
Melatonin Enhances the Mitochondrial Functionality of Brown Adipose Tissue in Obese—Diabetic Rats

 ,
,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Reagents
2.3. Animals and Experimental Protocols
2.3.1. Mitochondrial Preparation
2.3.2. Mitochondrial High-Resolution Respirometry
2.3.3. ATP Concentration
2.3.4. Determination of Mitochondrial Nitrites
2.3.5. Measurement of Superoxide Dismutase (SOD) Mitochondrial Activity
2.3.6. Measurement of Calcium Retention Capacity and mPTP Activity
2.4. Statistical Analysis
3. Results
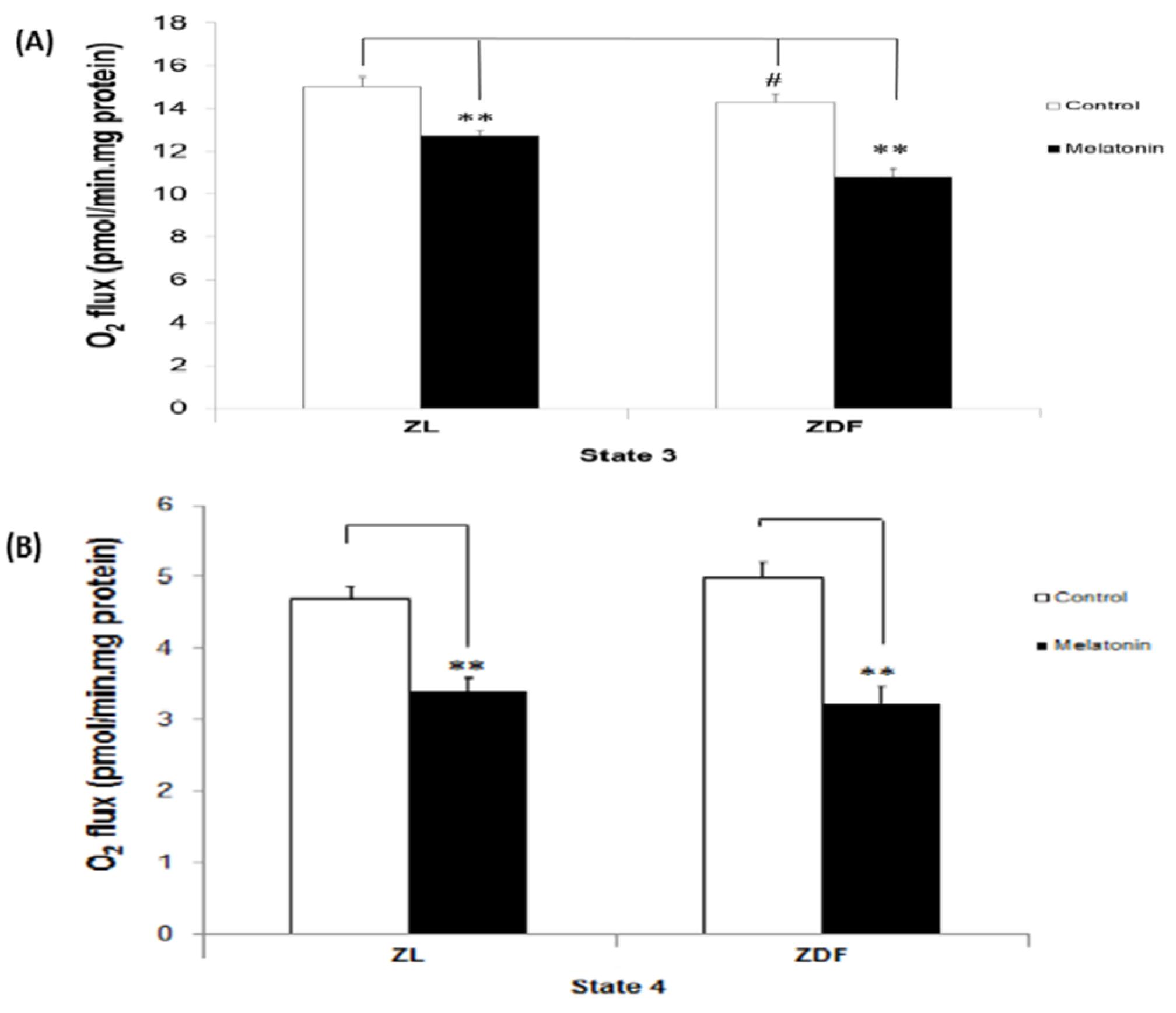
3.1. The Effect of Melatonin Treatment on Mitochondrial Respiratory (States) in BAT-Isolated Mitochondria
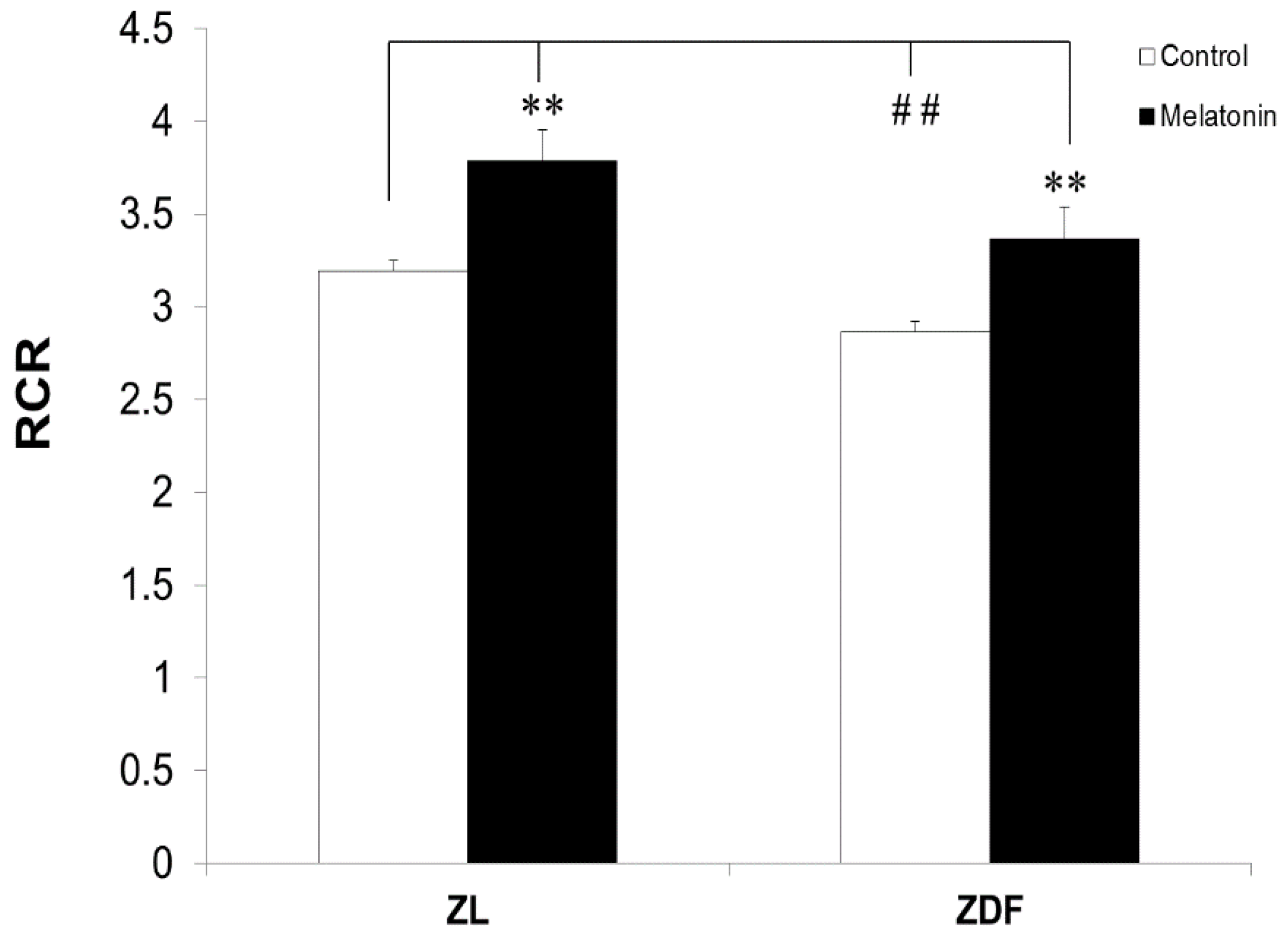
3.2. The Effect of Melatonin Treatment on RCR of Mitochondria from BAT
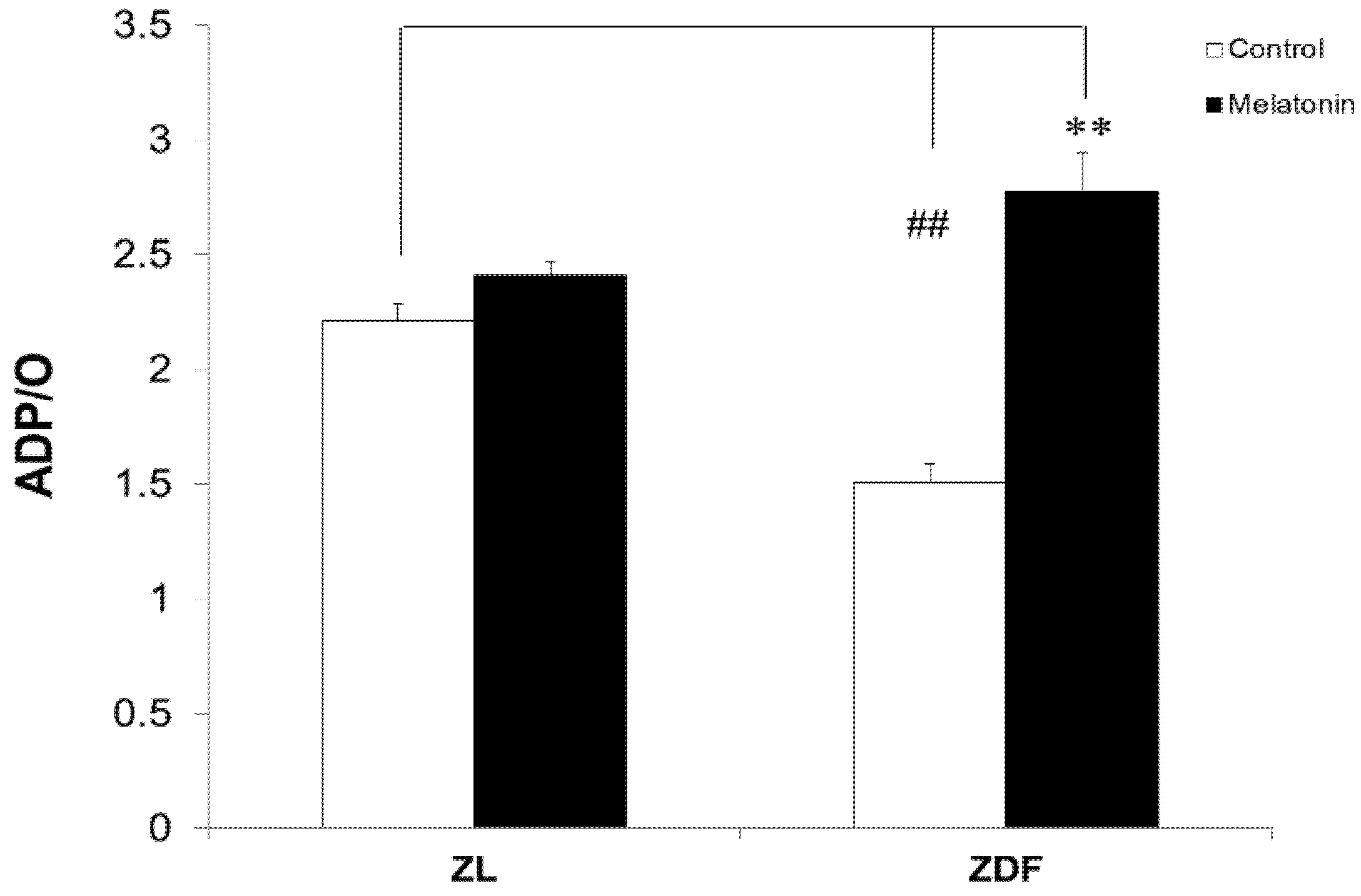
3.3. Melatonin Increased the Respiratory Phosphorylation Coefficient (ADP/O Ratio) of BAT-Isolated Mitochondria
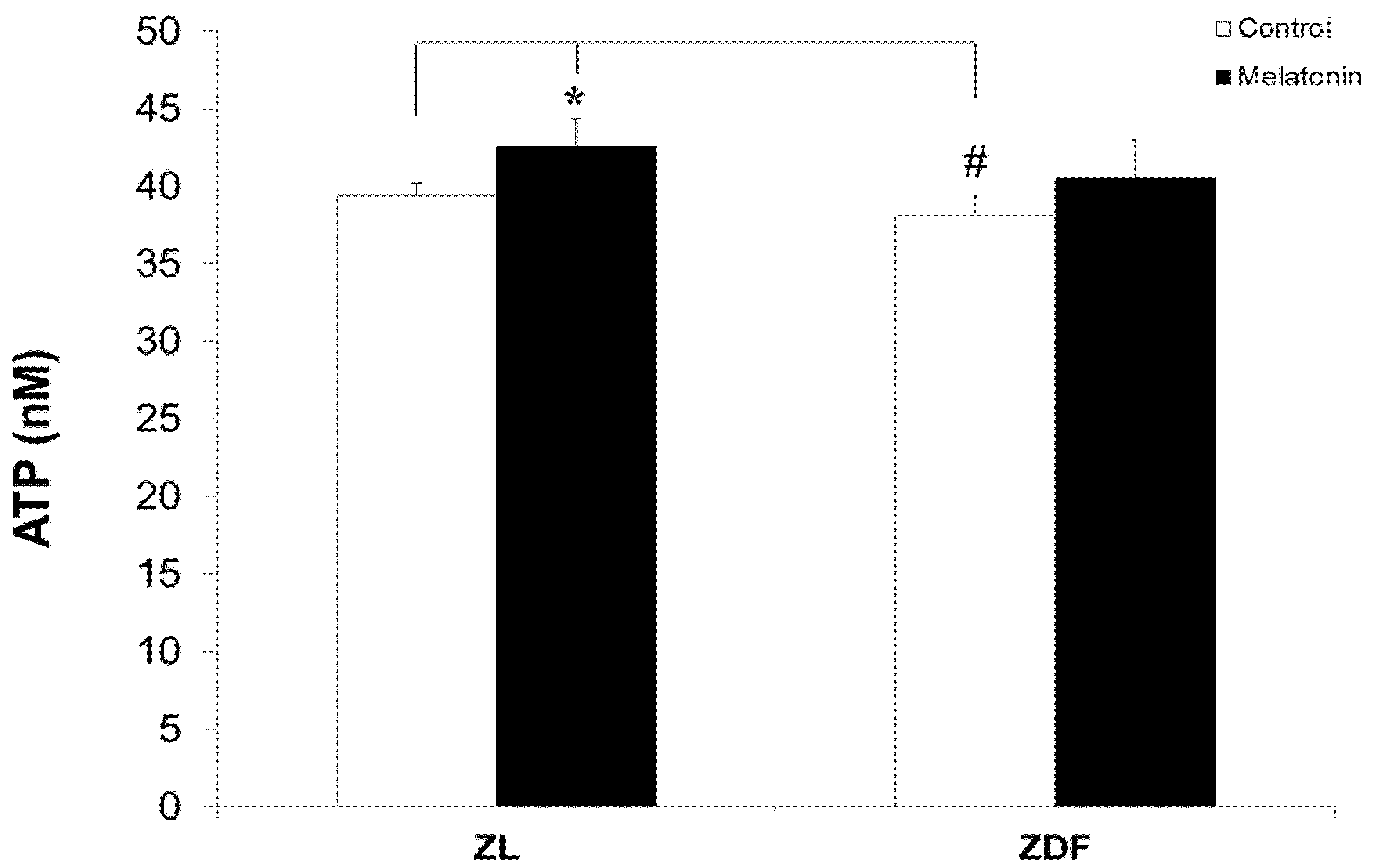
3.4. Melatonin Nonsignificantly Increased ATP Levels of Mitochondria from BAT
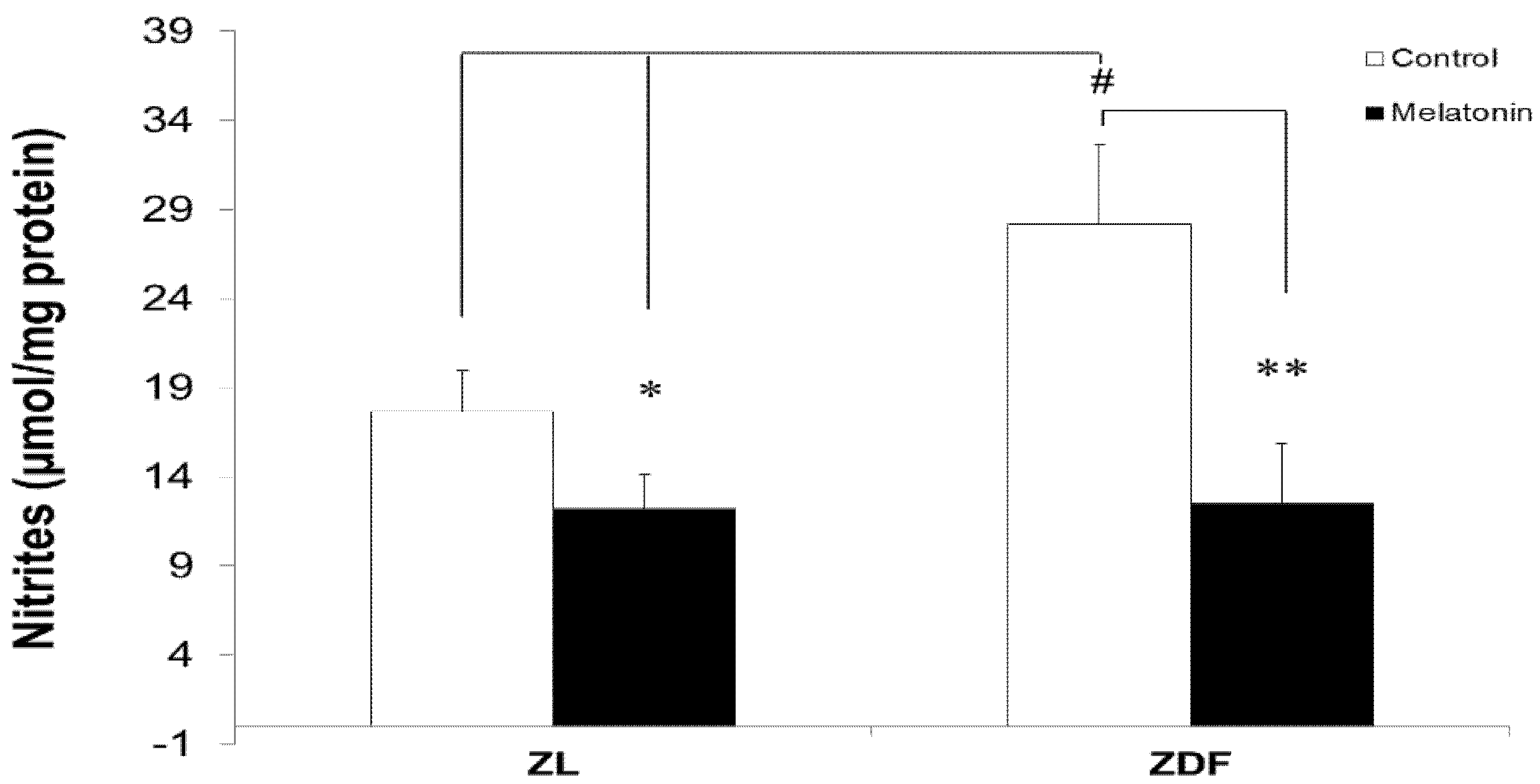
3.5. Melatonin Reduced Levels of Mitochondrial Nitrites from Brown Fat Depots
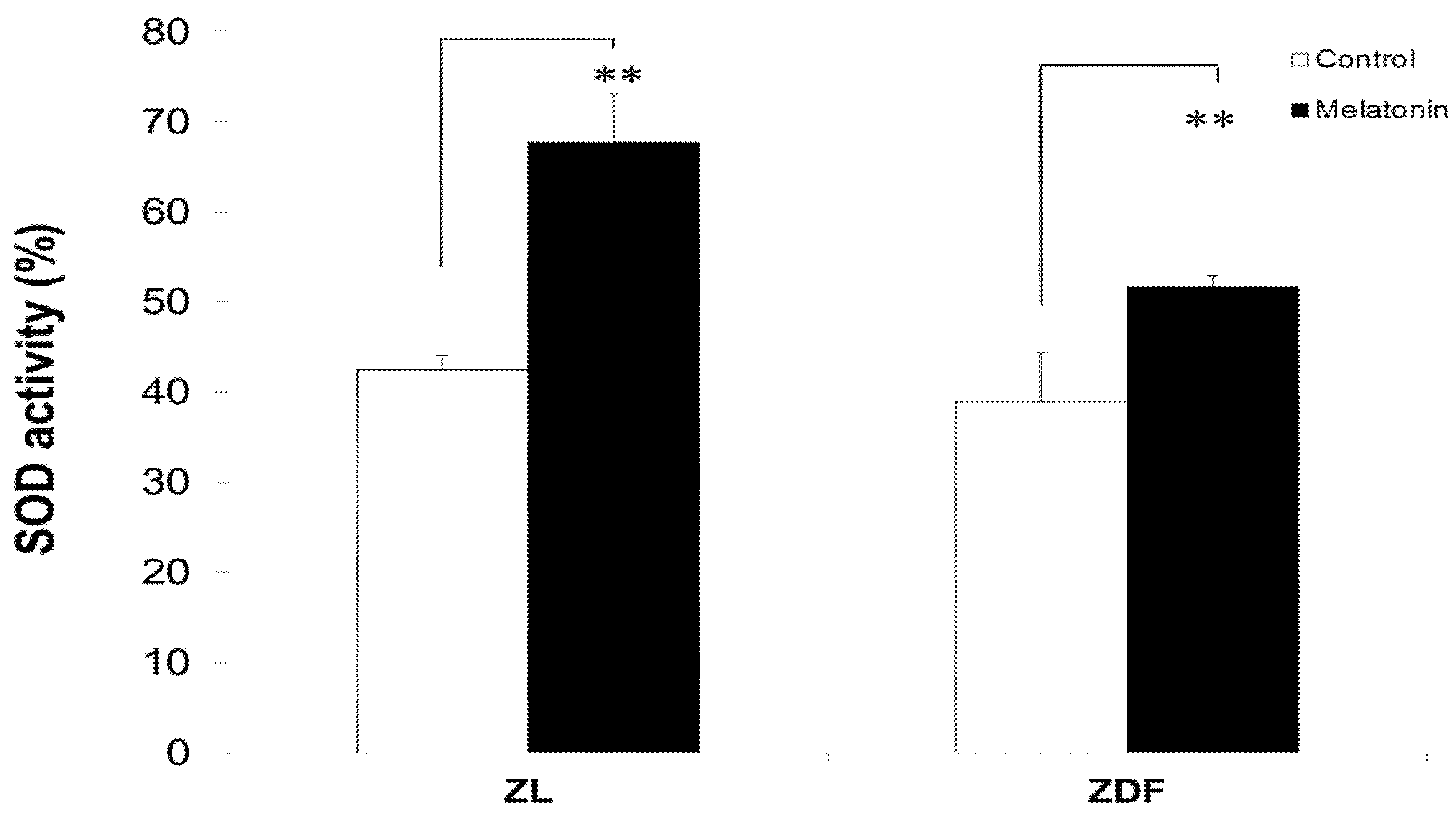
3.6. Melatonin Increased SOD Activity of BAT-Derived Mitochondria
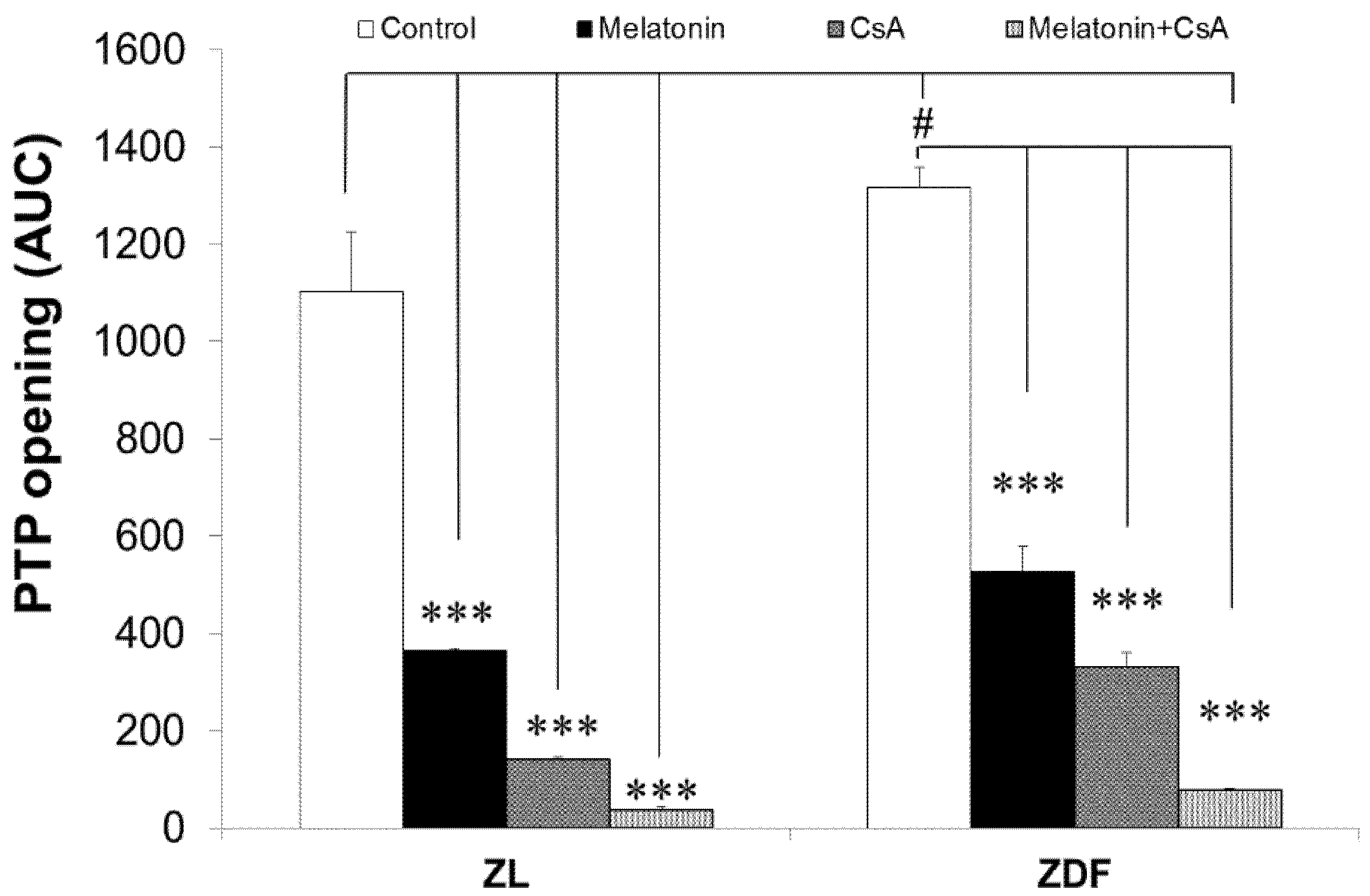
3.7. Melatonin Treatment Altered Calcium Retention Capacity and Sensitivity of mPTP Opening
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H. Mitochondrial deficiency is associated with insulin resistance. Diabetes 2013, 62, 1032–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Aranda, A.; Fernandez-Vazquez, G.; Campos, D.; Tassi, M.; Velasco-Perez, L.; Tan, D.X.; Reiter, R.J.; Agil, A. Melatonin induces browning of inguinal white adipose tissue in Zucker diabetic fatty rats. J. Pineal Res. 2013, 55, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; Romijn, J.A.; Heine, R.J. Fatty acid-induced mitochondrial uncoupling in adipocytes as a key protective factor against insulin resistance and beta cell dysfunction: A new concept in the pathogenesis of obesity-associated type 2 diabetes mellitus. Diabetologia 2007, 50, 2036–2041. [Google Scholar] [CrossRef] [Green Version]
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am. J. Pathol. 2009, 175, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Stehle, J.H.; Saade, A.; Rawashdeh, O.; Ackermann, K.; Jilg, A.; Sebesteny, T.; Maronde, E. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J. Pineal Res. 2011, 51, 17–43. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Diaz-Casado, M.E.; Lima-Cabello, E.; Lopez, L.C.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef]
- Hardeland, R.; Madrid, J.A.; Tan, D.X.; Reiter, R.J. Melatonin, the circadian multioscillator system and health: The need for detailed analyses of peripheral melatonin signaling. J. Pineal Res. 2012, 52, 139–166. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal Res. 2012, 52, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.-X.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Reiter, R. Melatonin: A potent endogenous hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Reiter, R.J.; Paredes, S.D.; Manchester, L.C.; Tan, D.X. Reducing oxidative/nitrosative stress: A newly-discovered genre for melatonin. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 2013, 54, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont-Rousselot, D.; Collin, F. Melatonin: Action as antioxidant and potential applications in human disease and aging. Toxicology 2010, 278, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Reiter, R.J.; Ruggiero, F.M. Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J. Pineal Res. 2010, 48, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; Ozturk, G.; Bano-Otalora, B.; Pozo, M.J.; Madrid, J.A.; Reiter, R.J.; Serrano, E.; Concepcion, M.; Acuna-Castroviejo, D. Exercise and melatonin in humans: Reciprocal benefits. J. Pineal Res. 2012, 52, 1–11. [Google Scholar] [CrossRef]
- Agil, A.; Chayah, M.; Visiedo, L.; Navarro-Alarcon, M.; Rodriguez Ferrer, J.M.; Tassi, M.; Reiter, R.J.; Fernandez-Vazquez, G. Melatonin Improves Mitochondrial Dynamics and Function in the Kidney of Zucker Diabetic Fatty Rats. J. Clin. Med. 2020, 9, 2916. [Google Scholar] [CrossRef]
- Navarro-Alarcon, M.; Ruiz-Ojeda, F.J.; Blanca-Herrera, R.M.; A-Serrano, M.M.; Acuna-Castroviejo, D.; Fernandez-Vazquez, G.; Agil, A. Melatonin and metabolic regulation: A review. Food Funct. 2014, 5, 2806–2832. [Google Scholar] [CrossRef] [PubMed]
- Elmahallawy, E.K.; Luque, J.O.; Aloweidi, A.S.; Gutierrez-Fernandez, J.; Sampedro-Martinez, A.; Rodriguez-Granger, J.; Kaki, A.; Agil, A. Potential Relevance of Melatonin Against Some Infectious Agents: A Review and Assessment of Recent Research. Curr. Med. Chem. 2015, 22, 3848–3861. [Google Scholar] [CrossRef]
- Wolden-Hanson, T.; Mitton, D.R.; McCants, R.L.; Yellon, S.M.; Wilkinson, C.W.; Matsumoto, A.M.; Rasmussen, D.D. Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology 2000, 141, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Prunet-Marcassus, B.; Desbazeille, M.; Bros, A.; Louche, K.; Delagrange, P.; Renard, P.; Casteilla, L.; Penicaud, L. Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology 2003, 144, 5347–5352. [Google Scholar] [CrossRef] [Green Version]
- Raskind, M.A.; Burke, B.L.; Crites, N.J.; Tapp, A.M.; Rasmussen, D.D. Olanzapine-induced weight gain and increased visceral adiposity is blocked by melatonin replacement therapy in rats. Neuropsychopharmacology 2007, 32, 284–288. [Google Scholar] [CrossRef]
- Agil, A.; Navarro-Alarcon, M.; Ruiz, R.; Abuhamadah, S.; El-Mir, M.Y.; Vazquez, G.F. Beneficial effects of melatonin on obesity and lipid profile in young Zucker diabetic fatty rats. J. Pineal Res. 2011, 50, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Terron, M.P.; Delgado-Adamez, J.; Pariente, J.A.; Barriga, C.; Paredes, S.D.; Rodriguez, A.B. Melatonin reduces body weight gain and increases nocturnal activity in male Wistar rats. Physiol. Behav. 2013, 118, 8–13. [Google Scholar] [CrossRef]
- Fernández Vázquez, G.; Reiter, R.J.; Agil, A. Melatonin increases brown adipose tissue mass and function in Zücker diabetic fatty rats: Implications for obesity control. J. Pineal Res. 2018, 64, e12472. [Google Scholar] [CrossRef] [PubMed]
- Acuna Castroviejo, D.; Lopez, L.C.; Escames, G.; Lopez, A.; Garcia, J.A.; Reiter, R.J. Melatonin-mitochondria interplay in health and disease. Curr. Top. Med. Chem. 2011, 11, 221–240. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Furio, A.M.; Brusco, L.I. Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr. Neuropharmacol. 2010, 8, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, U.; Yilmaz, B.; Ugur, M.; Yuksel, A.; Reiter, R.J.; Hermann, D.M.; Kilic, E. Evidence that membrane-bound G protein-coupled melatonin receptors MT1 and MT2 are not involved in the neuroprotective effects of melatonin in focal cerebral ischemia. J. Pineal Res. 2012, 52, 228–235. [Google Scholar] [CrossRef]
- Zavodnik, I.B.; Lapshina, E.A.; Cheshchevik, V.T.; Dremza, I.K.; Kujawa, J.; Zabrodskaya, S.V.; Reiter, R.J. Melatonin and succinate reduce rat liver mitochondrial dysfunction in diabetes. J. Physiol. Pharmacol. 2011, 62, 421–427. [Google Scholar]
- Subramanian, P.; Mirunalini, S.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Melatonin treatment improves the antioxidant status and decreases lipid content in brain and liver of rats. Eur. J. Pharmacol. 2007, 571, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Contini Mdel, C.; Millen, N.; Mahieu, S.T. Role of melatonin in the oxidative damage prevention at different times of hepatic regeneration. Cell Biochem. Funct. 2012, 30, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Bruck, R.; Aeed, H.; Avni, Y.; Shirin, H.; Matas, Z.; Shahmurov, M.; Avinoach, I.; Zozulya, G.; Weizman, N.; Hochman, A. Melatonin inhibits nuclear factor kappa B activation and oxidative stress and protects against thioacetamide induced liver damage in rats. J. Hepatol. 2004, 40, 86–93. [Google Scholar] [CrossRef]
- Agil, A.; El-Hammadi, M.; Jimenez-Aranda, A.; Tassi, M.; Abdo, W.; Fernandez-Vazquez, G.; Reiter, R.J. Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J. Pineal Res. 2015, 59, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Colantuono, G.; Moro, N.; Ruggiero, F.M.; Tiravanti, E.; Di Venosa, N.; Fiore, T.; Paradies, G. Melatonin protects against heart ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am. J. Physiol. Circ. Physiol. 2009, 297, H1487–H1493. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.I.; Escames, G.; Lopez, L.C.; Garcia, J.A.; Ortiz, F.; Lopez, A.; Acuna-Castroviejo, D. Melatonin administration prevents cardiac and diaphragmatic mitochondrial oxidative damage in senescence-accelerated mice. J. Endocrinol. 2007, 194, 637–643. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Duan, W.; Jin, Z.; Yi, W.; Yan, J.; Zhang, S.; Wang, N.; Liang, Z.; Li, Y.; Chen, W.; et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J. Pineal Res. 2013, 55, 275–286. [Google Scholar] [CrossRef]
- Wang, W.Z.; Fang, X.H.; Stephenson, L.L.; Zhang, X.; Khiabani, K.T.; Zamboni, W.A. Melatonin attenuates I/R-induced mitochondrial dysfunction in skeletal muscle. J. Surg. Res. 2011, 171, 108–113. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Sayeed, I.; Siemen, D.; Wolf, G.; Horn, T.F. Direct inhibition of the mitochondrial permeability transition pore: A possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004, 18, 869–871. [Google Scholar] [CrossRef] [Green Version]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Aranda, A.; Fernandez-Vazquez, G.; Mohammad, A.S.M.; Reiter, R.J.; Agil, A. Melatonin improves mitochondrial function in inguinal white adipose tissue of Zucker diabetic fatty rats. J. Pineal Res. 2014, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E.; Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Steurer, W.; Margreiter, R. Mitochondria in the cold. In Life in the Cold; Springer: Berlin/Heidelberg, Germany, 2000; pp. 431–442. [Google Scholar]
- Haller, T.; Ortner, M.; Gnaiger, E. A respirometer for investigating oxidative cell metabolism: Toward optimization of respiratory studies. Anal. Biochem. 1994, 218, 338–342. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360. [Google Scholar]
- Montgomery, M.K. Mitochondrial Dysfunction and Diabetes: Is Mitochondrial Transfer a Friend or Foe? Biology 2019, 8, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agil, A.; Rosado, I.; Ruiz, R.; Figueroa, A.; Zen, N.; Fernandez-Vazquez, G. Melatonin improves glucose homeostasis in young Zucker diabetic fatty rats. J. Pineal Res. 2012, 52, 203–210. [Google Scholar] [CrossRef]
- Cheshchevik, V.T.; Dremza, I.K.; Lapshina, E.A.; Zabrodskaya, S.V.; Kujawa, J.; Zavodnik, I.B. Corrections by melatonin of liver mitochondrial disorders under diabetes and acute intoxication in rats. Cell Biochem. Funct. 2011, 29, 481–488. [Google Scholar] [CrossRef]
- Okatani, Y.; Wakatsuki, A.; Reiter, R.J.; Miyahara, Y. Hepatic mitochondrial dysfunction in senescence-accelerated mice: Correction by long-term, orally administered physiological levels of melatonin. J. Pineal Res. 2002, 33, 127–133. [Google Scholar] [CrossRef]
- Leon, J.; Acuña-Castroviejo, D.; Escames, G.; Tan, D.X.; Reiter, R.J. Melatonin mitigates mitochondrial malfunction. J. Pineal Res. 2005, 38, 1–9. [Google Scholar] [CrossRef]
- Patki, G.; Lau, Y.-S. Melatonin protects against neurobehavioral and mitochondrial deficits in a chronic mouse model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2011, 99, 704–711. [Google Scholar] [CrossRef] [Green Version]
- Rosales-Corral, S.; Acuna-Castroviejo, D.; Tan, D.X.; Lopez-Armas, G.; Cruz-Ramos, J.; Munoz, R.; Melnikov, V.G.; Manchester, L.C.; Reiter, R.J. Accumulation of exogenous amyloid-beta peptide in hippocampal mitochondria causes their dysfunction: A protective role for melatonin. Oxidative Med. Cell. Longev. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Afanas’Ev, I. Signaling of reactive oxygen and nitrogen species in Diabetes mellitus. Oxid. Med. Cell Longev. 2010, 3, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria: I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria: III. Transitional Ca2+ release. Arch. Biochem. Biophys. 1979, 195, 468–477. [Google Scholar] [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Moulin, M. Physiological roles of the permeability transition pore. Circ. Res. 2012, 111, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Elmahallawy, E.K.; Jimenez-Aranda, A.; Martinez, A.S.; Rodriguez-Granger, J.; Navarro-Alarcon, M.; Gutierrez-Fernandez, J.; Agil, A. Activity of melatonin against Leishmania infantum promastigotes by mitochondrial dependent pathway. Chem. Biol. Interact. 2014, 220, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Molkentin, J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agil, A.; Navarro-Alarcon, M.; Ali, F.A.Z.; Albrakati, A.; Salagre, D.; Campoy, C.; Elmahallawy, E.K. Melatonin Enhances the Mitochondrial Functionality of Brown Adipose Tissue in Obese—Diabetic Rats. Antioxidants 2021, 10, 1482. https://doi.org/10.3390/antiox10091482
Agil A, Navarro-Alarcon M, Ali FAZ, Albrakati A, Salagre D, Campoy C, Elmahallawy EK. Melatonin Enhances the Mitochondrial Functionality of Brown Adipose Tissue in Obese—Diabetic Rats. Antioxidants. 2021; 10(9):1482. https://doi.org/10.3390/antiox10091482
Chicago/Turabian StyleAgil, Ahmad, Miguel Navarro-Alarcon, Fatma Abo Zakaib Ali, Ashraf Albrakati, Diego Salagre, Cristina Campoy, and Ehab Kotb Elmahallawy. 2021. "Melatonin Enhances the Mitochondrial Functionality of Brown Adipose Tissue in Obese—Diabetic Rats" Antioxidants 10, no. 9: 1482. https://doi.org/10.3390/antiox10091482
APA StyleAgil, A., Navarro-Alarcon, M., Ali, F. A. Z., Albrakati, A., Salagre, D., Campoy, C., & Elmahallawy, E. K. (2021). Melatonin Enhances the Mitochondrial Functionality of Brown Adipose Tissue in Obese—Diabetic Rats. Antioxidants, 10(9), 1482. https://doi.org/10.3390/antiox10091482