
Mitochondrial Uncoupling Proteins (UCPs) as Key Modulators of ROS Homeostasis: A Crosstalk between Diabesity and Male Infertility?

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Mitochondrial Uncoupling Proteins (UCPs) Expression and Function
2.1. UCP1
2.2. UCP2
2.3. UCP3
2.4. UCP4
2.5. UCP5
2.6. UCP6
3. UCPs Are Key Regulators of ROS Production and Redox Homeostasis
4. UCPs Dysregulation Leads to Diabesity
5. The Potential Role of UCPs on Diabesity-Induced Male Infertility
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Nuertey, B.D.; Alhassan, A.I.; Nuertey, A.D.; Mensah, I.A.; Adongo, V.; Kabutey, C.; Addai, J.; Biritwum, R.B. Prevalence of obesity and overweight and its associated factors among registered pensioners in Ghana; A cross sectional studies. BMC Obes. 2017, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Vincent, H.K.; Taylor, A.G. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int. J. Obes. 2006, 30, 400–418. [Google Scholar] [CrossRef]
- Ferramosca, A.; Zara, V. Bioenergetics of mammalian sperm capacitation. Biomed. Res. Int. 2014, 2014, 902953. [Google Scholar] [CrossRef]
- Carrageta, D.F.; Guerra-Carvalho, B.; Sousa, M.; Barros, A.; Oliveira, P.F.; Monteiro, M.P.; Alves, M.G. Mitochondrial Activation and Reactive Oxygen-Species Overproduction during Sperm Capacitation are Independent of Glucose Stimuli. Antioxidants 2020, 9, 750. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Ho, P.W.; Ho, J.W.; Liu, H.F.; So, D.H.; Tse, H.M.; Chan, K.H.; Ho, S.L. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): Structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012, 2, 468–478. [Google Scholar] [CrossRef]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Chan, S.L.; Liu, D.; Kyriazis, G.A.; Bagsiyao, P.; Ouyang, X.; Mattson, M.P. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J. Biol. Chem. 2006, 281, 37391–37403. [Google Scholar] [CrossRef]
- Miwa, S.; Brand, M.D. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem. Soc. Trans. 2003, 31, 1300–1301. [Google Scholar] [CrossRef]
- Pecqueur, C.; Alves-Guerra, M.C.; Gelly, C.; Levi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J. Biol. Chem. 2001, 276, 8705–8712. [Google Scholar] [CrossRef]
- Hinz, W.; Gruninger, S.; de Pover, A.; Chiesi, M. Properties of the human long and short isoforms of the uncoupling protein-3 expressed in yeast cells. FEBS Lett. 1999, 462, 411–415. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- Sanchis, D.; Fleury, C.; Chomiki, N.; Goubern, M.; Huang, Q.; Neverova, M.; Gregoire, F.; Easlick, J.; Raimbault, S.; Levi-Meyrueis, C.; et al. BMCP1, a novel mitochondrial carrier with high expression in the central nervous system of humans and rodents, and respiration uncoupling activity in recombinant yeast. J. Biol. Chem. 1998, 273, 34611–34615. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Yu, X.X.; Zhong, A.; Li, W.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett. 1999, 443, 326–330. [Google Scholar] [CrossRef]
- Eckerskorn, C.; Klingenberg, M. In the uncoupling protein from brown adipose tissue the C-terminus protrudes to the c-side of the membrane as shown by tryptic cleavage. FEBS Lett. 1987, 226, 166–170. [Google Scholar] [CrossRef]
- Miroux, B.; Frossard, V.; Raimbault, S.; Ricquier, D.; Bouillaud, F. The topology of the brown adipose tissue mitochondrial uncoupling protein determined with antibodies against its antigenic sites revealed by a library of fusion proteins. EMBO J. 1993, 12, 3739–3745. [Google Scholar] [CrossRef]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.; Smith, M.D.; Jelokhani-Niaraki, M. Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4, and UCP5. Biochemistry 2012, 51, 4004–4014. [Google Scholar] [CrossRef]
- Liu, D.; Chan, S.L.; de Souza-Pinto, N.C.; Slevin, J.R.; Wersto, R.P.; Zhan, M.; Mustafa, K.; de Cabo, R.; Mattson, M.P. Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromol. Med. 2006, 8, 389–414. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Tiraby, C.; Tavernier, G.; Lefort, C.; Larrouy, D.; Bouillaud, F.; Ricquier, D.; Langin, D. Acquirement of brown fat cell features by human white adipocytes. J. Biol. Chem. 2003, 278, 33370–33376. [Google Scholar] [CrossRef]
- Mori, S.; Yoshizuka, N.; Takizawa, M.; Takema, Y.; Murase, T.; Tokimitsu, I.; Saito, M. Expression of uncoupling proteins in human skin and skin-derived cells. J. Investig. Dermatol. 2008, 128, 1894–1900. [Google Scholar] [CrossRef]
- Matthias, A.; Ohlson, K.B.; Fredriksson, J.M.; Jacobsson, A.; Nedergaard, J.; Cannon, B. Thermogenic responses in brown fat cells are fully UCP1-dependent. UCP2 or UCP3 do not substitute for UCP1 in adrenergically or fatty scid-induced thermogenesis. J. Biol. Chem. 2000, 275, 25073–25081. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar] [CrossRef]
- Fujita, H.; Habuta, M.; Hattori, T.; Kubota, S.; Kumon, H.; Ohuchi, H. UCP1 expression in the mouse adrenal gland is not upregulated by thermogenic conditions. Biochem. Biophys. Res. Commun. 2021, 566, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Jakus, P.B.; Sipos, K.; Kispal, G.; Sandor, A. Opposite regulation of uncoupling protein 1 and uncoupling protein 3 in vivo in brown adipose tissue of cold-exposed rats. FEBS Lett. 2002, 519, 210–214. [Google Scholar] [CrossRef]
- Sepa-Kishi, D.M.; Jani, S.; da Eira, D.; Ceddia, R.B. Cold acclimation enhances UCP1 content, lipolysis, and triacylglycerol resynthesis, but not mitochondrial uncoupling and fat oxidation, in rat white adipocytes. Am. J. Physiol. Cell Physiol. 2019, 316, C365–C376. [Google Scholar] [CrossRef]
- Nigro, M.; de Sanctis, C.; Formisano, P.; Stanzione, R.; Forte, M.; Capasso, G.; Gigliotti, G.; Rubattu, S.; Viggiano, D. Cellular and subcellular localization of uncoupling protein 2 in the human kidney. J. Mol. Histol. 2018, 49, 437–445. [Google Scholar] [CrossRef]
- Haguenauer, A.; Raimbault, S.; Masscheleyn, S.; Gonzalez-Barroso Mdel, M.; Criscuolo, F.; Plamondon, J.; Miroux, B.; Ricquier, D.; Richard, D.; Bouillaud, F.; et al. A new renal mitochondrial carrier, KMCP1, is up-regulated during tubular cell regeneration and induction of antioxidant enzymes. J. Biol. Chem. 2005, 280, 22036–22043. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.J.; Hodyl, N.A.; Butler, M.; Clifton, V.L. Localisation and characterisation of uncoupling protein-2 (UCP2) in the human preterm placenta. Placenta 2012, 33, 1020–1025. [Google Scholar] [CrossRef]
- Wang, X.; Qian, H.; Huang, X.; Li, J.; Zhang, J.; Zhu, N.; Chen, H.; Zhu, C.; Wang, J.; Zhang, P.; et al. UCP2 Mitigates the Loss of Human Spermatozoa Motility by Promoting mROS Elimination. Cell Physiol. Biochem. 2018, 50, 952–962. [Google Scholar] [CrossRef]
- Boss, O.; Samec, S.; Paoloni-Giacobino, A.; Rossier, C.; Dulloo, A.; Seydoux, J.; Muzzin, P.; Giacobino, J.P. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997, 408, 39–42. [Google Scholar] [CrossRef]
- Chan, C.B.; de Leo, D.; Joseph, J.W.; McQuaid, T.S.; Ha, X.F.; Xu, F.; Tsushima, R.G.; Pennefather, P.S.; Salapatek, A.M.; Wheeler, M.B. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: Mechanism of action. Diabetes 2001, 50, 1302–1310. [Google Scholar] [CrossRef]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Krauss, S.; Zhang, C.Y.; Lowell, B.B. A significant portion of mitochondrial proton leak in intact thymocytes depends on expression of UCP2. Proc. Natl. Acad. Sci. USA 2002, 99, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Qiu, W.; Zhou, Y.; Wen, P.; Fang, L.; Cao, H.; Zen, K.; He, W.; Zhang, C.; Dai, C.; et al. A microRNA-30e/mitochondrial uncoupling protein 2 axis mediates TGF-beta1-induced tubular epithelial cell extracellular matrix production and kidney fibrosis. Kidney Int. 2013, 84, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Rousset, S.; Alves-Guerra, M.C.; Ouadghiri-Bencherif, S.; Kozak, L.P.; Miroux, B.; Richard, D.; Bouillaud, F.; Ricquier, D.; Cassard-Doulcier, A.M. Uncoupling protein 2, but not uncoupling protein 1, is expressed in the female mouse reproductive tract. J. Biol. Chem. 2003, 278, 45843–45847. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Dong, Y.; Tang, R.; Chen, J.; Zhang, C.Y.; Zen, K. Mitochondrial uncoupling protein 2 protects splenocytes from oxidative stress-induced apoptosis during pathogen activation. Cell Immunol. 2013, 286, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, D.; Clavel, S.; Sanchis, D.; Plamondon, J.; Huang, Q.; Ricquier, D.; Rouger, L.; Richard, D. Induction of Ucp2 expression in brain phagocytes and neurons following murine toxoplasmosis: An essential role of IFN-gamma and an association with negative energy balance. J. Neuroimmunol. 2007, 186, 121–132. [Google Scholar] [CrossRef]
- Motloch, L.J.; Larbig, R.; Gebing, T.; Reda, S.; Schwaiger, A.; Leitner, J.; Wolny, M.; Eckardt, L.; Hoppe, U.C. By Regulating Mitochondrial Ca2+-Uptake UCP2 Modulates Intracellular Ca2+. PLoS ONE 2016, 11, e0148359. [Google Scholar] [CrossRef]
- Zhang, K.; Shang, Y.; Liao, S.; Zhang, W.; Nian, H.; Liu, Y.; Chen, Q.; Han, C. Uncoupling protein 2 protects testicular germ cells from hyperthermia-induced apoptosis. Biochem. Biophys. Res. Commun. 2007, 360, 327–332. [Google Scholar] [CrossRef]
- Diao, J.; Allister, E.M.; Koshkin, V.; Lee, S.C.; Bhattacharjee, A.; Tang, C.; Giacca, A.; Chan, C.B.; Wheeler, M.B. UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc. Natl. Acad. Sci. USA 2008, 105, 12057–12062. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Barger, J.L.; Boyer, B.B.; Brand, M.D.; Pan, G.; Adams, S.H. Impact of endotoxin on UCP homolog mRNA abundance, thermoregulation, and mitochondrial proton leak kinetics. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E433–E446. [Google Scholar] [CrossRef]
- Rousset, S.; Emre, Y.; Join-Lambert, O.; Hurtaud, C.; Ricquier, D.; Cassard-Doulcier, A.M. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine 2006, 35, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, K.; Chen, J.; Guo, J.; Yin, Y.; Cai, X.; Guo, X.; Wang, G.; Yang, R.; Zhu, L.; et al. In vitro evidence suggests that miR-133a-mediated regulation of uncoupling protein 2 (UCP2) is an indispensable step in myogenic differentiation. J. Biol. Chem. 2009, 284, 5362–5369. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.; Fasching, A.; Hansell, P.; Nordquist, L.; Palm, F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim. Biophys. Acta 2008, 1777, 935–940. [Google Scholar] [CrossRef]
- Larrouy, D.; Laharrague, P.; Carrera, G.; Viguerie-Bascands, N.; Levi-Meyrueis, C.; Fleury, C.; Pecqueur, C.; Nibbelink, M.; Andre, M.; Casteilla, L.; et al. Kupffer cells are a dominant site of uncoupling protein 2 expression in rat liver. Biochem. Biophys. Res. Commun. 1997, 235, 760–764. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.A.; Pandolfi, M.; Bernardo, A.; de Nuccio, C.; Minghetti, L.; Visentin, S. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J. Neurochem. 2015, 135, 147–156. [Google Scholar] [CrossRef]
- Horvath, T.L.; Warden, C.H.; Hajos, M.; Lombardi, A.; Goglia, F.; Diano, S. Brain uncoupling protein 2: Uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J. Neurosci. 1999, 19, 10417–10427. [Google Scholar] [CrossRef]
- Teshima, Y.; Akao, M.; Jones, S.P.; Marban, E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 2003, 93, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S.; Buckingham, J.A.; Samec, S.; Seydoux, J.; Din, N.; Dulloo, A.G.; Brand, M.D. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Lett. 1999, 462, 257–260. [Google Scholar] [CrossRef]
- Amaral, S.; Mota, P.; Rodrigues, A.S.; Martins, L.; Oliveira, P.J.; Ramalho-Santos, J. Testicular aging involves mitochondrial dysfunction as well as an increase in UCP2 levels and proton leak. FEBS Lett. 2008, 582, 4191–4196. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.B.; MacDonald, P.E.; Saleh, M.C.; Johns, D.C.; Marban, E.; Wheeler, M.B. Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes 1999, 48, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Alan, L.; Smolkova, K.; Kronusova, E.; Santorova, J.; Jezek, P. Absolute levels of transcripts for mitochondrial uncoupling proteins UCP2, UCP3, UCP4, and UCP5 show different patterns in rat and mice tissues. J. Bioenerg. Biomembr. 2009, 41, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Penicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.M.; Porter, R.K. Starvation-sensitive UCP 3 protein expression in thymus and spleen mitochondria. Biochim. Biophys. Acta 2004, 1700, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Maedler, K.; Shu, L.; Haataja, L. UCP-2 and UCP-3 proteins are differentially regulated in pancreatic beta-cells. PLoS ONE 2008, 3, e1397. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, M.K.; Greenhaff, P.L.; Constantin-Teodosiu, D.; Hultman, E.; Saris, W.H.; Nieuwlaat, R.; Schaart, G.; Kornips, E.; Schrauwen, P. Increased uncoupling protein 3 content does not affect mitochondrial function in human skeletal muscle in vivo. J. Clin. Investig. 2003, 111, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.; Solanes, G.; Grujic, D.; Flier, J.S.; Lowell, B.B. UCP3: An uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem. Biophys. Res. Commun. 1997, 235, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Hilse, K.E.; Kalinovich, A.V.; Rupprecht, A.; Smorodchenko, A.; Zeitz, U.; Staniek, K.; Erben, R.G.; Pohl, E.E. The expression of UCP3 directly correlates to UCP1 abundance in brown adipose tissue. Biochim. Biophys. Acta 2016, 1857, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kelly, O.M.; Porter, R.K. Absence of mitochondrial uncoupling protein 3: Effect on thymus and spleen in the fed and fasted mice. Biochim. Biophys. Acta 2011, 1807, 1064–1074. [Google Scholar] [CrossRef]
- O’Connor, E.B.; Munoz-Wolf, N.; Leon, G.; Lavelle, E.C.; Mills, K.H.G.; Walsh, P.T.; Porter, R.K. UCP3 reciprocally controls CD4+ Th17 and Treg cell differentiation. PLoS ONE 2020, 15, e0239713. [Google Scholar] [CrossRef]
- Boudina, S.; Han, Y.H.; Pei, S.; Tidwell, T.J.; Henrie, B.; Tuinei, J.; Olsen, C.; Sena, S.; Abel, E.D. UCP3 regulates cardiac efficiency and mitochondrial coupling in high fat-fed mice but not in leptin-deficient mice. Diabetes 2012, 61, 3260–3269. [Google Scholar] [CrossRef]
- Huang, Z.; Li, J.; Du, S.; Chen, G.; Qi, Y.; Huang, L.; Xiao, L.; Tong, P. Effects of UCP4 on the Proliferation and Apoptosis of Chondrocytes: Its Possible Involvement and Regulation in Osteoarthritis. PLoS ONE 2016, 11, e0150684. [Google Scholar] [CrossRef] [PubMed]
- Smorodchenko, A.; Rupprecht, A.; Sarilova, I.; Ninnemann, O.; Brauer, A.U.; Franke, K.; Schumacher, S.; Techritz, S.; Nitsch, R.; Schuelke, M.; et al. Comparative analysis of uncoupling protein 4 distribution in various tissues under physiological conditions and during development. Biochim. Biophys. Acta 2009, 1788, 2309–2319. [Google Scholar] [CrossRef]
- Smorodchenko, A.; Rupprecht, A.; Fuchs, J.; Gross, J.; Pohl, E.E. Role of mitochondrial uncoupling protein 4 in rat inner ear. Mol. Cell Neurosci. 2011, 47, 244–253. [Google Scholar] [CrossRef]
- Kitahara, T.; Li, H.S.; Balaban, C.D. Localization of the mitochondrial uncoupling protein family in the rat inner ear. Hear. Res. 2004, 196, 39–48. [Google Scholar] [CrossRef]
- Donhoffer, S.; Szegvari, G.; Jarai, I.; Farkas, M. Thermoregulatory heat production in the brain. Nature 1959, 184 (Suppl. 13), 993–994. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Fattal, R.; Pattani, S.; Yang, E.; Zaheer, S.; Santillan, D.A.; Santillan, M.K.; Zaheer, A. Mast Cells Release Chemokine CCL2 in Response to Parkinsonian Toxin 1-Methyl-4-Phenyl-Pyridinium (MPP(+)). Neurochem. Res. 2016, 41, 1042–1049. [Google Scholar] [CrossRef]
- Kitahara, T.; Li-Korotky, H.S.; Balaban, C.D. Regulation of mitochondrial uncoupling proteins in mouse inner ear ganglion cells in response to systemic kanamycin challenge. Neuroscience 2005, 135, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Lengacher, S.; Magistretti, P.J.; Pellerin, L. Quantitative rt-PCR analysis of uncoupling protein isoforms in mouse brain cortex: Methodological optimization and comparison of expression with brown adipose tissue and skeletal muscle. J. Cereb. Blood Flow Metab. 2004, 24, 780–788. [Google Scholar] [CrossRef]
- Yu, X.X.; Mao, W.; Zhong, A.; Schow, P.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. Characterization of novel UCP5/BMCP1 isoforms and differential regulation of UCP4 and UCP5 expression through dietary or temperature manipulation. FASEB J. 2000, 14, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Kim-Han, J.S.; Reichert, S.A.; Quick, K.L.; Dugan, L.L. BMCP1: A mitochondrial uncoupling protein in neurons which regulates mitochondrial function and oxidant production. J. Neurochem. 2001, 79, 658–668. [Google Scholar] [CrossRef]
- Erden, Y.; Tekin, S.; Sandal, S.; Onalan, E.E.; Tektemur, A.; Kirbag, S. Effects of central irisin administration on the uncoupling proteins in rat brain. Neurosci. Lett. 2016, 618, 6–13. [Google Scholar] [CrossRef]
- Huang, P.S.; Son, J.H.; Abbott, L.C.; Winzer-Serhan, U.H. Regulated expression of neuronal SIRT1 and related genes by aging and neuronal beta2-containing nicotinic cholinergic receptors. Neuroscience 2011, 196, 189–202. [Google Scholar] [CrossRef]
- Vidal-Puig, A.J.; Grujic, D.; Zhang, C.Y.; Hagen, T.; Boss, O.; Ido, Y.; Szczepanik, A.; Wade, J.; Mootha, V.; Cortright, R.; et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J. Biol. Chem. 2000, 275, 16258–16266. [Google Scholar] [CrossRef]
- Gorgoglione, R.; Porcelli, V.; Santoro, A.; Daddabbo, L.; Vozza, A.; Monne, M.; di Noia, M.A.; Palmieri, L.; Fiermonte, G.; Palmieri, F. The human uncoupling proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) transport sulfur oxyanions, phosphate and dicarboxylates. Biochim. Biophys. Acta 2019, 1860, 724–733. [Google Scholar] [CrossRef]
- Ko, E.Y.; Sabanegh, E.S., Jr.; Agarwal, A. Male infertility testing: Reactive oxygen species and antioxidant capacity. Fertil. Steril. 2014, 102, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Okamatsu-Ogura, Y.; Fukano, K.; Tsubota, A.; Uozumi, A.; Terao, A.; Kimura, K.; Saito, M. Thermogenic ability of uncoupling protein 1 in beige adipocytes in mice. PLoS ONE 2013, 8, e84229. [Google Scholar] [CrossRef]
- Nibbelink, M.; Moulin, K.; Arnaud, E.; Duval, C.; Penicaud, L.; Casteilla, L. Brown fat UCP1 is specifically expressed in uterine longitudinal smooth muscle cells. J. Biol. Chem. 2001, 276, 47291–47295. [Google Scholar] [CrossRef]
- Carroll, A.M.; Haines, L.R.; Pearson, T.W.; Fallon, P.G.; Walsh, C.M.; Brennan, C.M.; Breen, E.P.; Porter, R.K. Identification of a functioning mitochondrial uncoupling protein 1 in thymus. J. Biol. Chem. 2005, 280, 15534–15543. [Google Scholar] [CrossRef] [PubMed]
- Frontini, A.; Rousset, S.; Cassard-Doulcier, A.M.; Zingaretti, C.; Ricquier, D.; Cinti, S. Thymus uncoupling protein 1 is exclusive to typical brown adipocytes and is not found in thymocytes. J. Histochem. Cytochem. 2007, 55, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef]
- Darley-Usmar, V. The powerhouse takes control of the cell; The role of mitochondria in signal transduction. Free Radic. Biol. Med. 2004, 37, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D.; Bouillaud, F.; Toumelin, P.; Mory, G.; Bazin, R.; Arch, J.; Penicaud, L. Expression of uncoupling protein mRNA in thermogenic or weakly thermogenic brown adipose tissue. Evidence for a rapid beta-adrenoreceptor-mediated and transcriptionally regulated step during activation of thermogenesis. J. Biol. Chem. 1986, 261, 13905–13910. [Google Scholar] [CrossRef]
- Busiello, R.A.; Savarese, S.; Lombardi, A. Mitochondrial uncoupling proteins and energy metabolism. Front. Physiol. 2015, 6, 36. [Google Scholar] [CrossRef]
- Rabelo, R.; Schifman, A.; Rubio, A.; Sheng, X.; Silva, J.E. Delineation of thyroid hormone-responsive sequences within a critical enhancer in the rat uncoupling protein gene. Endocrinology 1995, 136, 1003–1013. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Alvarez, R.; de Andres, J.; Yubero, P.; Vinas, O.; Mampel, T.; Iglesias, R.; Giralt, M.; Villarroya, F. A novel regulatory pathway of brown fat thermogenesis. Retinoic acid is a transcriptional activator of the mitochondrial uncoupling protein gene. J. Biol. Chem. 1995, 270, 5666–5673. [Google Scholar] [CrossRef]
- Gong, D.W.; He, Y.; Karas, M.; Reitman, M. Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, beta3-adrenergic agonists, and leptin. J. Biol. Chem. 1997, 272, 24129–24132. [Google Scholar] [CrossRef]
- Rial, E.; Muga, A.; Valpuesta, J.M.; Arrondo, J.L.; Goni, F.M. Infrared spectroscopic studies of detergent-solubilized uncoupling protein from brown-adipose-tissue mitochondria. Eur. J. Biochem. 1990, 188, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, R.E.; Dembski, M.; Weng, X.; Deng, N.; Shyjan, A.W.; Gimeno, C.J.; Iris, F.; Ellis, S.J.; Woolf, E.A.; Tartaglia, L.A. Cloning and characterization of an uncoupling protein homolog: A potential molecular mediator of human thermogenesis. Diabetes 1997, 46, 900–906. [Google Scholar] [CrossRef]
- Krauss, S.; Zhang, C.Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J. Clin. Investig. 2003, 112, 1831–1842. [Google Scholar] [CrossRef]
- Horimoto, M.; Fulop, P.; Derdak, Z.; Wands, J.R.; Baffy, G. Uncoupling protein-2 deficiency promotes oxidant stress and delays liver regeneration in mice. Hepatology 2004, 39, 386–392. [Google Scholar] [CrossRef]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef]
- Lee, K.U.; Lee, I.K.; Han, J.; Song, D.K.; Kim, Y.M.; Song, H.S.; Kim, H.S.; Lee, W.J.; Koh, E.H.; Song, K.H.; et al. Effects of recombinant adenovirus-mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ. Res. 2005, 96, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Alves-Guerra, M.C.; Rousset, S.; Pecqueur, C.; Mallat, Z.; Blanc, J.; Tedgui, A.; Bouillaud, F.; Cassard-Doulcier, A.M.; Ricquier, D.; Miroux, B. Bone marrow transplantation reveals the in vivo expression of the mitochondrial uncoupling protein 2 in immune and nonimmune cells during inflammation. J. Biol. Chem. 2003, 278, 42307–42312. [Google Scholar] [CrossRef] [PubMed]
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e41–e52. [Google Scholar] [CrossRef] [PubMed]
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003, 9, 1062–1068. [Google Scholar] [CrossRef]
- Horvath, B.; Spies, C.; Horvath, G.; Kox, W.J.; Miyamoto, S.; Barry, S.; Warden, C.H.; Bechmann, I.; Diano, S.; Heemskerk, J.; et al. Uncoupling protein 2 (UCP2) lowers alcohol sensitivity and pain threshold. Biochem. Pharmacol. 2002, 64, 369–374. [Google Scholar] [CrossRef]
- Nakase, T.; Yoshida, Y.; Nagata, K. Amplified expression of uncoupling proteins in human brain ischemic lesions. Neuropathology 2007, 27, 442–447. [Google Scholar] [CrossRef] [PubMed]
- McLeod, C.J.; Aziz, A.; Hoyt, R.F., Jr.; McCoy, J.P., Jr.; Sack, M.N. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J. Biol. Chem. 2005, 280, 33470–33476. [Google Scholar] [CrossRef]
- Kizaki, T.; Suzuki, K.; Hitomi, Y.; Taniguchi, N.; Saitoh, D.; Watanabe, K.; Onoe, K.; Day, N.K.; Good, R.A.; Ohno, H. Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. USA 2002, 99, 9392–9397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Baffy, G.; Perret, P.; Krauss, S.; Peroni, O.; Grujic, D.; Hagen, T.; Vidal-Puig, A.J.; Boss, O.; Kim, Y.B.; et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001, 105, 745–755. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; de Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Brand, M.D. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem. Sci. 2010, 35, 298–307. [Google Scholar] [CrossRef]
- Hurtaud, C.; Gelly, C.; Chen, Z.; Levi-Meyrueis, C.; Bouillaud, F. Glutamine stimulates translation of uncoupling protein 2mRNA. Cell. Mol. Life Sci. 2007, 64, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Shimabukuro, M.; Koyama, K.; Lee, Y.; Wang, M.Y.; Trieu, F.; Newgard, C.B.; Unger, R.H. Induction by leptin of uncoupling protein-2 and enzymes of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1997, 94, 6386–6390. [Google Scholar] [CrossRef]
- Brand, M.D.; Pamplona, R.; Portero-Otin, M.; Requena, J.R.; Roebuck, S.J.; Buckingham, J.A.; Clapham, J.C.; Cadenas, S. Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem. J. 2002, 368, 597–603. [Google Scholar] [CrossRef]
- Talbot, D.A.; Lambert, A.J.; Brand, M.D. Production of endogenous matrix superoxide from mitochondrial complex I leads to activation of uncoupling protein 3. FEBS Lett. 2004, 556, 111–115. [Google Scholar] [CrossRef]
- Vincent, A.M.; Olzmann, J.A.; Brownlee, M.; Sivitz, W.I.; Russell, J.W. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes 2004, 53, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S.; Echtay, K.S.; Harper, J.A.; Jekabsons, M.B.; Buckingham, J.A.; Grau, E.; Abuin, A.; Chapman, H.; Clapham, J.C.; Brand, M.D. The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. J. Biol. Chem. 2002, 277, 2773–2778. [Google Scholar] [CrossRef]
- Liu, Q.; Bai, C.; Chen, F.; Wang, R.; MacDonald, T.; Gu, M.; Zhang, Q.; Morsy, M.A.; Caskey, C.T. Uncoupling protein-3: A muscle-specific gene upregulated by leptin in ob/ob mice. Gene 1998, 207, 1–7. [Google Scholar] [CrossRef]
- Larkin, S.; Mull, E.; Miao, W.; Pittner, R.; Albrandt, K.; Moore, C.; Young, A.; Denaro, M.; Beaumont, K. Regulation of the third member of the uncoupling protein family, UCP3, by cold and thyroid hormone. Biochem. Biophys. Res. Commun. 1997, 240, 222–227. [Google Scholar] [CrossRef]
- Boss, O.; Samec, S.; Kuhne, F.; Bijlenga, P.; Assimacopoulos-Jeannet, F.; Seydoux, J.; Giacobino, J.P.; Muzzin, P. Uncoupling protein-3 expression in rodent skeletal muscle is modulated by food intake but not by changes in environmental temperature. J. Biol. Chem. 1998, 273, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Millet, L.; Vidal, H.; Andreelli, F.; Larrouy, D.; Riou, J.P.; Ricquier, D.; Laville, M.; Langin, D. Increased uncoupling protein-2 and -3 mRNA expression during fasting in obese and lean humans. J. Clin. Investig. 1997, 100, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Jing, X.; Tian, X.; Qin, M.C.; Xu, Z.H.; Wu, D.P.; Zhong, Z.G. Neuroprotective Properties of Panax notoginseng Saponins via Preventing Oxidative Stress Injury in SAMP8 Mice. Evid Based Complement. Alternat. Med. 2017, 2017, 8713561. [Google Scholar] [CrossRef]
- Xu, S.; Yang, X.; Qian, Y.; Xiao, Q. Parkinson’s disease-related DJ-1 modulates the expression of uncoupling protein 4 against oxidative stress. J. Neurochem. 2018, 145, 312–322. [Google Scholar] [CrossRef]
- Xu, Y.; Peng, S.; Cao, X.; Qian, S.; Shen, S.; Luo, J.; Zhang, X.; Sun, H.; Shen, W.L.; Jia, W.; et al. High doses of butyrate induce a reversible body temperature drop through transient proton leak in mitochondria of brain neurons. Life Sci. 2021, 278, 119614. [Google Scholar] [CrossRef]
- Viggiano, E.; Monda, V.; Messina, A.; Moscatelli, F.; Valenzano, A.; Tafuri, D.; Cibelli, G.; de Luca, B.; Messina, G.; Monda, M. Cortical spreading depression produces a neuroprotective effect activating mitochondrial uncoupling protein-5. Neuropsychiatr. Dis. Treat. 2016, 12, 1705–1710. [Google Scholar] [CrossRef]
- Pichiule, P.; Chavez, J.C.; LaManna, J.C. Oxygen and oxidative stress modulate the expression of uncoupling protein-5 in vitro and in vivo. Adv. Exp. Med. Biol. 2003, 540, 103–107. [Google Scholar] [CrossRef]
- Xie, Z.Z.; Liu, Y.; Bian, J.S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef] [PubMed]
- Nohara, K.; Tateishi, Y.; Suzuki, T.; Okamura, K.; Murai, H.; Takumi, S.; Maekawa, F.; Nishimura, N.; Kobori, M.; Ito, T. Late-onset increases in oxidative stress and other tumorigenic activities and tumors with a Ha-ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicol. Sci. 2012, 129, 293–304. [Google Scholar] [CrossRef]
- Mydin, R.; Okekpa, S.I. Reactive oxygen species, cellular redox homeostasis and cancer. In Homeostasis-An Integrated Vision; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Papa, S.; Skulachev, V.P. Reactive oxygen species, mitochondria, apoptosis and aging. Mol. Cell. Biochem. 1997, 174, 305–319. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Azzu, V.; Jastroch, M.; Divakaruni, A.S.; Brand, M.D. The regulation and turnover of mitochondrial uncoupling proteins. Biochim. Biophys. Acta 2010, 1797, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Dlaskova, A.; Clarke, K.J.; Porter, R.K. The role of UCP 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim. Biophys. Acta 2010, 1797, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Oelkrug, R.; Kutschke, M.; Meyer, C.W.; Heldmaier, G.; Jastroch, M. Uncoupling protein 1 decreases superoxide production in brown adipose tissue mitochondria. J. Biol. Chem. 2010, 285, 21961–21968. [Google Scholar] [CrossRef]
- Stier, A.; Bize, P.; Habold, C.; Bouillaud, F.; Massemin, S.; Criscuolo, F. Mitochondrial uncoupling prevents cold-induced oxidative stress: A case study using UCP1 knockout mice. J. Exp. Biol. 2014, 217, 624–630. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Petrovic, N.; Kramarova, T.V.; Hoeks, J.; Cannon, B.; Nedergaard, J. UCP1 and defense against oxidative stress. 4-Hydroxy-2-nonenal effects on brown fat mitochondria are uncoupling protein 1-independent. J. Biol. Chem. 2006, 281, 13882–13893. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vrbacky, M.; Pecinova, A.; Kalinovich, A.V.; Drahota, Z.; Houstek, J.; Mracek, T.; Cannon, B.; Nedergaard, J. ROS production in brown adipose tissue mitochondria: The question of UCP1-dependence. Biochim. Biophys. Acta 2014, 1837, 2017–2030. [Google Scholar] [CrossRef]
- Jun, Z.; Ibrahim, M.M.; Dezheng, G.; Bo, Y.; Qiong, W.; Yuan, Z. UCP2 protects against amyloid beta toxicity and oxidative stress in primary neuronal culture. Biomed. Pharmacother. 2015, 74, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, L.; Xu, X.; Vicaut, E.; Sercombe, R. Both ischemic preconditioning and ghrelin administration protect hippocampus from ischemia/reperfusion and upregulate uncoupling protein-2. BMC Physiol. 2009, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Wu, C.A.; Wu, K.L.; Ho, Y.H.; Chang, A.Y.; Chan, J.Y. Transcriptional upregulation of mitochondrial uncoupling protein 2 protects against oxidative stress-associated neurogenic hypertension. Circ. Res. 2009, 105, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Robson-Doucette, C.A.; Wheeler, M.B. Uncoupling protein 2 regulates reactive oxygen species formation in islets and influences susceptibility to diabetogenic action of streptozotocin. J. Endocrinol. 2009, 203, 33–43. [Google Scholar] [CrossRef][Green Version]
- Robson-Doucette, C.A.; Sultan, S.; Allister, E.M.; Wikstrom, J.D.; Koshkin, V.; Bhattacharjee, A.; Prentice, K.J.; Sereda, S.B.; Shirihai, O.S.; Wheeler, M.B. Beta-cell uncoupling protein 2 regulates reactive oxygen species production, which influences both insulin and glucagon secretion. Diabetes 2011, 60, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Daniel, K.W.; Liu, D.; Lyght, O.; Edelstein, D.; Brownlee, M.; Corkey, B.E.; Collins, S. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology 2009, 150, 3040–3048. [Google Scholar] [CrossRef]
- Blanc, J.; Alves-Guerra, M.C.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation 2003, 107, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Uncoupling protein-2 and cancer. Mitochondrion 2010, 10, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Adjeitey, C.N.; Harper, M.E. Genipin-induced inhibition of uncoupling protein-2 sensitizes drug-resistant cancer cells to cytotoxic agents. PLoS ONE 2010, 5, e13289. [Google Scholar] [CrossRef]
- Anderson, E.J.; Yamazaki, H.; Neufer, P.D. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J. Biol. Chem. 2007, 282, 31257–31266. [Google Scholar] [CrossRef] [PubMed]
- Talbot, D.A.; Brand, M.D. Uncoupling protein 3 protects aconitase against inactivation in isolated skeletal muscle mitochondria. Biochim. Biophys. Acta 2005, 1709, 150–156. [Google Scholar] [CrossRef][Green Version]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem. 2002, 277, 47129–47135. [Google Scholar] [CrossRef]
- Echtay, K.S.; Pakay, J.L.; Esteves, T.C.; Brand, M.D. Hydroxynonenal and uncoupling proteins: A model for protection against oxidative damage. Biofactors 2005, 24, 119–130. [Google Scholar] [CrossRef]
- Murphy, M.P.; Echtay, K.S.; Blaikie, F.H.; Asin-Cayuela, J.; Cocheme, H.M.; Green, K.; Buckingham, J.A.; Taylor, E.R.; Hurrell, F.; Hughes, G.; et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: Studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. J. Biol. Chem. 2003, 278, 48534–48545. [Google Scholar] [CrossRef]
- Toime, L.J.; Brand, M.D. Uncoupling protein-3 lowers reactive oxygen species production in isolated mitochondria. Free Radic. Biol. Med. 2010, 49, 606–611. [Google Scholar] [CrossRef]
- Ozcan, C.; Palmeri, M.; Horvath, T.L.; Russell, K.S.; Russell, R.R., 3rd. Role of uncoupling protein 3 in ischemia-reperfusion injury, arrhythmias, and preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1192–H1200. [Google Scholar] [CrossRef]
- MacLellan, J.D.; Gerrits, M.F.; Gowing, A.; Smith, P.J.; Wheeler, M.B.; Harper, M.E. Physiological increases in uncoupling protein 3 augment fatty acid oxidation and decrease reactive oxygen species production without uncoupling respiration in muscle cells. Diabetes 2005, 54, 2343–2350. [Google Scholar] [CrossRef]
- Jiang, N.; Zhang, G.; Bo, H.; Qu, J.; Ma, G.; Cao, D.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Upregulation of uncoupling protein-3 in skeletal muscle during exercise: A potential antioxidant function. Free Radic. Biol. Med. 2009, 46, 138–145. [Google Scholar] [CrossRef]
- Nabben, M.; Hoeks, J.; Briede, J.J.; Glatz, J.F.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. The effect of UCP3 overexpression on mitochondrial ROS production in skeletal muscle of young versus aged mice. FEBS Lett. 2008, 582, 4147–4152. [Google Scholar] [CrossRef]
- Goglia, F.; Skulachev, V.P. A function for novel uncoupling proteins: Antioxidant defense of mitochondrial matrix by translocating fatty acid peroxides from the inner to the outer membrane leaflet. FASEB J. 2003, 17, 1585–1591. [Google Scholar] [CrossRef]
- Carrageta, D.F.; Oliveira, P.F.; Alves, M.G.; Monteiro, M.P. Obesity and male hypogonadism: Tales of a vicious cycle. Obes. Rev. 2019, 20, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Betz, M.J.; Enerback, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, B.; Mirzaei, K.; Maghbooli, Z.; Keshavarz, S.A.; Hossein-Nezhad, A. Compare the resting metabolic rate status in the healthy metabolically obese with the unhealthy metabolically obese participants. J. Nutr. Intermed. Metab. 2016, 6, 48–53. [Google Scholar] [CrossRef][Green Version]
- Rolfe, D.F.; Brand, M.D. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am. J. Physiol. 1996, 271, C1380–C1389. [Google Scholar] [CrossRef]
- Harper, M.E.; Green, K.; Brand, M.D. The efficiency of cellular energy transduction and its implications for obesity. Annu. Rev. Nutr. 2008, 28, 13–33. [Google Scholar] [CrossRef]
- Abumrad, N.A. The Liver as a Hub in Thermogenesis. Cell Metab. 2017, 26, 454–455. [Google Scholar] [CrossRef]
- Enerback, S.; Jacobsson, A.; Simpson, E.M.; Guerra, C.; Yamashita, H.; Harper, M.E.; Kozak, L.P. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 1997, 387, 90–94. [Google Scholar] [CrossRef]
- Liu, X.; Rossmeisl, M.; McClaine, J.; Riachi, M.; Harper, M.E.; Kozak, L.P. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J. Clin. Investig. 2003, 111, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Kontani, Y.; Wang, Y.; Kimura, K.; Inokuma, K.I.; Saito, M.; Suzuki-Miura, T.; Wang, Z.; Sato, Y.; Mori, N.; Yamashita, H. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell 2005, 4, 147–155. [Google Scholar] [CrossRef]
- Feldmann, H.M.; Golozoubova, V.; Cannon, B.; Nedergaard, J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009, 9, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.A.; Maurya, S.K.; Bal, N.C.; Kozak, L.; Periasamy, M. Sarcolipin and uncoupling protein 1 play distinct roles in diet-induced thermogenesis and do not compensate for one another. Obesity 2016, 24, 1430–1433. [Google Scholar] [CrossRef] [PubMed]
- Von Essen, G.; Lindsund, E.; Cannon, B.; Nedergaard, J. Adaptive facultative diet-induced thermogenesis in wild-type but not in UCP1-ablated mice. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E515–E527. [Google Scholar] [CrossRef] [PubMed]
- Luijten, I.H.N.; Feldmann, H.M.; von Essen, G.; Cannon, B.; Nedergaard, J. In the absence of UCP1-mediated diet-induced thermogenesis, obesity is augmented even in the obesity-resistant 129S mouse strain. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E729–E740. [Google Scholar] [CrossRef] [PubMed]
- Almind, K.; Manieri, M.; Sivitz, W.I.; Cinti, S.; Kahn, C.R. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
- Winn, N.C.; Vieira-Potter, V.J.; Gastecki, M.L.; Welly, R.J.; Scroggins, R.J.; Zidon, T.M.; Gaines, T.L.; Woodford, M.L.; Karasseva, N.G.; Kanaley, J.A.; et al. Loss of UCP1 exacerbates Western diet-induced glycemic dysregulation independent of changes in body weight in female mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R74–R84. [Google Scholar] [CrossRef]
- Chondronikola, M.; Volpi, E.; Borsheim, E.; Porter, C.; Annamalai, P.; Enerback, S.; Lidell, M.E.; Saraf, M.K.; Labbe, S.M.; Hurren, N.M.; et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 2014, 63, 4089–4099. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Inokuma, K.; Ogura-Okamatsu, Y.; Toda, C.; Kimura, K.; Yamashita, H.; Saito, M. Uncoupling protein 1 is necessary for norepinephrine-induced glucose utilization in brown adipose tissue. Diabetes 2005, 54, 1385–1391. [Google Scholar] [CrossRef]
- Thoonen, R.; Ernande, L.; Cheng, J.; Nagasaka, Y.; Yao, V.; Miranda-Bezerra, A.; Chen, C.; Chao, W.; Panagia, M.; Sosnovik, D.E.; et al. Functional brown adipose tissue limits cardiomyocyte injury and adverse remodeling in catecholamine-induced cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 84, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Matsushita, M.; Yoneshiro, T.; Okamatsu-Ogura, Y. Brown Adipose Tissue, Diet-Induced Thermogenesis, and Thermogenic Food Ingredients: From Mice to Men. Front. Endocrinol. 2020, 11, 222. [Google Scholar] [CrossRef]
- Fernandez Vazquez, G.; Reiter, R.J.; Agil, A. Melatonin increases brown adipose tissue mass and function in Zucker diabetic fatty rats: Implications for obesity control. J. Pineal Res. 2018, 64, e12472. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, J.; Hodny, Z.; Rossmeisl, M.; Syrovy, I.; Kozak, L.P. Reduction of dietary obesity in aP2-Ucp transgenic mice: Physiology and adipose tissue distribution. Am. J. Physiol. 1996, 270, E768–E775. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.; Keipert, S.; Rossmeisl, M.; Kopecky, J. Augmenting energy expenditure by mitochondrial uncoupling: A role of AMP-activated protein kinase. Genes Nutr. 2012, 7, 369–386. [Google Scholar] [CrossRef]
- Li, B.; Nolte, L.A.; Ju, J.S.; Han, D.H.; Coleman, T.; Holloszy, J.O.; Semenkovich, C.F. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat. Med. 2000, 6, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Adjeitey, C.N.; Mailloux, R.J.; Dekemp, R.A.; Harper, M.E. Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E405–E415. [Google Scholar] [CrossRef]
- Keipert, S.; Ost, M.; Chadt, A.; Voigt, A.; Ayala, V.; Portero-Otin, M.; Pamplona, R.; Al-Hasani, H.; Klaus, S. Skeletal muscle uncoupling-induced longevity in mice is linked to increased substrate metabolism and induction of the endogenous antioxidant defense system. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E495–E506. [Google Scholar] [CrossRef]
- Neschen, S.; Katterle, Y.; Richter, J.; Augustin, R.; Scherneck, S.; Mirhashemi, F.; Schurmann, A.; Joost, H.G.; Klaus, S. Uncoupling protein 1 expression in murine skeletal muscle increases AMPK activation, glucose turnover, and insulin sensitivity in vivo. Physiol. Genom. 2008, 33, 333–340. [Google Scholar] [CrossRef][Green Version]
- Ost, M.; Werner, F.; Dokas, J.; Klaus, S.; Voigt, A. Activation of AMPKalpha2 is not crucial for mitochondrial uncoupling-induced metabolic effects but required to maintain skeletal muscle integrity. PLoS ONE 2014, 9, e94689. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Fink, B.D.; Dillon, J.S.; Sivitz, W.I. Effects of adenoviral overexpression of uncoupling protein-2 and -3 on mitochondrial respiration in insulinoma cells. Endocrinology 2001, 142, 249–256. [Google Scholar] [CrossRef][Green Version]
- Zhang, C.Y.; Parton, L.E.; Ye, C.P.; Krauss, S.; Shen, R.; Lin, C.T.; Porco, J.A., Jr.; Lowell, B.B. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 2006, 3, 417–427. [Google Scholar] [CrossRef]
- De Souza, C.T.; Araujo, E.P.; Stoppiglia, L.F.; Pauli, J.R.; Ropelle, E.; Rocco, S.A.; Marin, R.M.; Franchini, K.G.; Carvalheira, J.B.; Saad, M.J.; et al. Inhibition of UCP2 expression reverses diet-induced diabetes mellitus by effects on both insulin secretion and action. FASEB J. 2007, 21, 1153–1163. [Google Scholar] [CrossRef]
- Saleh, M.C.; Wheeler, M.B.; Chan, C.B. Endogenous islet uncoupling protein-2 expression and loss of glucose homeostasis in ob/ob mice. J. Endocrinol. 2006, 190, 659–667. [Google Scholar] [CrossRef]
- Pi, J.; Collins, S. Reactive oxygen species and uncoupling protein 2 in pancreatic beta-cell function. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 141–148. [Google Scholar] [CrossRef]
- Affourtit, C.; Brand, M.D. Uncoupling protein-2 contributes significantly to high mitochondrial proton leak in INS-1E insulinoma cells and attenuates glucose-stimulated insulin secretion. Biochem. J. 2008, 409, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Diano, S.; Horvath, T.L. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 2012, 18, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Affourtit, C.; Jastroch, M.; Brand, M.D. Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Clapham, J.C.; Arch, J.R.; Chapman, H.; Haynes, A.; Lister, C.; Moore, G.B.; Piercy, V.; Carter, S.A.; Lehner, I.; Smith, S.A.; et al. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 2000, 406, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Son, C.; Hosoda, K.; Ishihara, K.; Bevilacqua, L.; Masuzaki, H.; Fushiki, T.; Harper, M.E.; Nakao, K. Reduction of diet-induced obesity in transgenic mice overexpressing uncoupling protein 3 in skeletal muscle. Diabetologia 2004, 47, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Costford, S.R.; Chaudhry, S.N.; Salkhordeh, M.; Harper, M.E. Effects of the presence, absence, and overexpression of uncoupling protein-3 on adiposity and fuel metabolism in congenic mice. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1304–E1312. [Google Scholar] [CrossRef]
- Aguer, C.; Fiehn, O.; Seifert, E.L.; Bezaire, V.; Meissen, J.K.; Daniels, A.; Scott, K.; Renaud, J.M.; Padilla, M.; Bickel, D.R.; et al. Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete beta-oxidation. FASEB J. 2013, 27, 4213–4225. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.W.; Monemdjou, S.; Gavrilova, O.; Leon, L.R.; Marcus-Samuels, B.; Chou, C.J.; Everett, C.; Kozak, L.P.; Li, C.; Deng, C.; et al. Lack of obesity and normal response to fasting and thyroid hormone in mice lacking uncoupling protein-3. J. Biol. Chem. 2000, 275, 16251–16257. [Google Scholar] [CrossRef]
- Lomax, T.M.; Ashraf, S.; Yilmaz, G.; Harmancey, R. Loss of Uncoupling Protein 3 Attenuates Western Diet-Induced Obesity, Systemic Inflammation, and Insulin Resistance in Rats. Obesity 2020, 28, 1687–1697. [Google Scholar] [CrossRef]
- Choi, C.S.; Fillmore, J.J.; Kim, J.K.; Liu, Z.X.; Kim, S.; Collier, E.F.; Kulkarni, A.; Distefano, A.; Hwang, Y.J.; Kahn, M.; et al. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J. Clin. Investig. 2007, 117, 1995–2003. [Google Scholar] [CrossRef]
- Costford, S.R.; Chaudhry, S.N.; Crawford, S.A.; Salkhordeh, M.; Harper, M.E. Long-term high-fat feeding induces greater fat storage in mice lacking UCP3. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1018–E1024. [Google Scholar] [CrossRef]
- Senese, R.; Valli, V.; Moreno, M.; Lombardi, A.; Busiello, R.A.; Cioffi, F.; Silvestri, E.; Goglia, F.; Lanni, A.; de Lange, P. Uncoupling protein 3 expression levels influence insulin sensitivity, fatty acid oxidation, and related signaling pathways. Pflugers Arch. 2011, 461, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Hesselink, M.K.; Blaak, E.E.; Borghouts, L.B.; Schaart, G.; Saris, W.H.; Keizer, H.A. Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2001, 50, 2870–2873. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schrauwen, P.; Mensink, M.; Schaart, G.; Moonen-Kornips, E.; Sels, J.P.; Blaak, E.E.; Russell, A.P.; Hesselink, M.K. Reduced skeletal muscle uncoupling protein-3 content in prediabetic subjects and type 2 diabetic patients: Restoration by rosiglitazone treatment. J. Clin. Endocrinol. Metab. 2006, 91, 1520–1525. [Google Scholar] [CrossRef][Green Version]
- Mensink, M.; Hesselink, M.K.; Borghouts, L.B.; Keizer, H.; Moonen-Kornips, E.; Schaart, G.; Blaak, E.E.; Schrauwen, P. Skeletal muscle uncoupling protein-3 restores upon intervention in the prediabetic and diabetic state: Implications for diabetes pathogenesis? Diabetes Obes. Metab. 2007, 9, 594–596. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, J. The role of oxygen in the metabolism and motility of human spermatozoa. Am. J. Physiol. 1943, 138, 0512–0518. [Google Scholar] [CrossRef]
- Tremellen, K. Oxidative stress and male infertility—A clinical perspective. Hum. Reprod. Update 2008, 14, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Shekarriz, M.; Thomas, A.J., Jr.; Agarwal, A. Incidence and level of seminal reactive oxygen species in normal men. Urology 1995, 45, 103–107. [Google Scholar] [CrossRef]
- Ochsendorf, F.R.; Thiele, J.; Fuchs, J.; Schuttau, H.; Freisleben, H.J.; Buslau, M.; Milbradt, R. Chemiluminescence in semen of infertile men. Andrologia 1994, 26, 289–293. [Google Scholar] [CrossRef]
- Zini, A.; de Lamirande, E.; Gagnon, C. Reactive oxygen species in semen of infertile patients: Levels of superoxide dismutase- and catalase-like activities in seminal plasma and spermatozoa. Int. J. Androl. 1993, 16, 183–188. [Google Scholar] [CrossRef]
- Iwasaki, A.; Gagnon, C. Formation of reactive oxygen species in spermatozoa of infertile patients. Fertil. Steril. 1992, 57, 409–416. [Google Scholar] [CrossRef]
- Agarwal, A.; Said, T.M.; Bedaiwy, M.A.; Banerjee, J.; Alvarez, J.G. Oxidative stress in an assisted reproductive techniques setting. Fertil. Steril. 2006, 86, 503–512. [Google Scholar] [CrossRef]
- Agarwal, A.; Sharma, R.K.; Sharma, R.; Assidi, M.; Abuzenadah, A.M.; Alshahrani, S.; Durairajanayagam, D.; Sabanegh, E. Characterizing semen parameters and their association with reactive oxygen species in infertile men. Reprod. Biol. Endocrinol. 2014, 12, 33. [Google Scholar] [CrossRef]
- Agarwal, A.; Saleh, R.A.; Bedaiwy, M.A. Role of reactive oxygen species in the pathophysiology of human reproduction. Fertil. Steril. 2003, 79, 829–843. [Google Scholar] [CrossRef]
- Said, T.M.; Agarwal, A.; Sharma, R.K.; Mascha, E.; Sikka, S.C.; Thomas, A.J., Jr. Human sperm superoxide anion generation and correlation with semen quality in patients with male infertility. Fertil. Steril. 2004, 82, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.; Saleh, R.A.; Sharma, R.K.; Lewis-Jones, I.; Esfandiari, N.; Thomas, A.J., Jr.; Agarwal, A. Novel association between sperm reactive oxygen species production, sperm morphological defects, and the sperm deformity index. Fertil. Steril. 2004, 81, 349–354. [Google Scholar] [CrossRef]
- Moustafa, M.H.; Sharma, R.K.; Thornton, J.; Mascha, E.; Abdel-Hafez, M.A.; Thomas, A.J., Jr.; Agarwal, A. Relationship between ROS production, apoptosis and DNA denaturation in spermatozoa from patients examined for infertility. Hum. Reprod. 2004, 19, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Kaune, H.; Parodi, D.; Madariaga, M.; Rios, R.; Morales, I.; Castro, A. Increased sperm DNA damage in patients with varicocele: Relationship with seminal oxidative stress. Hum. Reprod. 2006, 21, 986–993. [Google Scholar] [CrossRef]
- De Lamirande, E.; Jiang, H.; Zini, A.; Kodama, H.; Gagnon, C. Reactive oxygen species and sperm physiology. Rev. Reprod. 1997, 2, 48–54. [Google Scholar] [CrossRef]
- Du Plessis, S.S.; Agarwal, A.; Halabi, J.; Tvrda, E. Contemporary evidence on the physiological role of reactive oxygen species in human sperm function. J. Assist. Reprod. Genet. 2015, 32, 509–520. [Google Scholar] [CrossRef]
- De Lamirande, E.; Gagnon, C. Human sperm hyperactivation and capacitation as parts of an oxidative process. Free Radic. Biol. Med. 1993, 14, 157–166. [Google Scholar] [CrossRef]
- Aitken, R.J.; Irvine, D.S.; Wu, F.C. Prospective analysis of sperm-oocyte fusion and reactive oxygen species generation as criteria for the diagnosis of infertility. Am. J. Obstet. Gynecol. 1991, 164, 542–551. [Google Scholar] [CrossRef]
- Dias, T.R.; Martin-Hidalgo, D.; Silva, B.M.; Oliveira, P.F.; Alves, M.G. Endogenous and exogenous antioxidants as a tool to ameliorate male infertility induced by reactive oxygen species. Antioxid. Redox Signal. 2020, 33, 767–785. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, G.; d’Agostino, G.; Mele, E.; Cardito, C.; Esposito, R.; Cimmino, A.; Giarra, A.; Trifuoggi, M.; Raimondo, S.; Notari, T.; et al. Discovery of the Involvement in DNA Oxidative Damage of Human Sperm Nuclear Basic Proteins of Healthy Young Men Living in Polluted Areas. Int. J. Mol. Sci. 2020, 21, 4198. [Google Scholar] [CrossRef]
- Lettieri, G.; Marra, F.; Moriello, C.; Prisco, M.; Notari, T.; Trifuoggi, M.; Giarra, A.; Bosco, L.; Montano, L.; Piscopo, M. Molecular alterations in spermatozoa of a family case living in the land of fires. A first look at possible transgenerational effects of pollutants. Int. J. Mol. Sci. 2020, 21, 6710. [Google Scholar] [CrossRef] [PubMed]
- Martin-Hidalgo, D.; Bragado, M.J.; Batista, A.R.; Oliveira, P.F.; Alves, M.G. Antioxidants and Male Fertility: From Molecular Studies to Clinical Evidence. Antioxidants 2019, 8, 89. [Google Scholar] [CrossRef]
- Athayde, K.S.; Cocuzza, M.; Agarwal, A.; Krajcir, N.; Lucon, A.M.; Srougi, M.; Hallak, J. Development of normal reference values for seminal reactive oxygen species and their correlation with leukocytes and semen parameters in a fertile population. J. Androl. 2007, 28, 613–620. [Google Scholar] [CrossRef]
- Hosseinzadeh Colagar, A.; Karimi, F.; Jorsaraei, S.G. Correlation of sperm parameters with semen lipid peroxidation and total antioxidants levels in astheno- and oligoasheno-teratospermic men. Iran. Red Crescent Med. J. 2013, 15, 780–785. [Google Scholar] [CrossRef]
- De Lamirande, E.; Gagnon, C. Reactive oxygen species and human spermatozoa. I. Effects on the motility of intact spermatozoa and on sperm axonemes. J. Androl. 1992, 13, 368–378. [Google Scholar]
- De Lamirande, E.; Gagnon, C. Reactive oxygen species and human spermatozoa. II. Depletion of adenosine triphosphate plays an important role in the inhibition of sperm motility. J. Androl. 1992, 13, 379–386. [Google Scholar] [PubMed]
- Kurkowska, W.; Bogacz, A.; Janiszewska, M.; Gabrys, E.; Tiszler, M.; Bellanti, F.; Kasperczyk, S.; Machon-Grecka, A.; Dobrakowski, M.; Kasperczyk, A. Oxidative Stress is Associated with Reduced Sperm Motility in Normal Semen. Am. J. Men’s Health 2020, 14, 1557988320939731. [Google Scholar] [CrossRef]
- Lewis, S.E.; Simon, L. Clinical implications of sperm DNA damage. Hum. Fertil. (Camb.) 2010, 13, 201–207. [Google Scholar] [CrossRef]
- Tarozzi, N.; Bizzaro, D.; Flamigni, C.; Borini, A. Clinical relevance of sperm DNA damage in assisted reproduction. Reprod. Biomed. Online 2007, 14, 746–757. [Google Scholar] [CrossRef]
- Cissen, M.; Wely, M.V.; Scholten, I.; Mansell, S.; Bruin, J.P.; Mol, B.W.; Braat, D.; Repping, S.; Hamer, G. Measuring Sperm DNA Fragmentation and Clinical Outcomes of Medically Assisted Reproduction: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0165125. [Google Scholar] [CrossRef]
- La Vignera, S.; Condorelli, R.; Vicari, E.; D’Agata, R.; Calogero, A.E. Diabetes mellitus and sperm parameters. J. Androl. 2012, 33, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.F.; Sousa, M.; Silva, B.M.; Monteiro, M.P.; Alves, M.G. Obesity, energy balance and spermatogenesis. Reproduction 2017, 153, R173–R185. [Google Scholar] [CrossRef]
- Garolla, A.; Torino, M.; Miola, P.; Caretta, N.; Pizzol, D.; Menegazzo, M.; Bertoldo, A.; Foresta, C. Twenty-four-hour monitoring of scrotal temperature in obese men and men with a varicocele as a mirror of spermatogenic function. Hum. Reprod. 2015, 30, 1006–1013. [Google Scholar] [CrossRef]
- Mieusset, R.; Bujan, L.; Mondinat, C.; Mansat, A.; Pontonnier, F.; Grandjean, H. Association of scrotal hyperthermia with impaired spermatogenesis in infertile men. Fertil. Steril. 1987, 48, 1006–1011. [Google Scholar] [CrossRef]
- Håkonsen, L.B.; Thulstrup, A.M.; Aggerholm, A.S.; Olsen, J.; Bonde, J.P.; Andersen, C.Y.; Bungum, M.; Ernst, E.H.; Hansen, M.L.; Ernst, E.H. Does weight loss improve semen quality and reproductive hormones? Results from a cohort of severely obese men. Reprod. Health 2011, 8, 1–8. [Google Scholar] [CrossRef]
- Crisóstomo, L.; Videira, R.A.; Jarak, I.; Starčević, K.; Mašek, T.; Rato, L.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. Diet during early life defines testicular lipid content and sperm quality in adulthood. Am. J. Physiol.-Endocrinol. Metab. 2020, 319, E1061–E1073. [Google Scholar] [CrossRef] [PubMed]
- Crisóstomo, L.; Rato, L.; Jarak, I.; Silva, B.M.; Raposo, J.F.; Batterham, R.L.; Oliveira, P.F.; Alves, M.G. A switch from high-fat to normal diet does not restore sperm quality but prevents metabolic syndrome. Reproduction 2019, 158, 377–387. [Google Scholar] [CrossRef]
- Maresch, C.C.; Stute, D.C.; Alves, M.G.; Oliveira, P.F.; de Kretser, D.M.; Linn, T. Diabetes-induced hyperglycemia impairs male reproductive function: A systematic review. Hum. Reprod. Update 2018, 24, 86–105. [Google Scholar] [CrossRef]
- Alves, M.G.; Martins, A.D.; Rato, L.; Moreira, P.I.; Socorro, S.; Oliveira, P.F. Molecular mechanisms beyond glucose transport in diabetes-related male infertility. Biochim. Biophys. Acta 2013, 1832, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Leisegang, K.; Sengupta, P.; Agarwal, A.; Henkel, R. Obesity and male infertility: Mechanisms and management. Andrologia 2021, 53, e13617. [Google Scholar] [CrossRef] [PubMed]
- Amaral, S.; Oliveira, P.J.; Ramalho-Santos, J. Diabetes and the impairment of reproductive function: Possible role of mitochondria and reactive oxygen species. Curr. Diabetes Rev. 2008, 4, 46–54. [Google Scholar] [CrossRef]
- Kreiter, J.; Rupprecht, A.; Zimmermann, L.; Moschinger, M.; Rokitskaya, T.I.; Antonenko, Y.N.; Gille, L.; Fedorova, M.; Pohl, E.E. Molecular Mechanisms Responsible for Pharmacological Effects of Genipin on Mitochondrial Proteins. Biophys. J. 2019, 117, 1845–1857. [Google Scholar] [CrossRef] [PubMed]



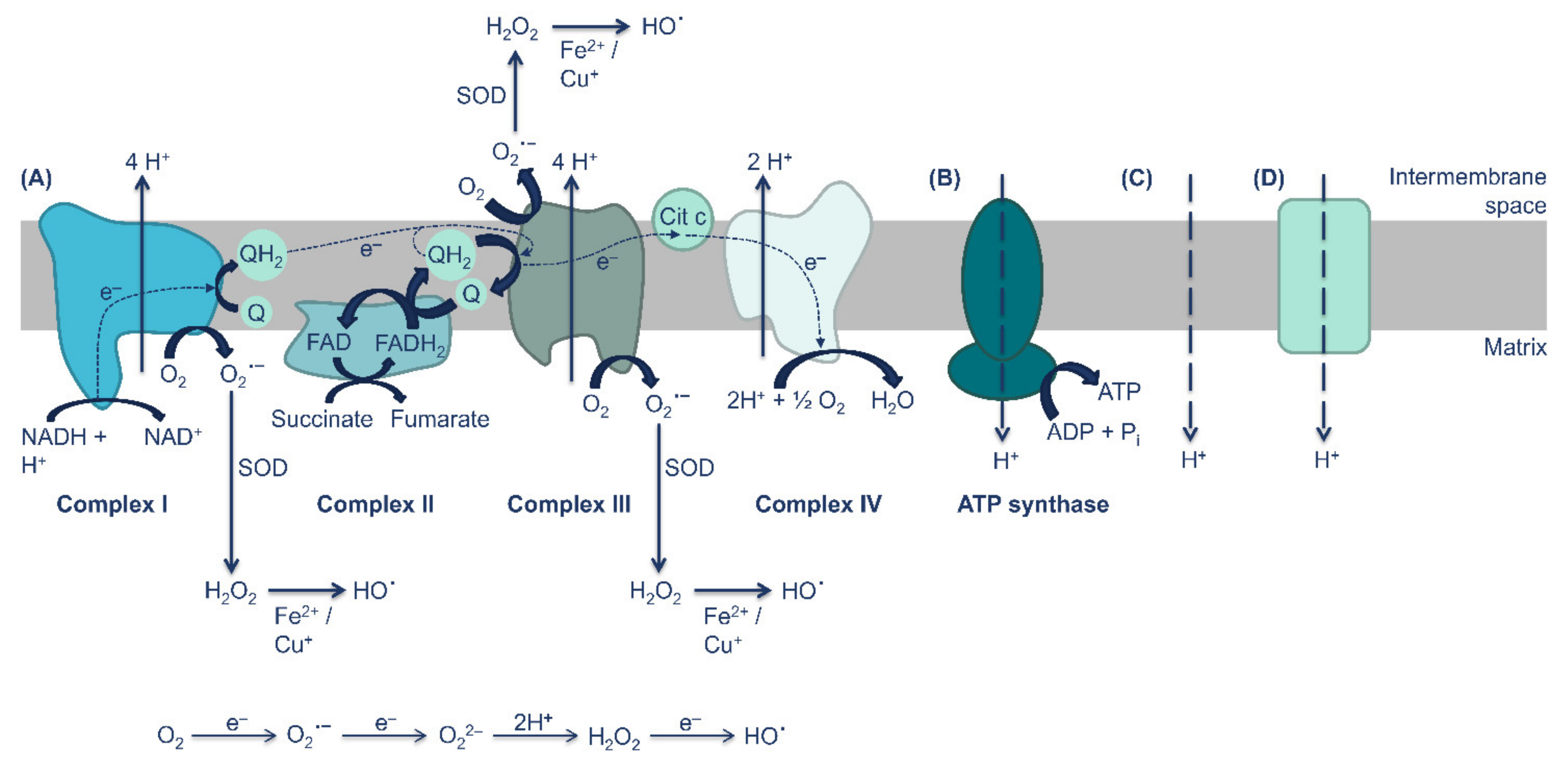{kind=link}
{kind=link}
| Isoform | Localization | Putative Function | References |
|---|---|---|---|
| UCP1 | Human brown adipose tissue, white adipose tissue, keratinocytes, sweat glands, sebum glands, hair follicles, and granular layer of the epidermis | Non-shivering thermogenesis, metabolic and bioenergetic regulation, modulation of ROS production | [21,22,23] |
| Mouse brown adipose tissue, white adipose tissue, and adrenal gland | [24,25,26] | ||
| Rat brown adipose tissue and white adipose tissue | [27,28] | ||
| UCP2 | Human proximal tubular cells, cytotrophoblasts, syncytiotrophoblast, lungs *, keratinocytes *, skin fibroblasts *, spermatozoa, white adipose tissue *, skeletal muscle *, brown adipose tissue *, and pancreatic β-cells *, brain | Modulation of ROS production, insulin sensitivity and secretion, lipid and glucose metabolism, control of mitochondrial Ca2+-uptake, inflammation, immunomodulation | [23,29,30,31,32,33,34,35] |
| Mouse thymocytes, tubular epithelial cells, granulosa cells *, theca cells *, endometrium glandular epithelium cells *, uterine glands *, oviduct mucosa epithelial cells *, mammary gland, stomach, macrophages, splenocytes, B lymphocytes, T lymphocytes, dendritic cells, neutrophils, microglial cells *, neurons *, cardiomyocytes, lungs, testicular germ cells, testicular interstitial cells, white adipose tissue *, skeletal muscle *, brown adipose tissue *, pancreatic β-cells *, pancreatic α-cells, and hepatocytes * | [13,34,35,36,37,38,39,40,41,42,43,44,45,46] | ||
| Rat proximal tubular cells, Kupffer cells *, microglial cells, neurons, cardiomyocytes, skeletal muscle *, testis, brown adipose tissue *, white adipose tissue *, pancreatic β-cells, lungs *, spleen *, and thymus * | [33,47,48,49,50,51,52,53,54,55,56] | ||
| UCP3 | Human skeletal muscle, heart *, spleen *, thymus *, keratinocytes *, skin fibroblasts, sweat glands, hair follicles, stratum basale of the epidermis, pancreatic β-cells, thyroid *, and bone marrow * | Modulation of ROS production, insulin secretion, lipid metabolism, protection against lipotoxicity, control of mitochondrial Ca2+-uptake, immunomodulation | [23,33,57,58,59,60] |
| Mouse skeletal muscle, brown adipose tissue, brain *, kidney *, colon *, liver *, heart, white adipose tissue, thymocytes, splenic lymphocytes, and peripheral naive CD4+ T cells * | [60,61,62,63] | ||
| Rat skeletal muscle, brown adipose tissue *, white adipose tissue *, kidney *, spleen reticulocytes, spleen monocytes, spleen lymphocytes, thymocytes, and heart | [33,52,57,64] | ||
| UCP4 | Human Purkinje cells, most of the brain tissues *, and cartilage * | Regulation of oxidative stress, thermoregulation, protection against mitochondrial Ca2+ overload | [7,9,15,65] |
| Rat Merkel cells, modiolus ear (fibrocyte, satellite cells), organ of Corti (spiral ganglion neurons, supporting cells, hair cells), brain (pyramidal cells of hippocampus and cortex, Purkinje cells of cerebellum, neurons of hippocampus, substantia nigra, striatum, neocortex *), chondrocytes, vestibular ganglion *, heart *, lungs *, skeletal muscle *, kidney *, and liver * | [7,55,65,66,67,68,69] | ||
| Mouse brain (neurons, astrocytes, cortex, brainstem, cerebellum), spinal cord, mast cells, spiral ganglion *, vestibular ganglion *, kidney *, heart *, white adipose tissue *, brown adipose tissue *, skeletal muscle *, and liver * | [55,66,70,71,72] | ||
| UCP5 | Human kidney *, heart *, lungs *, stomach *, liver *, spleen *, skeletal muscle *, brain (cerebellum, cortex, medulla, occipital pole, frontal lobe, putamen, amygdala, caudate nucleus, hippocampus, substantia nigra, thalamus, corpus callosum) *, spinal cord *, pituitary *, uterus *, and testis * | Regulation of oxidative stress, thermoregulation, transport of metabolites, regulation of mitochondrial metabolism | [14,73,74] |
| Rat heart *, lungs *, adrenals *, kidney *, gonadal fat *, ovary *, brain (striatum, cortex, hippocampus, cerebellum) *, skeletal muscle *, and liver * | [14,55,75] | ||
| Mouse heart, kidney, skeletal muscle, white adipose tissue, spinal cord, brain (neurons, astrocytes, cortex, thalamus, hippocampus, substantia nigra, cerebellum, basal ganglia, hypothalamus *, amygdala *, neocortex *, caudate putamen *), spiral ganglion *, vestibular ganglion *, hepatocytes*, brown adipose tissue *, spleen *, intestine *, lungs *, testis *, uterus *, and periovarian fat * | [14,30,44,71,72,74,76,77,78] | ||
| UCP6 | Mouse kidney (proximal tubules, distal tubules, surrounding nephron segments, glomeruli, medullary part of the loop of Henle, collecting duct) *, white adipose tissue *, brown adipose tissue *, brain *, heart *, muscle *, liver *, lungs *, spleen *, and testis * | Regulation of oxidative stress, transport of metabolites, regulation of mitochondrial metabolism | [30,74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteiro, B.S.; Freire-Brito, L.; Carrageta, D.F.; Oliveira, P.F.; Alves, M.G. Mitochondrial Uncoupling Proteins (UCPs) as Key Modulators of ROS Homeostasis: A Crosstalk between Diabesity and Male Infertility? Antioxidants 2021, 10, 1746. https://doi.org/10.3390/antiox10111746
Monteiro BS, Freire-Brito L, Carrageta DF, Oliveira PF, Alves MG. Mitochondrial Uncoupling Proteins (UCPs) as Key Modulators of ROS Homeostasis: A Crosstalk between Diabesity and Male Infertility? Antioxidants. 2021; 10(11):1746. https://doi.org/10.3390/antiox10111746
Chicago/Turabian StyleMonteiro, Bruno S., Laís Freire-Brito, David F. Carrageta, Pedro F. Oliveira, and Marco G. Alves. 2021. "Mitochondrial Uncoupling Proteins (UCPs) as Key Modulators of ROS Homeostasis: A Crosstalk between Diabesity and Male Infertility?" Antioxidants 10, no. 11: 1746. https://doi.org/10.3390/antiox10111746
APA StyleMonteiro, B. S., Freire-Brito, L., Carrageta, D. F., Oliveira, P. F., & Alves, M. G. (2021). Mitochondrial Uncoupling Proteins (UCPs) as Key Modulators of ROS Homeostasis: A Crosstalk between Diabesity and Male Infertility? Antioxidants, 10(11), 1746. https://doi.org/10.3390/antiox10111746