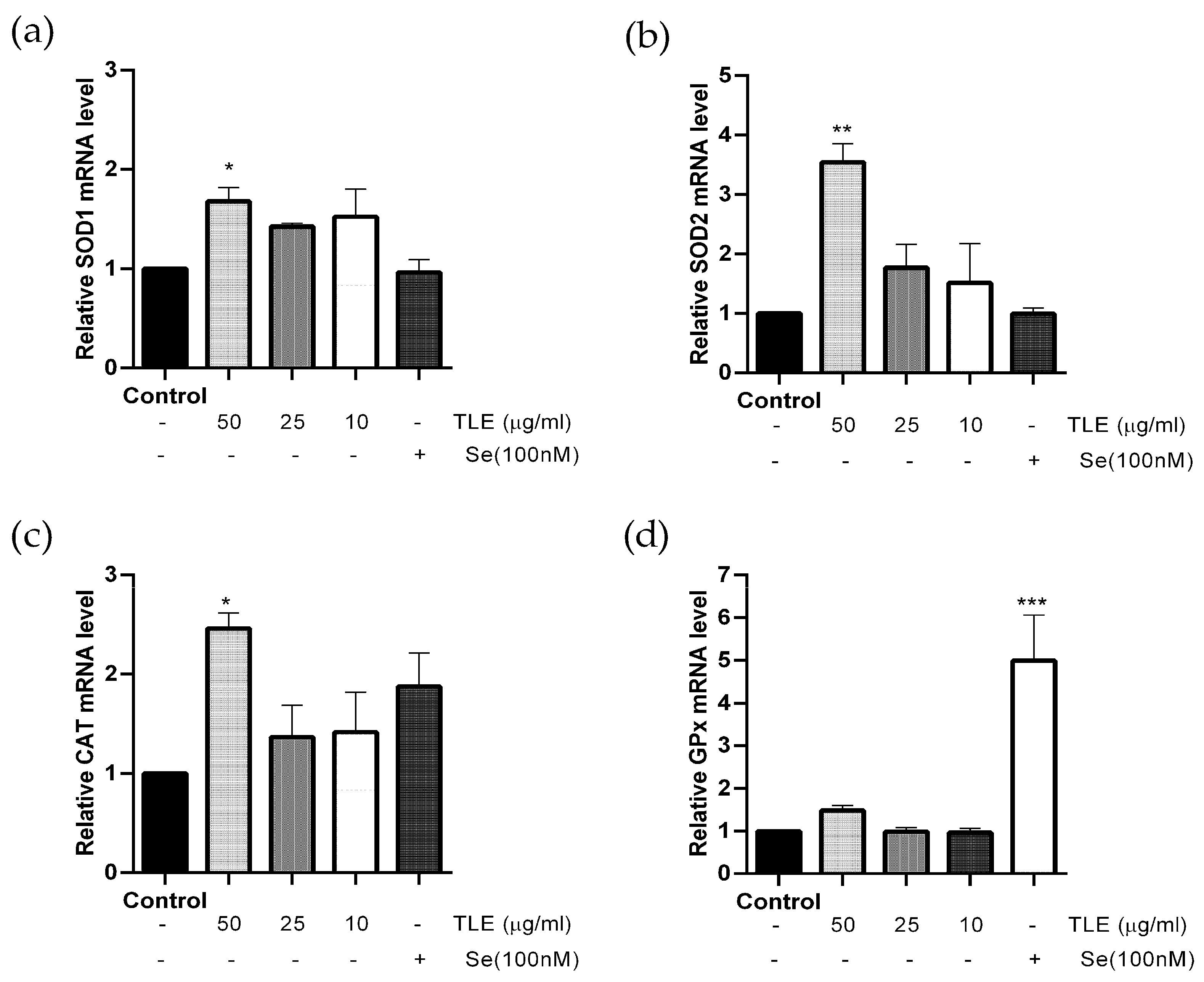
3.5. TLE Upregulates the mRNA Expression Level of Antioxidant Enzyme Genes
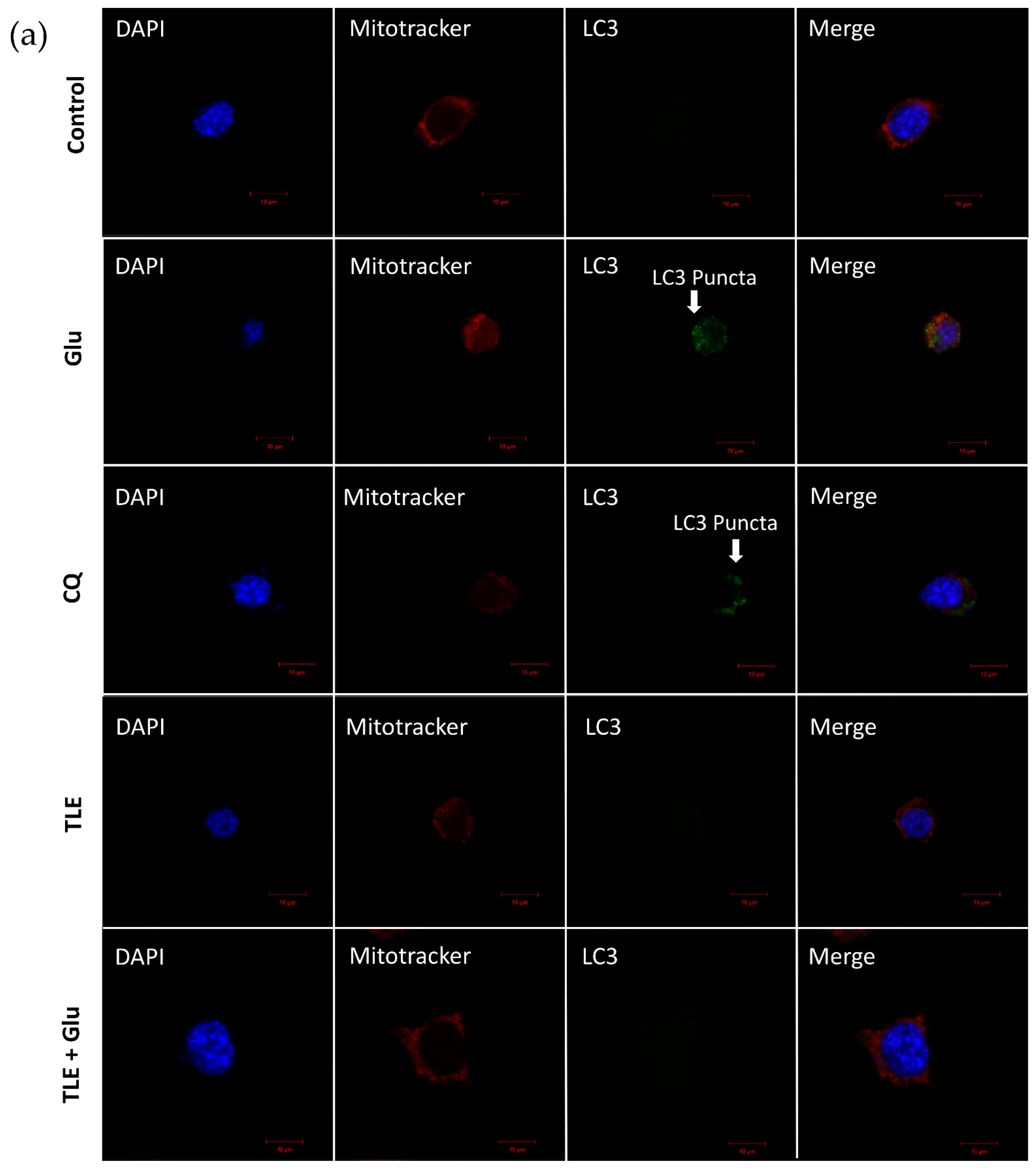
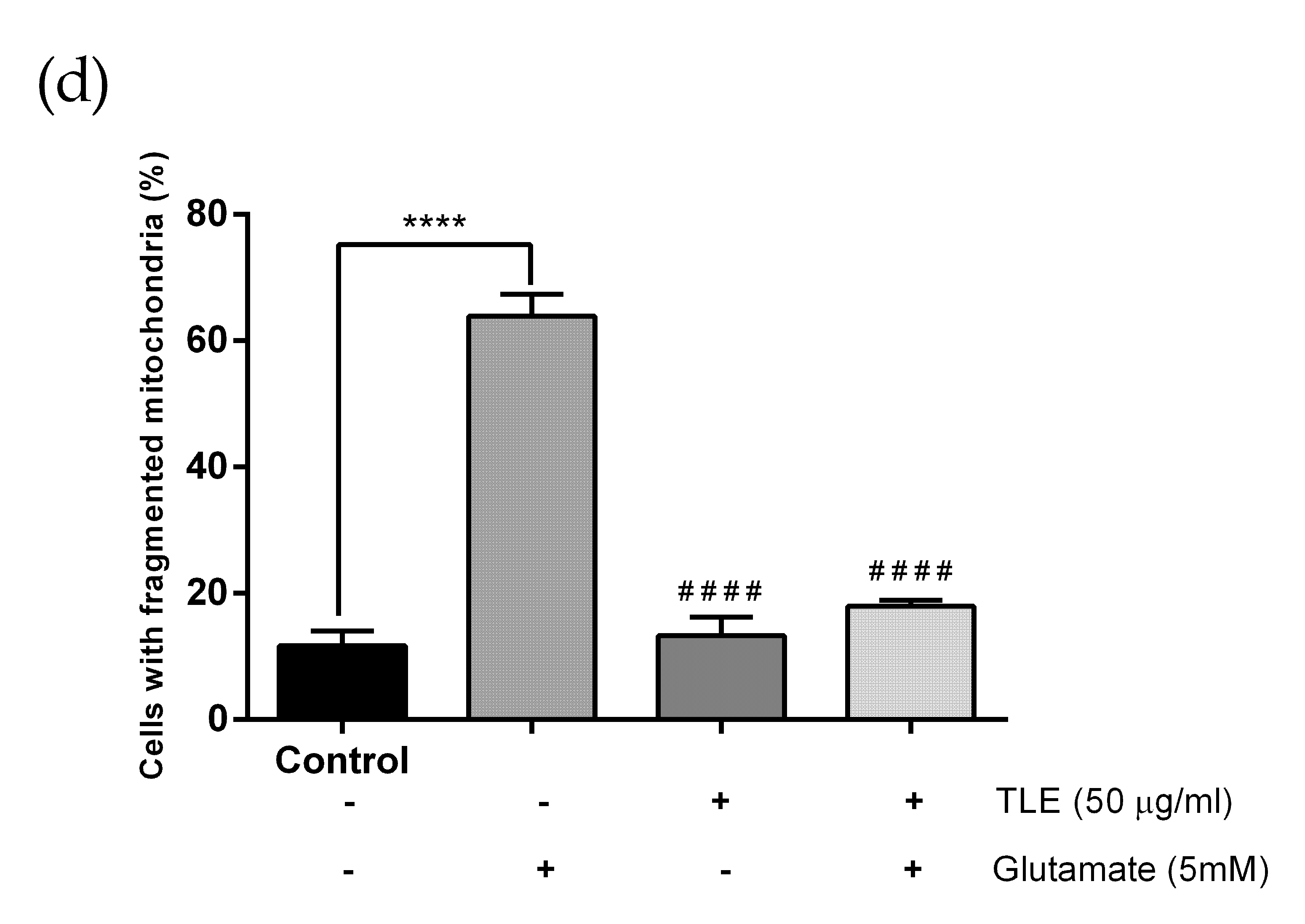
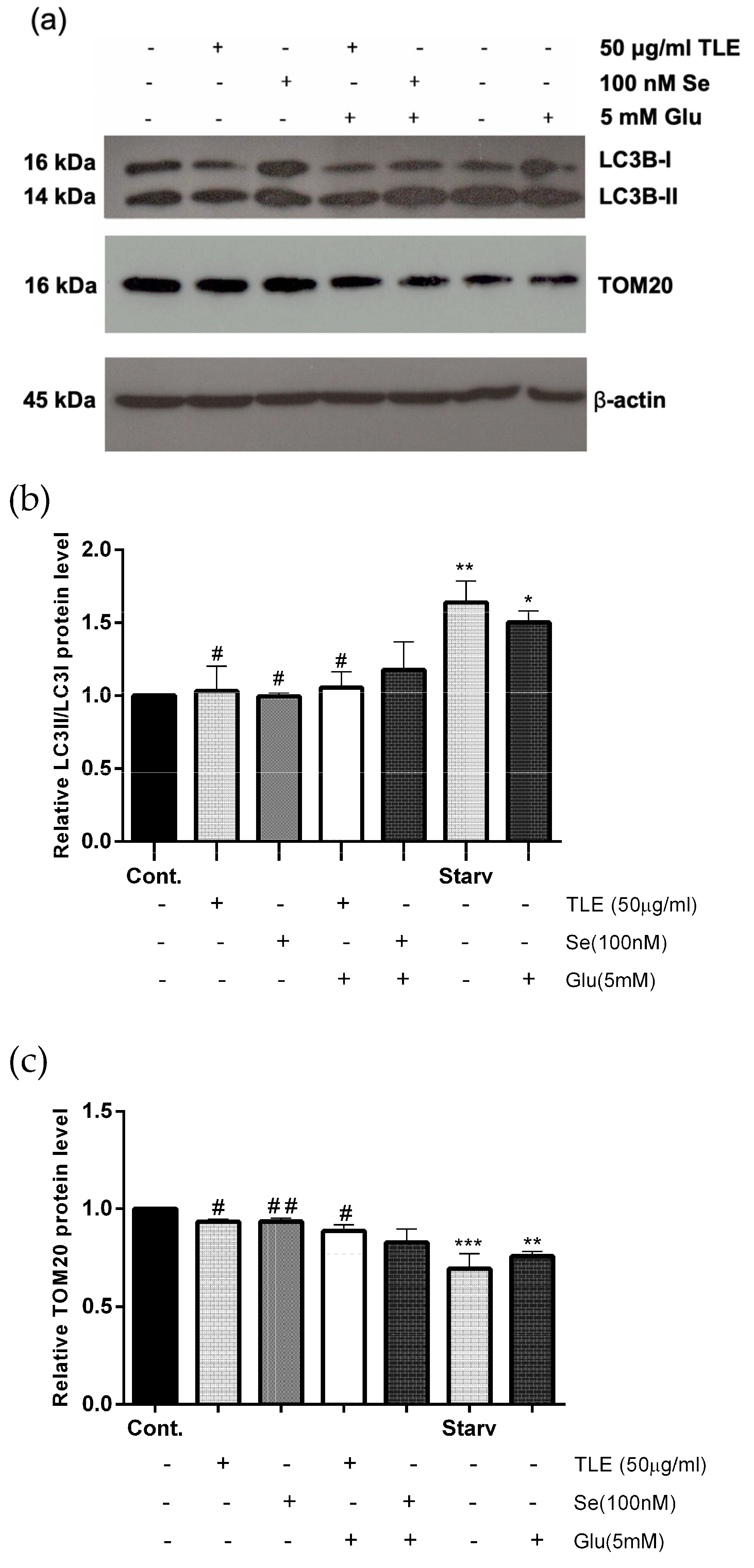
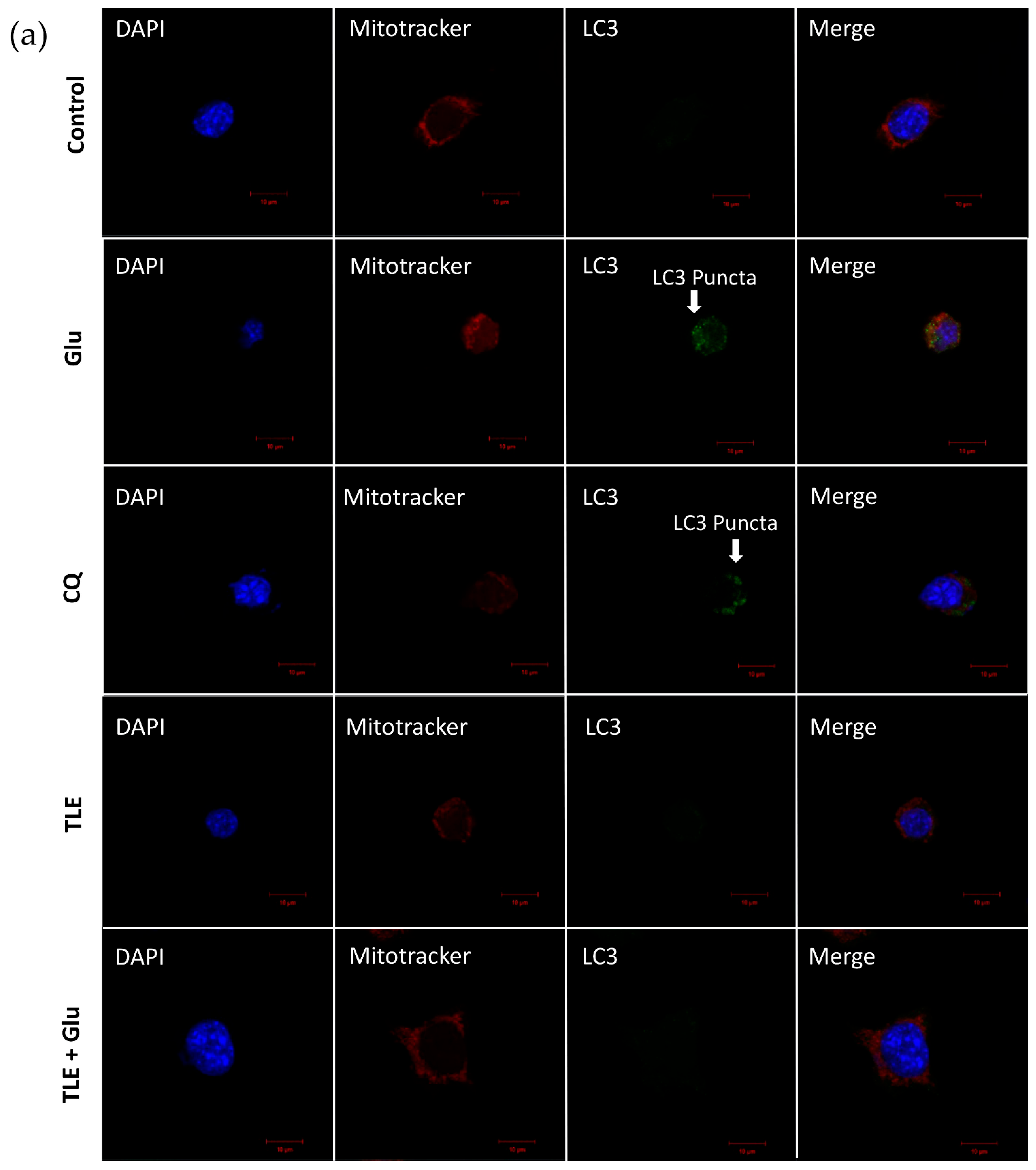
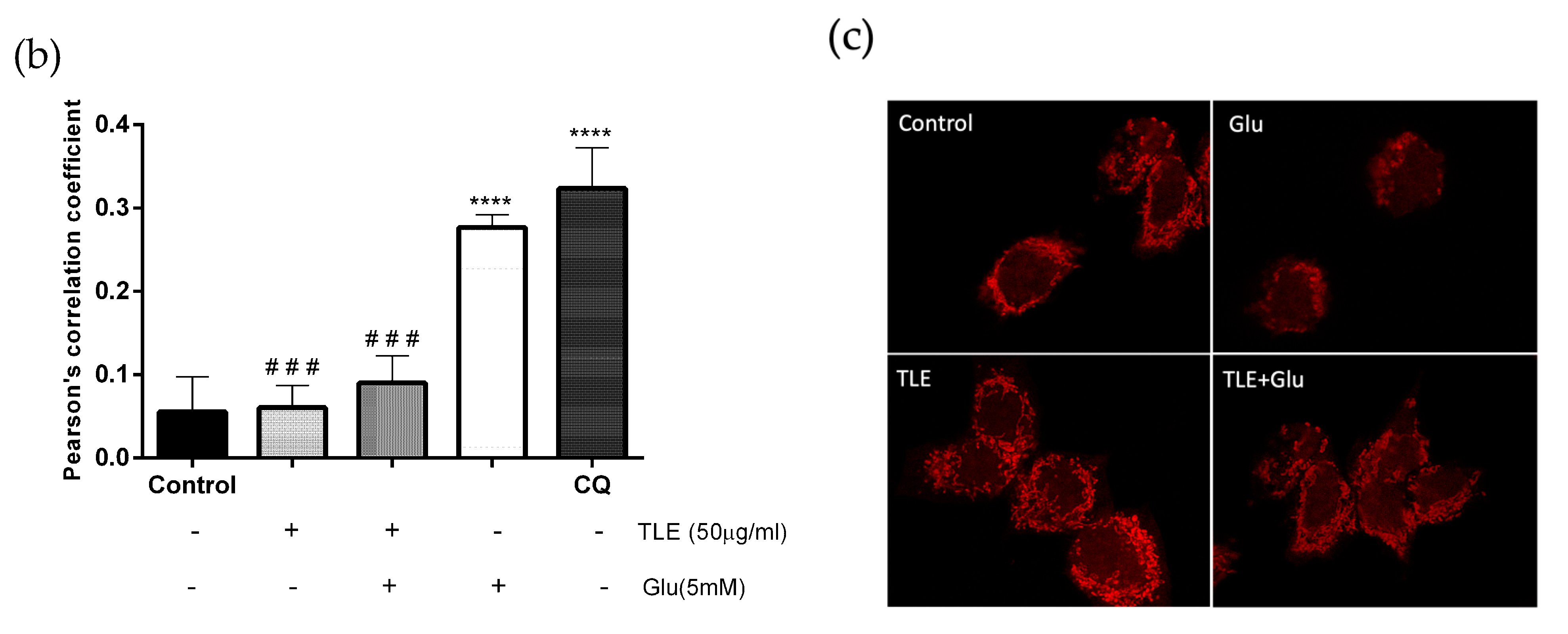
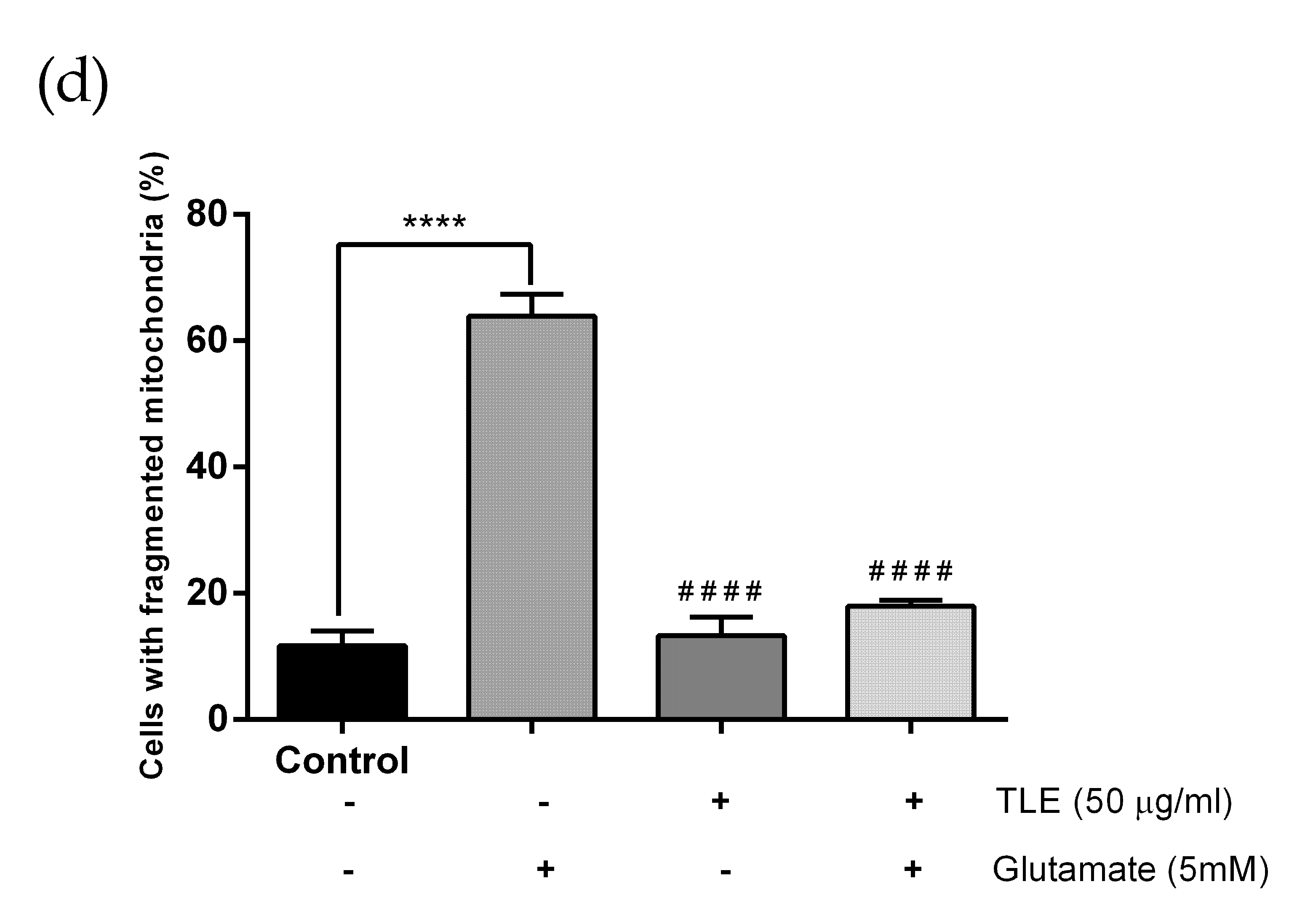
3.6. TLE Inhibits Glutamate-Induced Excessive Mitophagy in HT-22 Cells
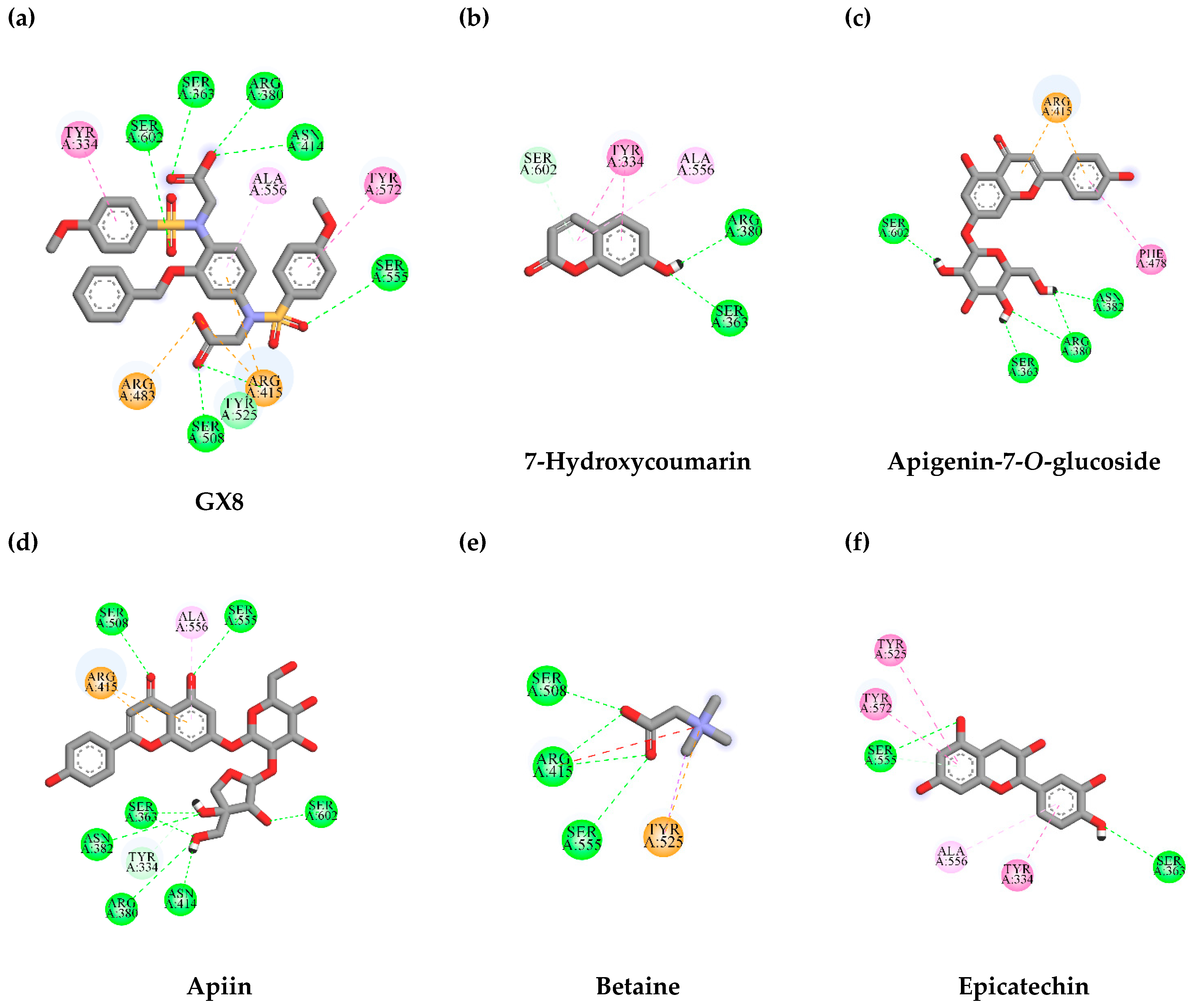
3.7. In Silico Virtual Screening of Binding Affinity between TLE-Identified Compounds and Mitophagy Protein Markers
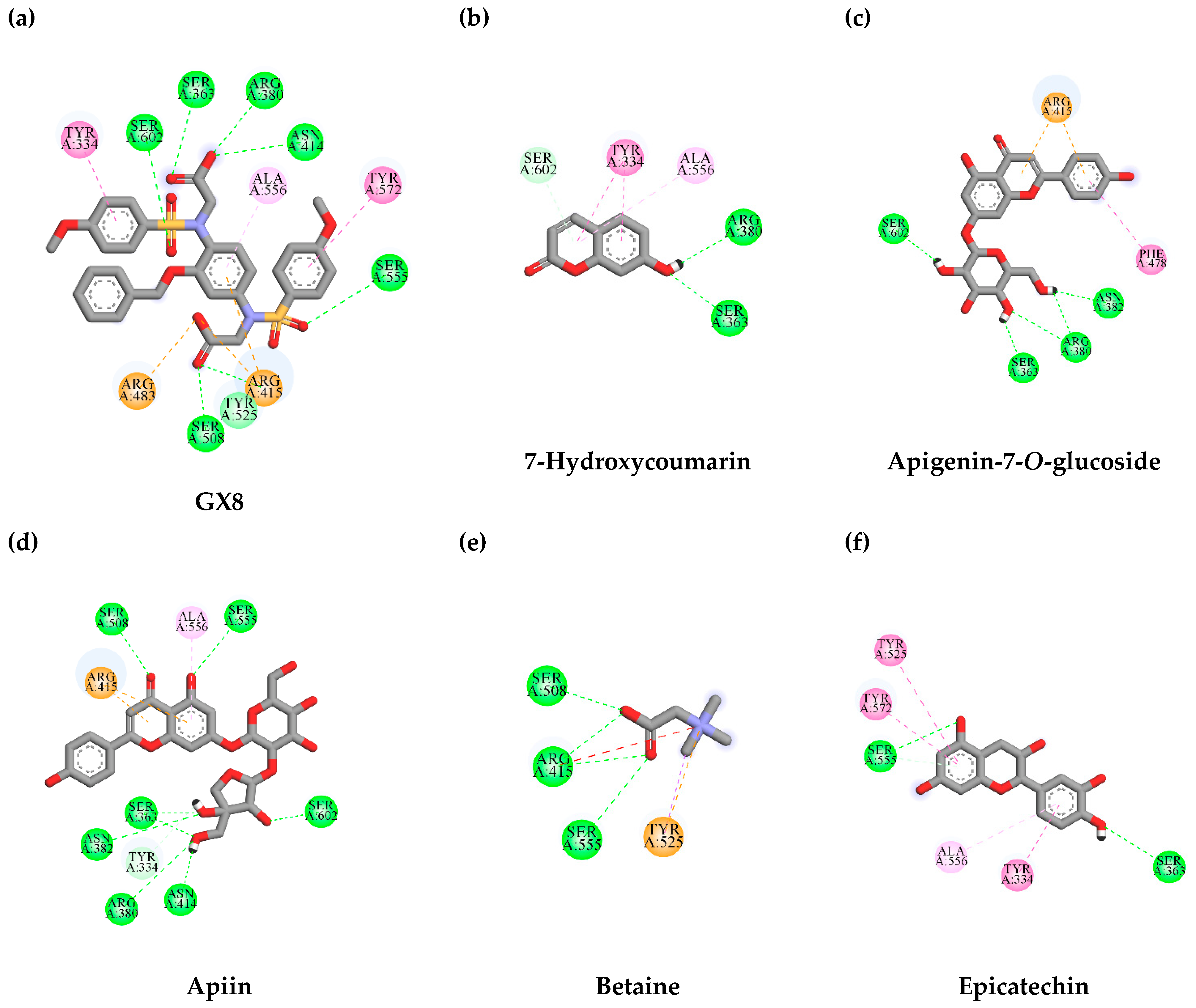
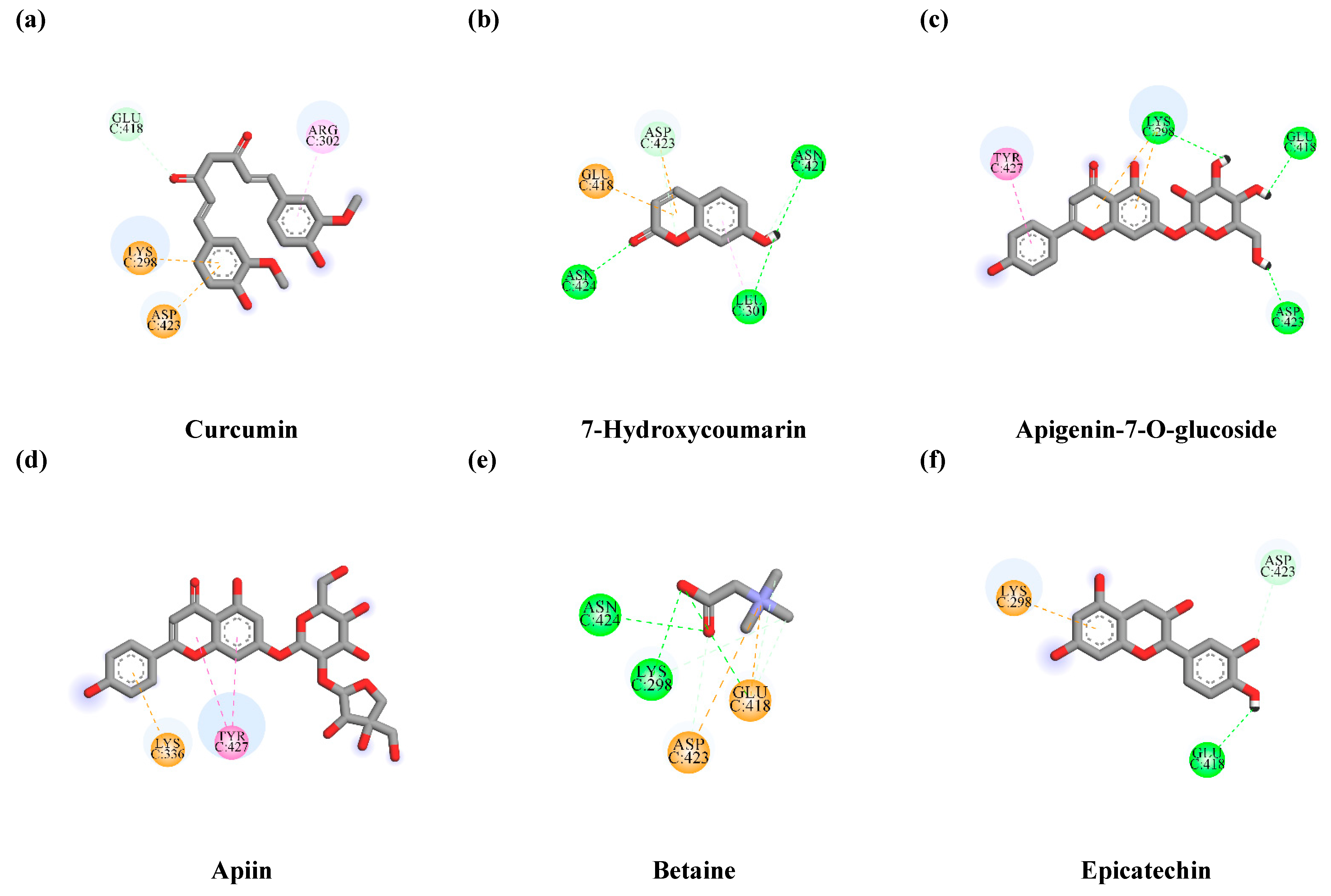
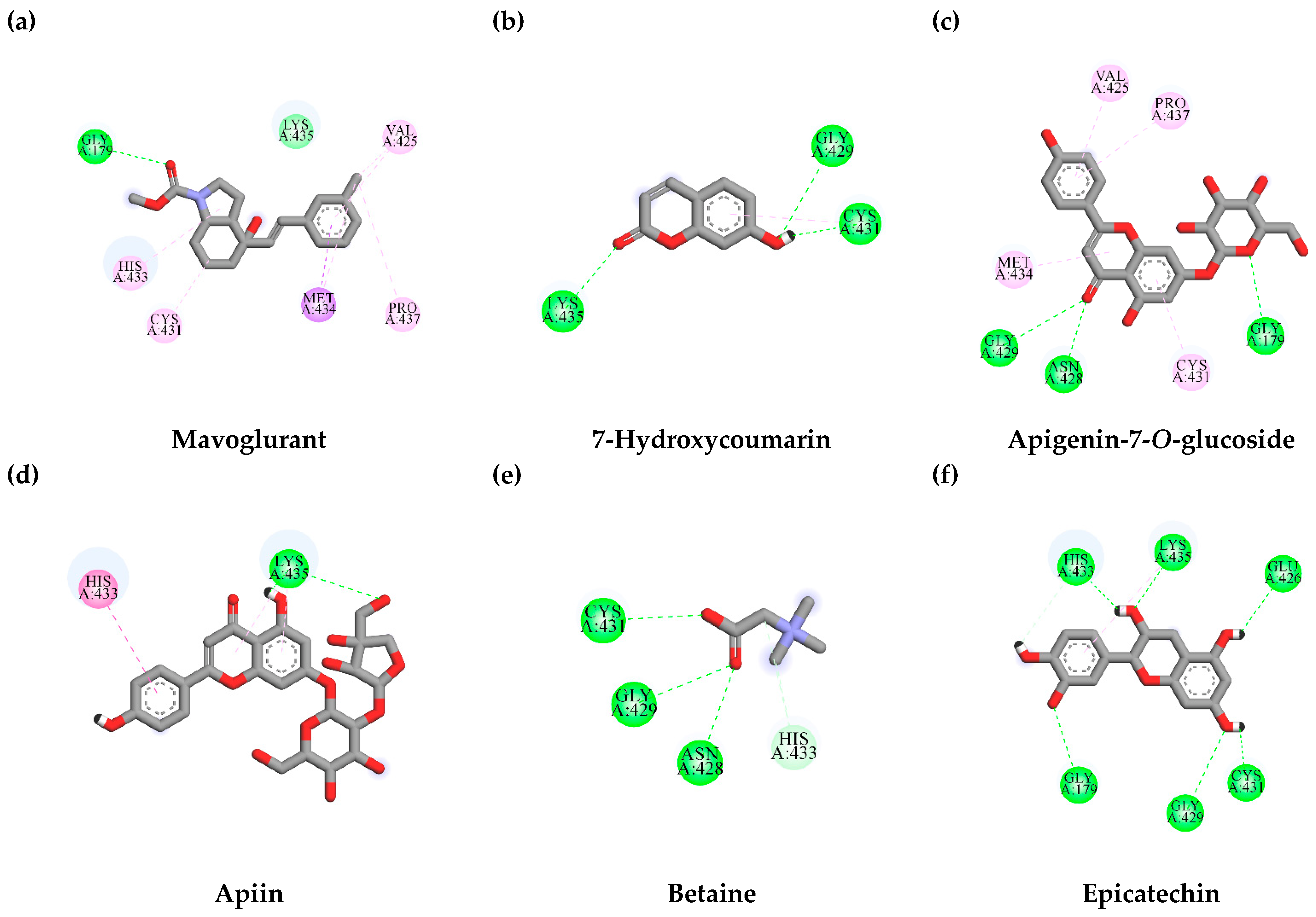
3.7.1. Interaction between TLE-Identified Compounds and PINK1
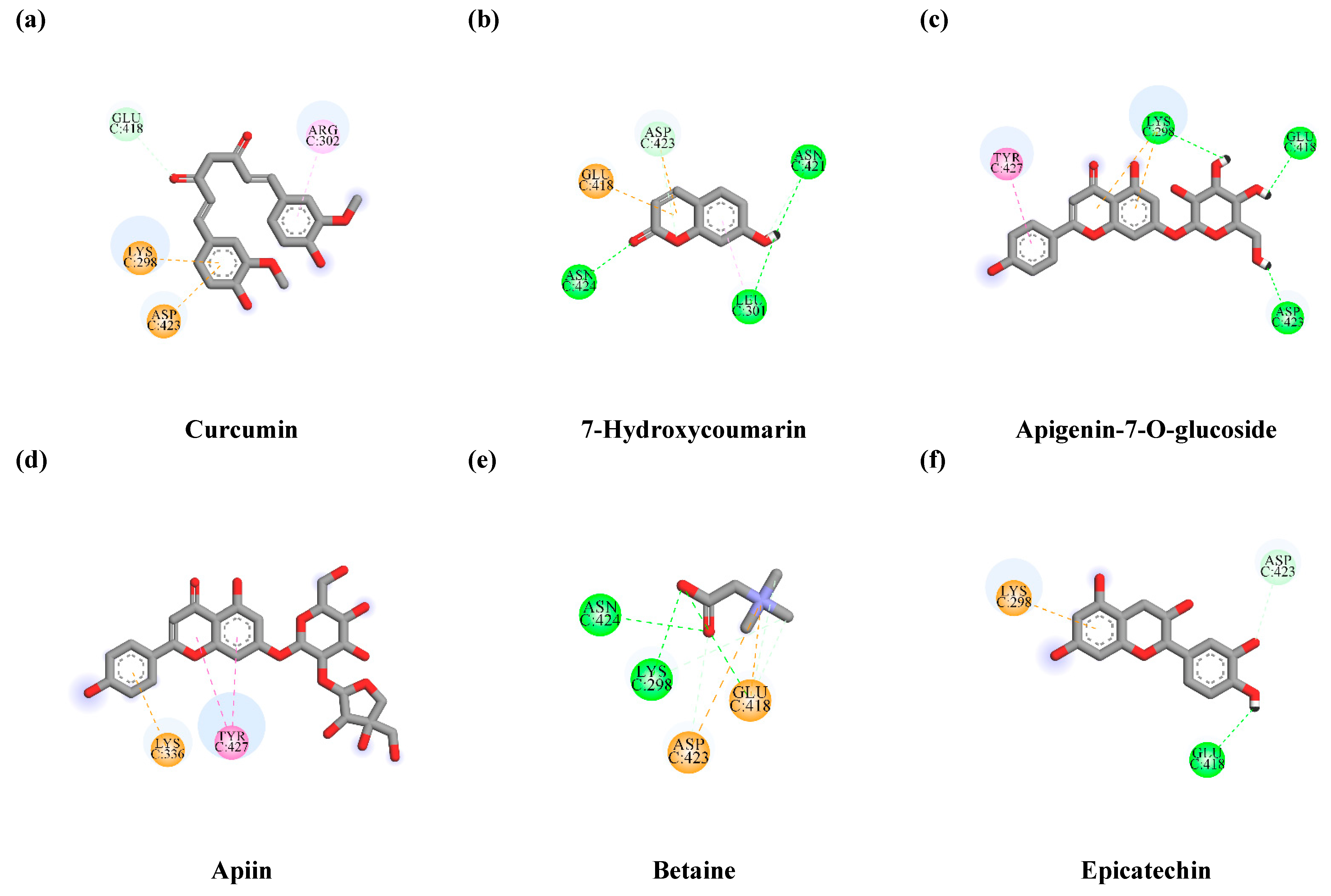
3.7.2. Interaction between TLE-Identified Compounds and E3 Ubiquitin-Protein Ligase Parkin
3.8. Lipinski’s Rule of Five Parameters and ADMET Properties of TLE Phytochemical Compounds
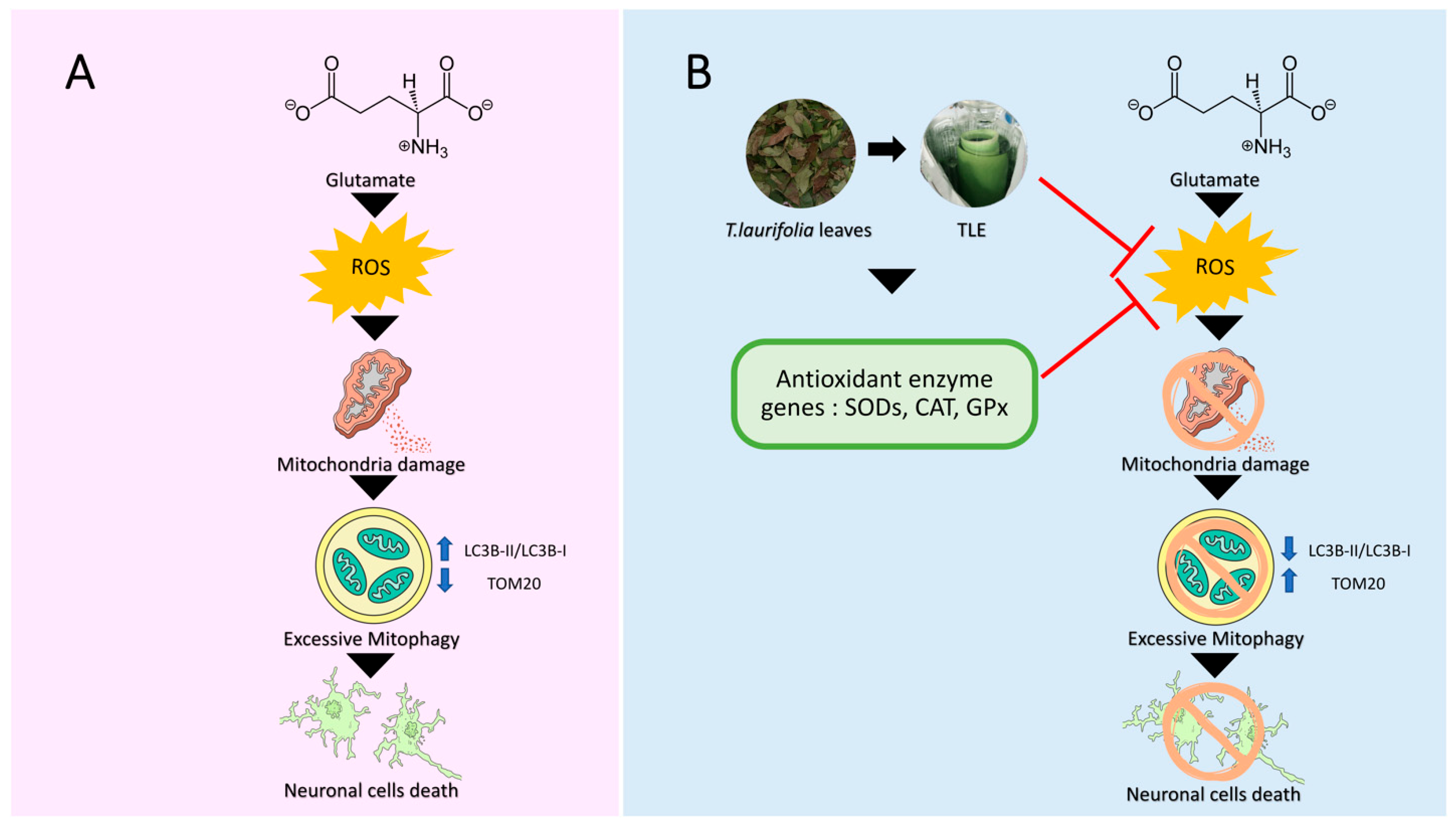
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; O, W.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. Pract. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A. Chapter 1 - Neurodegenerative Disorders: Why Do We Need New Therapies? In Drug Discovery Approaches for the Treatment of Neurodegenerative Disorders; Adejare, A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 1–16. [Google Scholar] [CrossRef]
- Albarracin, S.L.; Stab, B.; Casas, Z.; Sutachan, J.J.; Samudio, I.; Gonzalez, J.; Gonzalo, L.; Capani, F.; Morales, L.; Barreto, G.E. Effects of natural antioxidants in neurodegenerative disease. Nutr. Neurosci. 2012, 15, 1–9. [Google Scholar] [CrossRef]
- Amato, A.; Terzo, S.; Mulè, F. Natural Compounds as Beneficial Antioxidant Agents in Neurodegenerative Disorders: A Focus on Alzheimer’s Disease. Antioxidants 2019, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Khadrawyb Ya, E.H.S.A. Glutamate Excitotoxicity and Neurodegeneration. J. Mol. Genet. Med. 2014, 8, 4. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid ß peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol. 2013, 1, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Gu, L.; Smerin, D.; Mao, S.; Xiong, X. The Interrelation between Reactive Oxygen Species and Autophagy in Neurological Disorders. Oxidative Med. Cell. Longev. 2017, 2017, 8495160. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 612757. [Google Scholar] [CrossRef] [PubMed]
- Junsi, M.; Siripongvutikorn, S. Thunbergia laurifolia, a traditional herbal tea of Thailand: Botanical, chemical composition, biological properties and processing influence. Int. Food Res. J. 2016, 23, 923–927. [Google Scholar]
- Prasansuklab, A.; Meemon, K.; Sobhon, P.; Tencomnao, T. Ethanolic extract of Streblus asper leaves protects against glutamate-induced toxicity in HT22 hippocampal neuronal cells and extends lifespan of Caenorhabditis elegans. BMC Complement. Altern. Med. 2017, 17, 551. [Google Scholar] [CrossRef] [Green Version]
- Sukprasansap, M.; Chanvorachote, P.; Tencomnao, T. Cyanidin-3-glucoside activates Nrf2-antioxidant response element and protects against glutamate-induced oxidative and endoplasmic reticulum stress in HT22 hippocampal neuronal cells. BMC Complementary Med. Ther. 2020, 20, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, A.F.; Gladkova, C.; Pardon, E.; Wagstaff, J.L.; Freund, S.M.V.; Steyaert, J.; Maslen, S.L.; Komander, D. Structure of PINK1 in complex with its substrate ubiquitin. Nature 2017, 552, 51–56. [Google Scholar] [CrossRef]
- Kumar, A.; Aguirre, J.D.; Condos, T.E.C.; Martinez-Torres, R.J.; Chaugule, V.K.; Toth, R.; Sundaramoorthy, R.; Mercier, P.; Knebel, A.; Spratt, D.E.; et al. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 2015, 34, 2506–2521. [Google Scholar] [CrossRef]
- Rangsinth, P.; Duangjan, C.; Sillapachaiyaporn, C.; Isidoro, C.; Prasansuklab, A.; Tencomnao, T. Caesalpinia mimosoides Leaf Extract Promotes Neurite Outgrowth and Inhibits BACE1 Activity in Mutant APP-Overexpressing Neuronal Neuro2a Cells. Pharmaceuticals 2021, 14, 901. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.S.; Yu, Z.P.; Xia, J.Y.; Zhang, X.M.; Liu, K.C.; Sik, A.; Jin, M. Anti-Parkinson’s disease activity of phenolic acids from Eucommia ulmoides Oliver leaf extracts and their autophagy activation mechanism. Food Funct. 2020, 11, 1425–1440. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.-UK 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings (Reprinted from Advanced Drug Delivery Reviews, vol 23, pg 3-25, 1997). Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Vongthip, W.; Sukprasansap, M.; Tencomnao, T. Antioxidant activity and effect of Thunbergia laurifolia leaf crude extract in HT-22 mouse hippocampal cell line. In Proceedings of the 16th KU-KPS National Conference, Nakhon Pathom, Thailand, 3–4 December 2019; pp. 2917–2926. [Google Scholar]
- Kumari, S.; Mehta, S.L.; Li, P.A. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: Preventive effects of selenium. PLoS ONE 2012, 7, e39382. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [Green Version]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death—Where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, jcs215152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budina, A.; Hontz, R.; Pimkina, J.; Murphy, M. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A Short Mitochondrial Form of p19ARF Induces Autophagy and Caspase-Independent Cell Death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.; Zhu, B. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L.; Suo, W.Z. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009, 84, 267–271. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitophagy in neurodegeneration and aging. Front. Genet. 2012, 3, 297. [Google Scholar] [CrossRef] [Green Version]
- Arif, N.; Subhani, A.; Hussain, W.; Rasool, N. Computer-aided Analysis of Selective Phytochemicals as Potent Inhibitors of Parkin: Major Biological Target of Parkinson’s disease. Glob. J. Biotechnol. Biomater. Sci. 2020, 6, 13–23. [Google Scholar] [CrossRef]
- Petrov, D.; Pedros, I.; de Lemos, M.L.; Pallas, M.; Canudas, A.M.; Lazarowski, A.; Beas-Zarate, C.; Auladell, C.; Folch, J.; Camins, A. Mavoglurant as a treatment for Parkinson’s disease. Expert Opin. Investig. Drug 2014, 23, 1165–1179. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Ma, S. The Roles of Mitochondria in Autophagic Cell Death. Cancer Biother. Radiopharm. 2016, 31, 269–276. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The Ambiguous Relationship of Oxidative Stress, Tau Hyperphosphorylation, and Autophagy Dysfunction in Alzheimer’s Disease. Oxidative Med. Cell Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, Y.; Kotake, Y.; Komasaka, K.; Hakozaki, H.; Taguchi, R.; Kume, T.; Akaike, A.; Ohta, S. Glutamate Excitotoxicity Is Involved in Cell Death Caused by Tributyltin in Cultured Rat Cortical Neurons. Toxicol. Sci. 2006, 89, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-d.; Jin, M.-f.; Zhao, D.-j.; Ni, H. Reduction of Mitophagy-Related Oxidative Stress and Preservation of Mitochondria Function Using Melatonin Therapy in an HT22 Hippocampal Neuronal Cell Model of Glutamate-Induced Excitotoxicity. Front. Endocrinol. 2019, 10, 550. [Google Scholar] [CrossRef] [Green Version]
- Higdon, J.; Frei, B. Tea Catechins and Polyphenols: Health Effects, Metabolism, and Antioxidant Functions. Crit. Rev. Food Sci. Nutr. 2003, 43, 89–143. [Google Scholar] [CrossRef] [PubMed]
- Reygaert, W.C. Green Tea Catechins: Their Use in Treating and Preventing Infectious Diseases. Biomed. Res. Int. 2018, 2018, 9105261. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Wang, W.; Li, Q.; Han, X.; Xing, J.; Qi, C.; Lan, X.; Wan, J.; Potts, A.; Guan, F.; et al. Cerebroprotection of flavanol (-)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways. Free Radic. Biol. Med. 2016, 92, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, Z.; Li, R.-C.; Ahmad, A.; Kensler, T.; Tamamoto, M.; Biswal, S.; Dore, S. The Flavanol (−)-Epicatechin Prevents Stroke Damage through the Nrf2/HO1 Pathway. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Lee, J.-H.; Seo, Y.; Jang, J.-H.; Jeong, C.-H.; Lee, S.; Jeong, G.-S.; Park, B. Epicatechin Prevents Methamphetamine-Induced Neuronal Cell Death via Inhibition of ER Stress. Biomol. Ther. 2018, 27, 145–151. [Google Scholar] [CrossRef]
- Shay, J.; Elbaz, H.A.; Lee, I.; Zielske, S.P.; Malek, M.H.; Hüttemann, M. Molecular Mechanisms and Therapeutic Effects of (−)-Epicatechin and Other Polyphenols in Cancer, Inflammation, Diabetes, and Neurodegeneration. Oxidative Med. Cell. Longev. 2015, 2015, 181260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramiro-Puig, E.; Casadesus, G.; Lee, H.-g.; Zhu, X.; McShea, A.; Perry, G.; Pérez-Cano, F.; Castell, M. Neuroprotective effect of cocoa flavonids on in vitro oxidative stress. Eur. J. Nutr. 2008, 48, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Smiljkovic, M.; Stanisavljevic, D.; Stojkovic, D.; Petrovic, I.; Marjanovic Vicentic, J.; Popovic, J.; Golic Grdadolnik, S.; Markovic, D.; Sankovic-Babice, S.; Glamoclija, J.; et al. Apigenin-7-O-glucoside versus apigenin: Insight into the modes of anticandidal and cytotoxic actions. Excli J. 2017, 16, 795–807. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Huang, Y.; Wen, X.; Wu, Y.; Zhao, Y.; Ni, Y.Y. Extraction, Purification, and Hydrolysis Behavior of Apigenin-7-O-Glucoside from Chrysanthemum Morifolium Tea. Molecules 2018, 23, 2933. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.-S.; Kim, O.-S.; Kim, B.-Y.; Jeong, S.-J. Apigetrin from Scutellaria baicalensis Georgi Inhibits Neuroinflammation in BV-2 Microglia and Exerts Neuroprotective Effect in HT22 Hippocampal Cells. J. Med. Food 2016, 19, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.; Kannan, A.; Stojković, D.S.; Glamočlija, J.; Calhelha, R.C.; Ferreira, I.C.F.R.; Sanglard, D.; Soković, M. Flavones, Flavonols, and Glycosylated Derivatives-Impact on Candida albicans Growth and Virulence, Expression of CDR1 and ERG11, Cytotoxicity. Pharmaceuticals 2020, 14, 27. [Google Scholar] [CrossRef]
- Sim, M.-O.; Lee, H.-I.; Ham, J.R.; Seo, K.-I.; Kim, M.-J.; Lee, M.-K. Anti-inflammatory and antioxidant effects of umbelliferone in chronic alcohol-fed rats. Nutr. Res. Pract. 2015, 9, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazimba, O. Umbelliferone: Sources, chemistry and bioactivities review. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 223–232. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Ellis, E. Neuroprotective effects of umbelliferone and esculetin in a mouse model of Parkinson’s disease. J. Neurosci. Res. 2013, 91, 453–461. [Google Scholar] [CrossRef]
- DemİRkaya, A.; Gündoğdu, G.; Karakaya, S.; Yilmaz, Ş.; Nalci, K.; Hacimuftuoglu, A. Does Umbelliferone Protect Primary Cortical Neuron Cells Against Glutamate Excitotoxicity. Kafkas Univ. Vet. Fak. Derg. 2021, 27, 339–346. [Google Scholar] [CrossRef]
- Li, P.; Jia, J.; Zhang, D.; Xie, J.; Xu, X.; Wei, D. In vitro and in vivo antioxidant activities of a flavonoid isolated from celery (Apium graveolens L. var. dulce). Food Funct. 2013, 5, 50–56. [Google Scholar] [CrossRef]
- Alirezaei, M.; Jelodar, G.; Ghayemi, Z. Antioxidant Defense of Betaine Against Oxidative Stress Induced by Ethanol in the Rat Testes. Int. J. Pept. Res. Ther. 2012, 18, 239–247. [Google Scholar] [CrossRef]
- Alirezaei, M.; Gheisari, H.; Ranjbar, R.; Hajibemani, A. Betaine: A promising antioxidant agent for enhancement of broiler meat quality. Br. Poult. Sci. 2012, 53, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Kim, S.R.; Huh, H.; Jung, J.H.; Kim, Y.C. Betaine attenuates glutamate-induced neurotoxicity in primary cultured brain cells. Arch. Pharmacal Res. 1994, 17, 343–347. [Google Scholar] [CrossRef]
- Massaad, C.A.; Washington, T.M.; Pautler, R.G.; Klann, E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13576–13581. [Google Scholar] [CrossRef] [Green Version]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.-Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in Mitochondrial Superoxide Dismutase Modulates Alzheimer’s Disease-Like Pathology and Accelerates the Onset of Behavioral Changes in Human Amyloid Precursor Protein Transgenic Mice. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Son, T.G.; Kawamoto, E.M.; Yu, Q.S.; Greig, N.H.; Mattson, M.P.; Camandola, S. Naphthazarin protects against glutamate-induced neuronal death via activation of the Nrf2/ARE pathway. Biochem. Biophys. Res. Commun. 2013, 433, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, A.R.; Dos Santos, N.B.; Scavone, C.; Munhoz, C.D. Nrf2/ARE Pathway Modulation by Dietary Energy Regulation in Neurological Disorders. Front. Pharmacol. 2019, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell. Signal. 2013, 25, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Chapter 8 Monitoring Starvation-Induced Reactive Oxygen Species Formation. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Volume 452, pp. 119–130. [Google Scholar]
- Kim, H.; Choi, J.; Ryu, J.; Park, S.; Cho, S.; Park, B.C.; Lee, D. Activation of autophagy during glutamate-induced HT22 cell death. Biochem. Biophys. Res. Commun. 2009, 388, 339–344. [Google Scholar] [CrossRef]
- Jin, M.F.; Ni, H.; Li, L.L. Leptin Maintained Zinc Homeostasis Against Glutamate-Induced Excitotoxicity by Preventing Mitophagy-Mediated Mitochondrial Activation in HT22 Hippocampal Neuronal Cells. Front. Neurol. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Chen, G.; Chen, L.; Zhang, W.; Feng, D.; Liu, L.; Chen, Q. Monitoring mitophagy in mammalian cells. Methods Enzymol. 2014, 547, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Anne Stetler, R.; Leak, R.K.; Gao, Y.; Chen, J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, O.; Dekker Nitert, M.; Gallo, L.A.; Vejzovic, M.; Fisher, J.J.; Perkins, A.V. Review: Placental mitochondrial function and structure in gestational disorders. Placenta 2017, 54, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Kogot-Levin, A.; Saada, A.; Leibowitz, G.; Soiferman, D.; Douiev, L.; Raz, I.; Weksler-Zangen, S. Upregulation of Mitochondrial Content in Cytochrome c Oxidase Deficient Fibroblasts. PLoS ONE 2016, 11, e0165417. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Gene Accession Number | Sequence of Primer |
|---|---|---|
| SOD1 forward | NM_011434 | 5′-CAGGACCTCATTTTAATCCTCAC-3′ |
| SOD1 reverse | NM_011434 | 5′-CCCAGGTCTCCAACATGC-3′ |
| SOD2 forward | NM_013671 | 5′-CTGGACAAACCTGAGCCCTA-3′ |
| SOD2 reverse | NM_013671 | 5′-TGATAGCCTCCAGCAACTCTC-3′ |
| CAT forward | NM_009804 | 5′-CAGCGACCAGATGAAGCA-3′ |
| CAT reverse | NM_009804 | 5′-CTCCGGTGGTCAGGACAT-3′ |
| GPx forward | NM_008160 | 5′-ACAGTCCACCGTGTATGCCTTC-3′ |
| GPx reverse | NM_008160 | 5′-CTCTTCATTCTTGCCATTCTCCTG-3′ |
| β-actin forward | NM_007393 | 5′-GGCTGTATTCCCCTCCATCG-3′ |
| β-actin reverse | NM_007393 | 5′-CCAGTTGGTAACAATGCCATGT-3′ |
| Ligand | Binding Energy (kcal/mol) | Amino Acid Interaction | ||
|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Electrostatic Bond | ||
| GX8 (reference ligand) | −8.6 | SER363 ARG380 ASN414 ARG415 ARG483 (2) SER508 (2) SER555 SER602 | TYR334 ALA556 TYR572 | ARG415 (2) ARG483 |
| 7-Hydroxycoumarin | −6.5 | SER363 ARG380 SER602 | TYR334 (2) ALA556 | - |
| Apigenin-7-O-glucoside | −8.7 | SER363 ARG380 (2) ASN382 SER602 | PHE478 | ARG415 (2) |
| Apiin | −8.4 | TYR334 SER363 (2) ARG380 ASN382 ASN414 SER508 SER555 SER602 | ALA556 | ARG415 (2) |
| Betaine | −3.9 | ARG415 (3) SER508 (2) SER555 | TYR525 | TYR525 |
| Epicatechin | −7.8 | SER363 SER555 (2) | TYR334 TYR525 ALA556 TYR572 | - |
| Ligand | Binding Energy (kcal/mol) | Amino Acid Interaction | ||
|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Electrostatic Bond | ||
| Curcumin (reference ligand) | −5.4 | LYS298 GLU418 | ARG302 | LYS298 ASP423 |
| 7-Hydroxycoumarin | −4.9 | LEU301 ASN421 (2) ASP423 ASN424 | LEU301 | GLU418 ASP423 |
| Apigenin-7-O-glucoside | −5.1 | LYS298 ASP423 GLU418 | TYR427 | LYS298 (2) |
| Apiin | −4.4 | - | TYR427 (2) | LYS336 |
| Betaine | −3.4 | LYS298 (2) GLU418 (3) ASP423 ASN424 | - | GLU418 ASP423 |
| Epicatechin | −4.6 | GLU418 ASP423 | - | LYS298 |
| Ligand | Binding Energy (kcal/mol) | Amino Acid Interaction | ||
|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Bond | Electrostatic Bond | ||
| Mavoglurant (reference ligand) | −5.4 | GLY179 | VAL425 (2) CYS431 HIS433 MET434 (2) PRO437 | - |
| 7-Hydroxycoumarin | −4.8 | GLY429 CYS431 LYS435 | CYS431 | - |
| Apigenin-7-O-glucoside | −6.2 | GLY179 ASN428 GLY429 | VAL425 CYS431 MET434 PRO437 | - |
| Apiin | 1.6 | LYS435 (2) | HIS433 LYS435 (2) | - |
| Betaine | −3.4 | ASN428 GLY429 CYS431 HIS433 (2) | - | - |
| Epicatechin | −6.6 | GLY179 GLU426 GLY429 CYS431 HIS433 (2) LYS435 | - | LYS435 |
| Compound | Molecular Weight (≤500) | #H-Bond Acceptors (≤10) | #H-Bond Donors (≤5) | MLOGP (≤4.15) | Lipinski #Violations (≤1) |
|---|---|---|---|---|---|
| 7-Hydroxycoumarin | 162.14 | 3 | 1 | 1.04 | 0 |
| Apigenin-7-O-glucoside | 432.38 | 10 | 6 | −1.61 | 1 |
| Apiin | 564.49 | 14 | 8 | −3.16 | 3 |
| Betaine | 117.15 | 2 | 0 | −3.67 | 0 |
| Epicatechin | 290.27 | 6 | 5 | 0.24 | 0 |
| Pharmacokinetic Property | 7-Hydroxycoumarin | Apigenin-7-O-Glucoside | Apiin | Betaine | Epicatechin |
|---|---|---|---|---|---|
| Absorption | |||||
| Water solubility (log mol/L) | −2.131 | −2.559 | −2.851 | 0.723 | −3.117 |
| Caco2 permeability (log Papp in 10-6 cm/s) | 1.206 | 0.33 | −0.966 | 1.44 | −0.283 |
| Intestinal absorption (human) (% Absorbed) | 94.551 | 37.609 | 17.411 | 100 | 68.829 |
| Skin permeability (log Kp) | −2.6 | −2.735 | −2.735 | −2.78 | −2.735 |
| P-glycoprotein substrate | No | Yes | Yes | Yes | Yes |
| P-glycoprotein I inhibitor | No | No | No | No | No |
| P-glycoprotein II inhibitor | No | No | No | No | No |
| Distribution | |||||
| VDss (human) (log L/kg) | 0.032 | 0.342 | 1.004 | −0.304 | 1.027 |
| Fraction unbound (human) (Fu) | 0.432 | 0.218 | 0.171 | 0.875 | 0.235 |
| BBB permeability (log BB) | −0.278 | −1.391 | −1.793 | −0.214 | −1.054 |
| CNS permeability (log PS) | −2.741 | −3.746 | −4.972 | −2.804 | −3.298 |
| Metabolism | |||||
| CYP2D6 substrate | No | No | No | No | No |
| CYP3A4 substrate | No | No | No | No | No |
| CYP1A2 inhibitior | Yes | No | No | No | No |
| CYP2C19 inhibitior | No | No | No | No | No |
| CYP2C9 inhibitior | No | No | No | No | No |
| CYP2D6 inhibitior | No | No | No | No | No |
| CYP3A4 inhibitior | No | No | No | No | No |
| Excretion | |||||
| Total Clearance (log ml/min/kg) | 0.706 | 0.547 | −0.054 | 0.326 | 0.183 |
| Renal OCT2 substrate | No | No | No | No | No |
| Toxicity | |||||
| AMES toxicity | No | No | No | No | No |
| Max. tolerated dose (human) (log mg/kg/day) | 0.689 | 0.515 | 0.446 | 0.838 | 0.438 |
| hERG I inhibitor | No | No | No | No | No |
| hERG II inhibitor | No | No | Yes | No | No |
| Oral rat acute toxicity (LD50) (mol/kg) | 2.047 | 2.595 | 2.49 | 1.654 | 2.428 |
| Oral rat chronic toxicity (LOAEL) (log mg/kg_bw/day) | 1.751 | 4.359 | 4.574 | 0.254 | 2.5 |
| Hepatotoxicity | Yes | No | No | No | No |
| Skin sensitization | No | No | No | Yes | No |
| T.Pyriformis toxicity (log ug/L) | 0.546 | 0.285 | 0.285 | −0.057 | 0.347 |
| Minnow toxicity (log mM) | 1.714 | 5.507 | 3.835 | 2.97 | 3.585 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vongthip, W.; Sillapachaiyaporn, C.; Kim, K.-W.; Sukprasansap, M.; Tencomnao, T. Thunbergia laurifolia Leaf Extract Inhibits Glutamate-Induced Neurotoxicity and Cell Death through Mitophagy Signaling. Antioxidants 2021, 10, 1678. https://doi.org/10.3390/antiox10111678
Vongthip W, Sillapachaiyaporn C, Kim K-W, Sukprasansap M, Tencomnao T. Thunbergia laurifolia Leaf Extract Inhibits Glutamate-Induced Neurotoxicity and Cell Death through Mitophagy Signaling. Antioxidants. 2021; 10(11):1678. https://doi.org/10.3390/antiox10111678
Chicago/Turabian StyleVongthip, Wudtipong, Chanin Sillapachaiyaporn, Kyu-Won Kim, Monruedee Sukprasansap, and Tewin Tencomnao. 2021. "Thunbergia laurifolia Leaf Extract Inhibits Glutamate-Induced Neurotoxicity and Cell Death through Mitophagy Signaling" Antioxidants 10, no. 11: 1678. https://doi.org/10.3390/antiox10111678
APA StyleVongthip, W., Sillapachaiyaporn, C., Kim, K.-W., Sukprasansap, M., & Tencomnao, T. (2021). Thunbergia laurifolia Leaf Extract Inhibits Glutamate-Induced Neurotoxicity and Cell Death through Mitophagy Signaling. Antioxidants, 10(11), 1678. https://doi.org/10.3390/antiox10111678