
Role of the Redox State of Human Peroxiredoxin-5 on Its TLR4-Activating DAMP Function

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
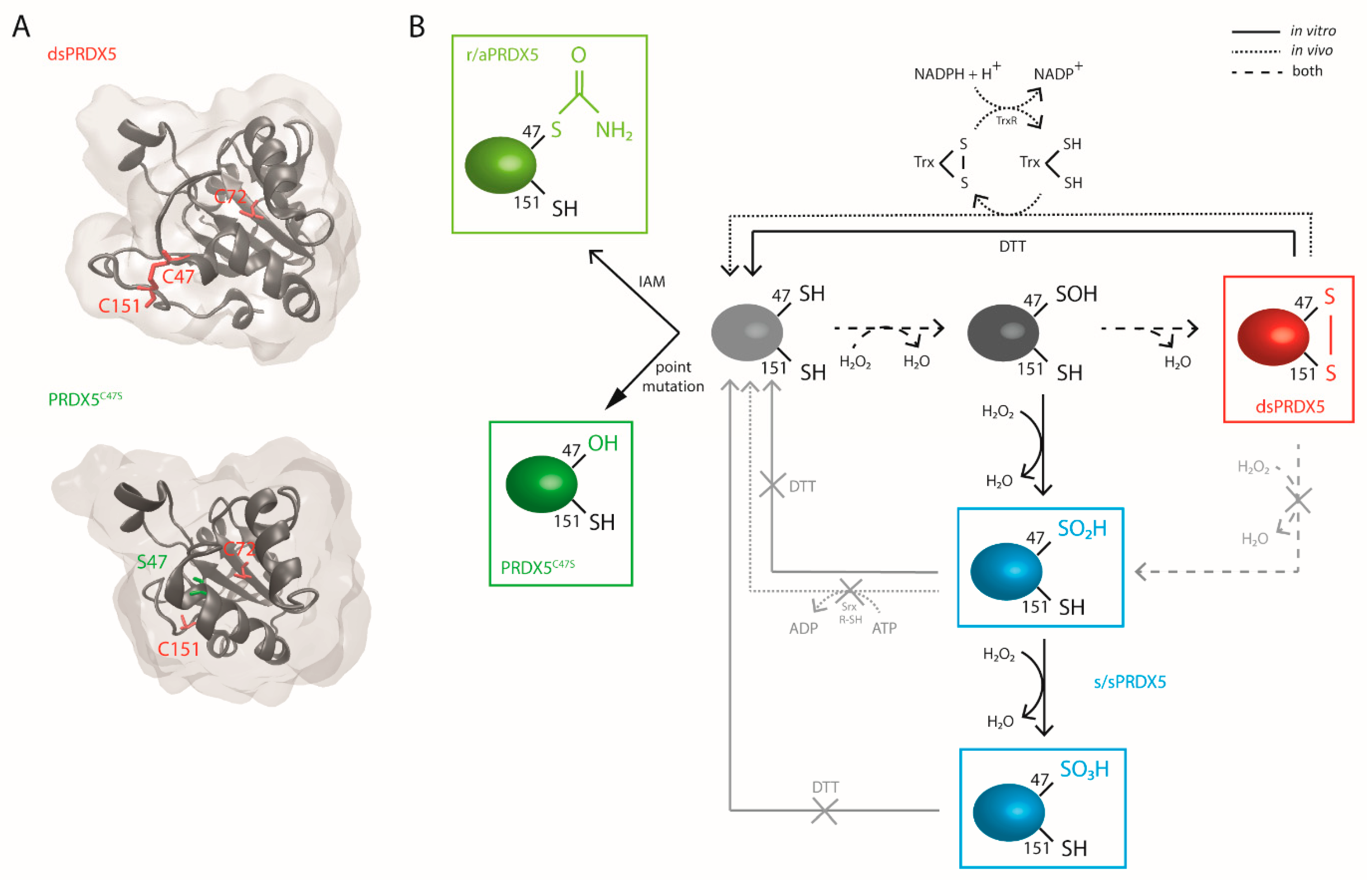
2.1. Production and Purification of Recombinant Human PRDX5 and C47S Mutant PRDX5
2.2. Preparation of the PRDX5 Redox Forms
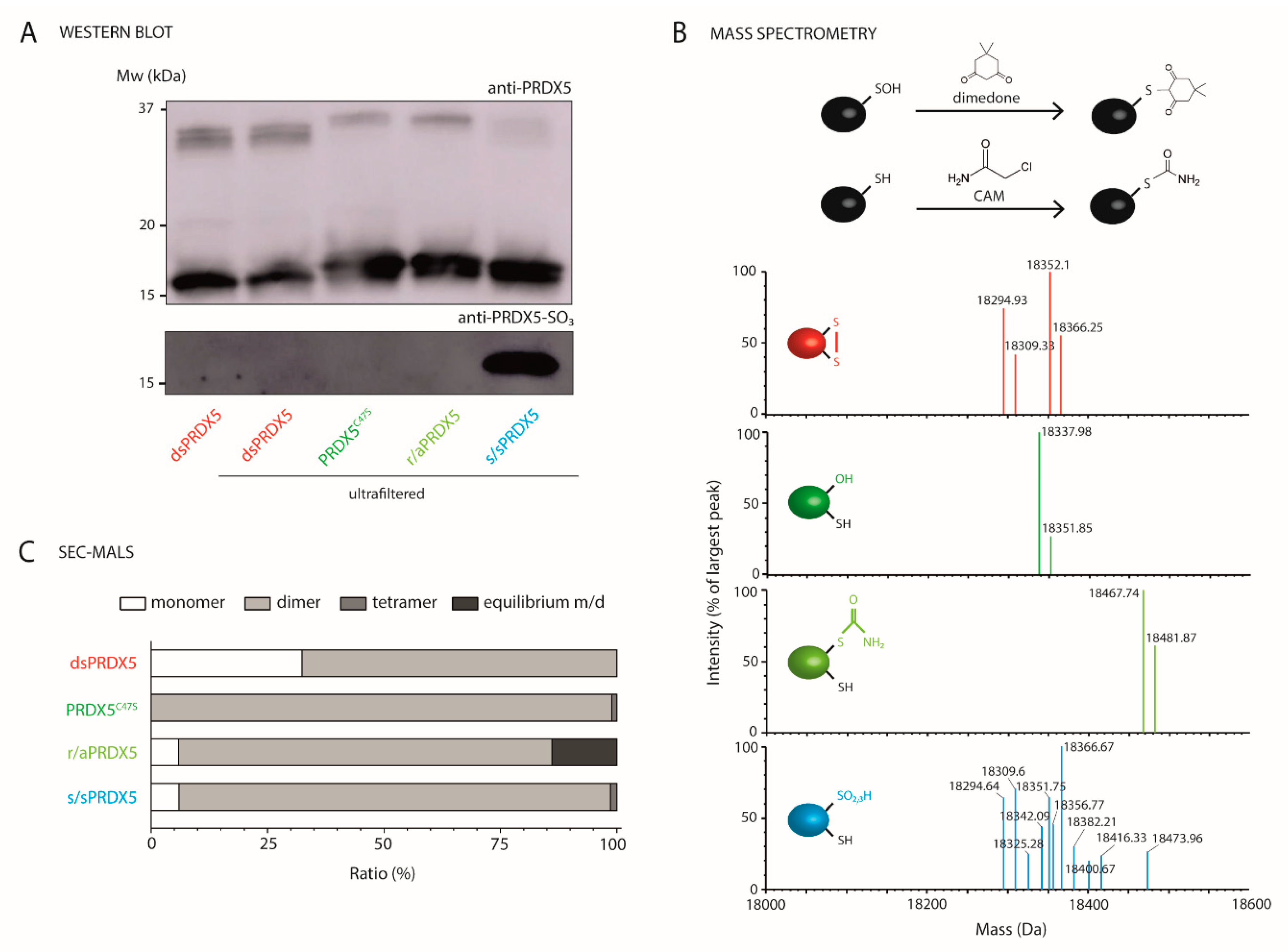
2.3. Western Blotting for Detection of Hyperoxidized PRDX5
2.4. Mass Spectrometric Characterization of the PRDX5 Cysteine Redox State
2.4.1. Blocking of Cysteine Redox States
2.4.2. Sample Preparation
2.4.3. ESI-Q-TOF Analysis of Intact Proteins by Direct Infusion
2.4.4. Maximum Entropy Deconvolution
2.5. SEC-MALS Analysis of the Oligomeric State
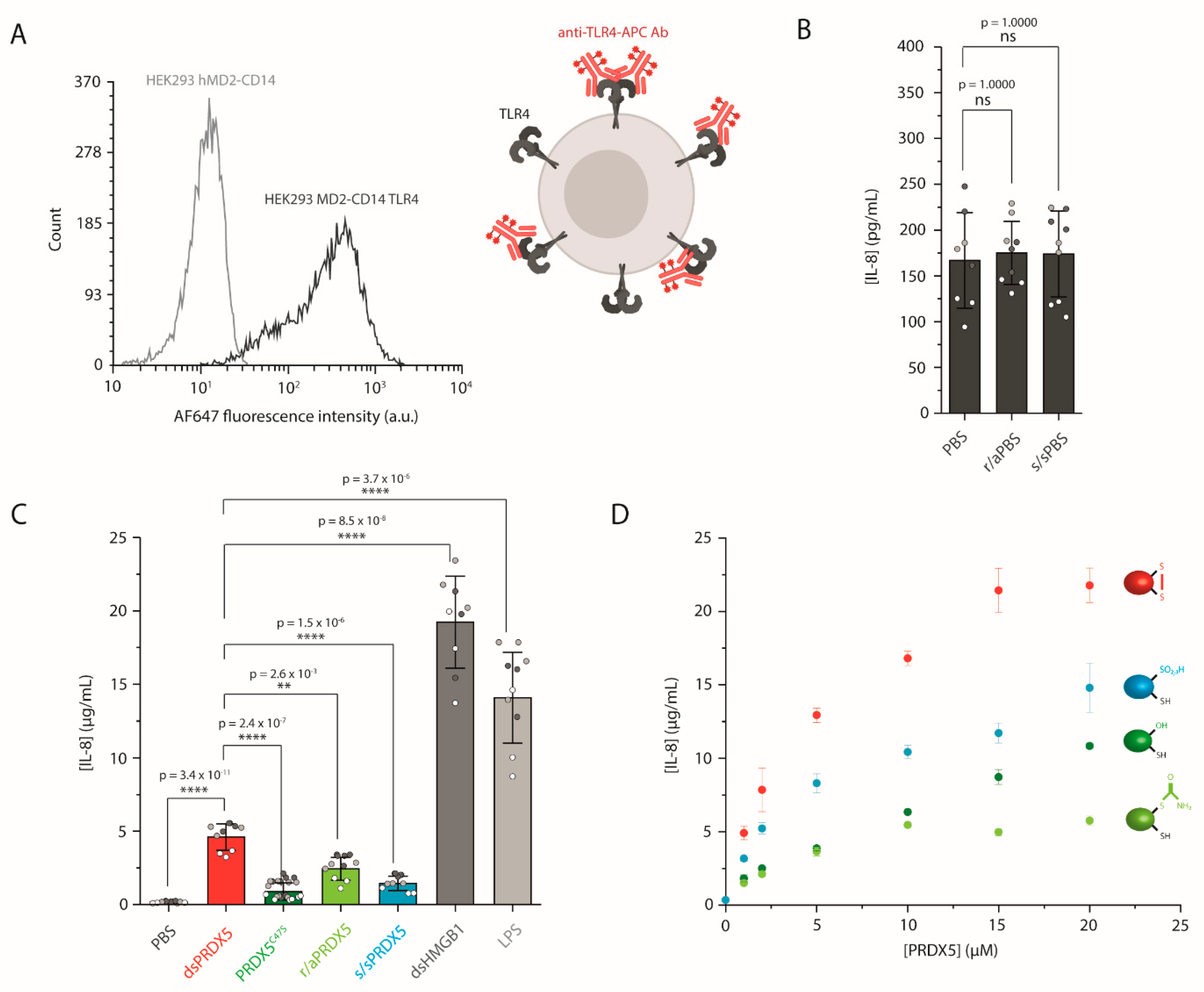
2.6. Generation and Culture of HEK293 Cell Line Expressing TLR4
2.7. Quantification of Human IL-8 Secretion
2.8. Functionalization of AFM Tips
2.8.1. Amino-Functionalization
2.8.2. Coupling of Aldehyde-PEG24-NHS Linker
2.9. Preparation of TLR4-Coated Surfaces
2.9.1. Alkanethiol SAM Formation
2.9.2. NHS/EDC Chemistry
2.10. FD-Curve Based AFM on TLR4-Coated Surfaces
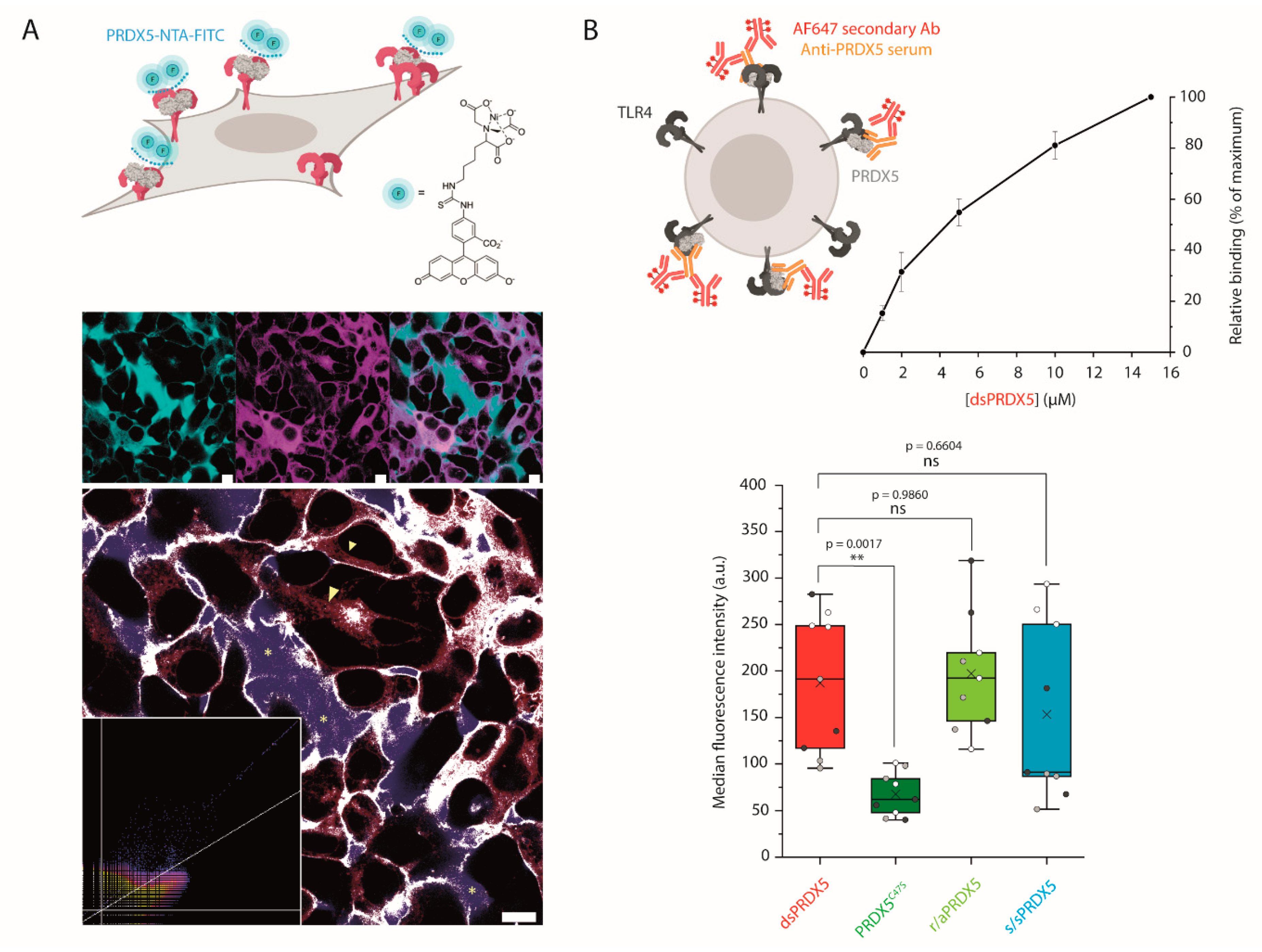
2.11. Confocal Microscopy Analysis of PRDX5 Binding to the Cell Surface
2.12. Flow Cytometry Binding Assay
2.13. Statistical Analysis
3. Results and Discussion
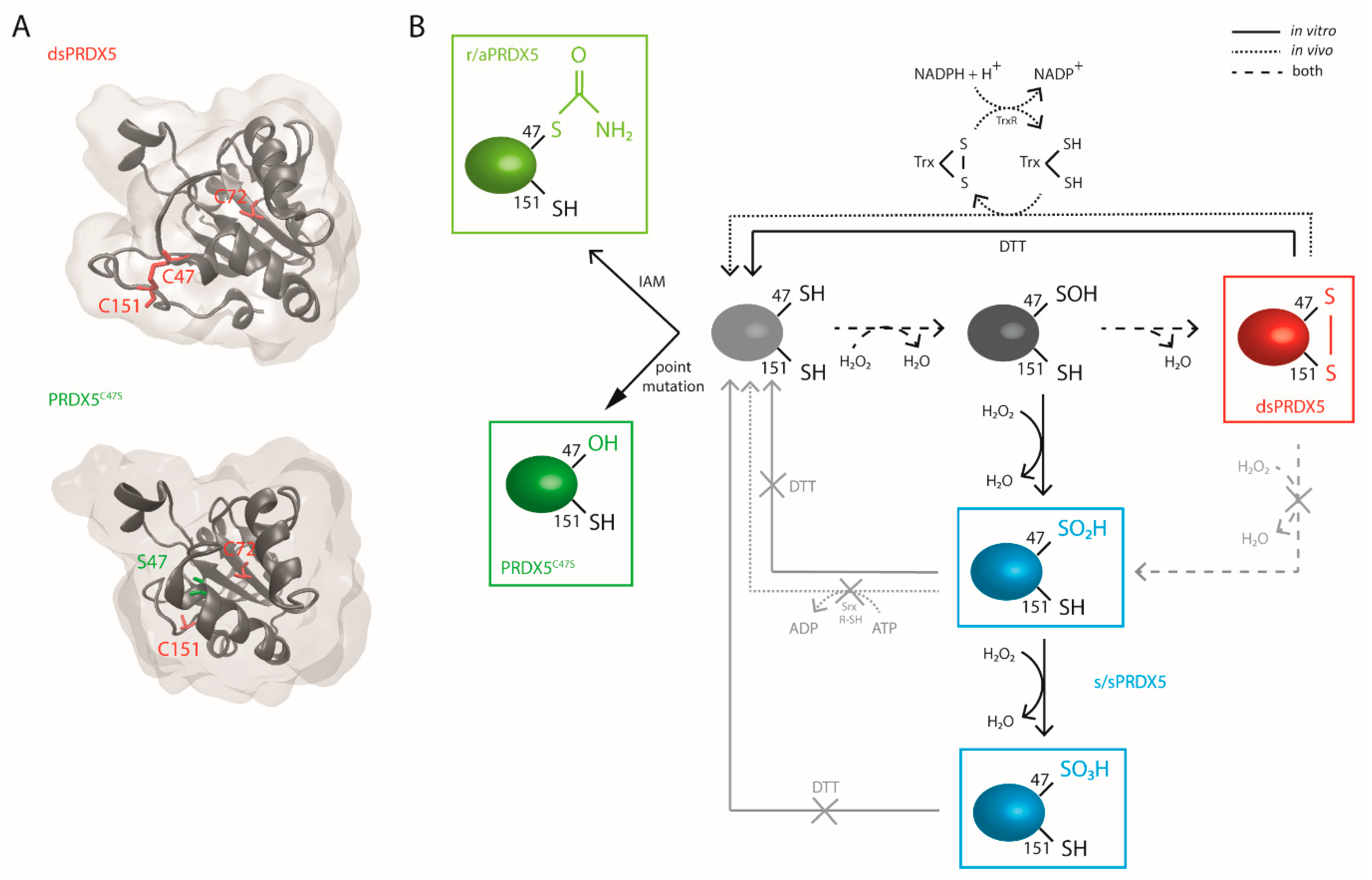
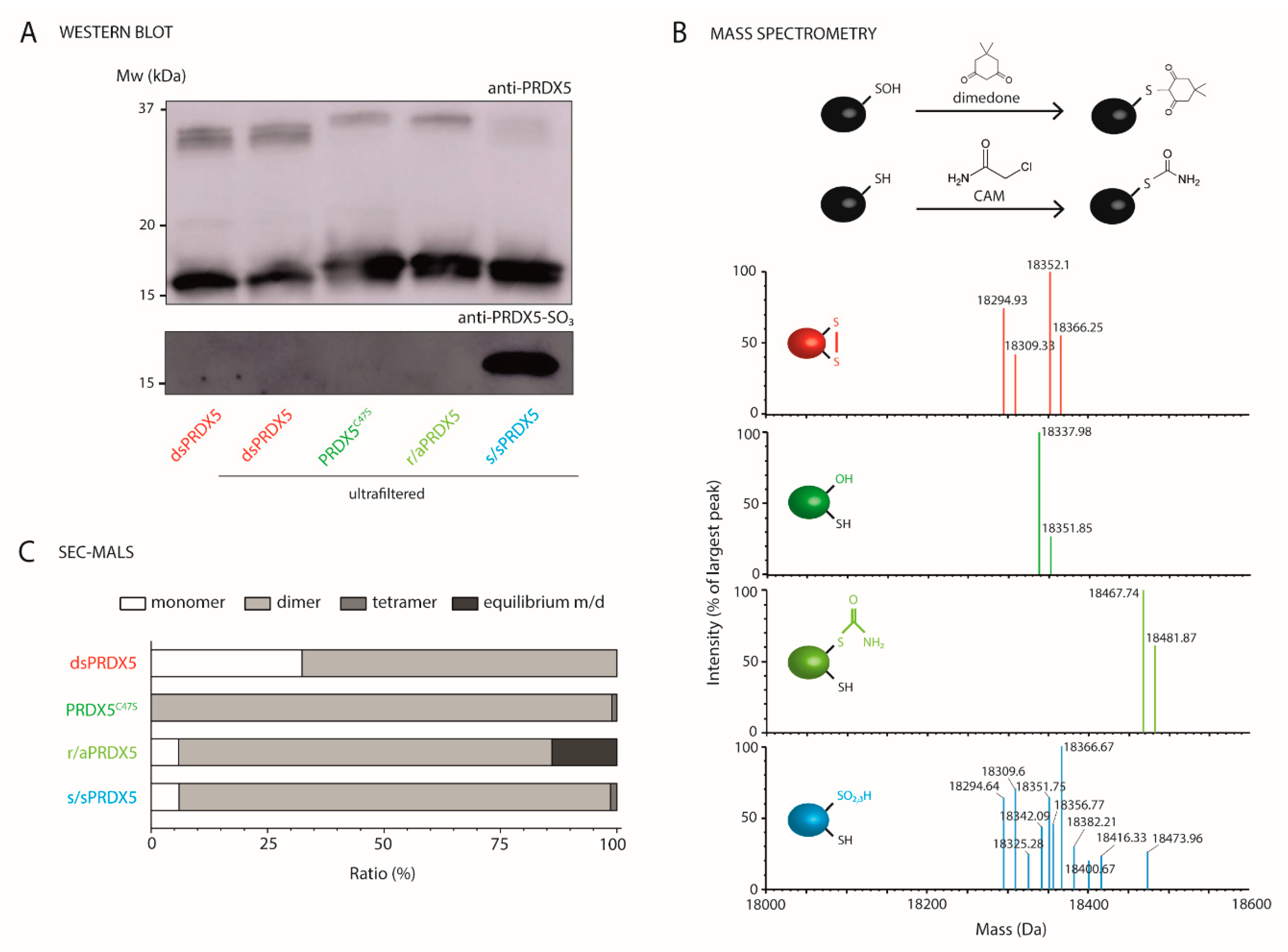
3.1. Production and Characterization of PRDX5 Redox Forms
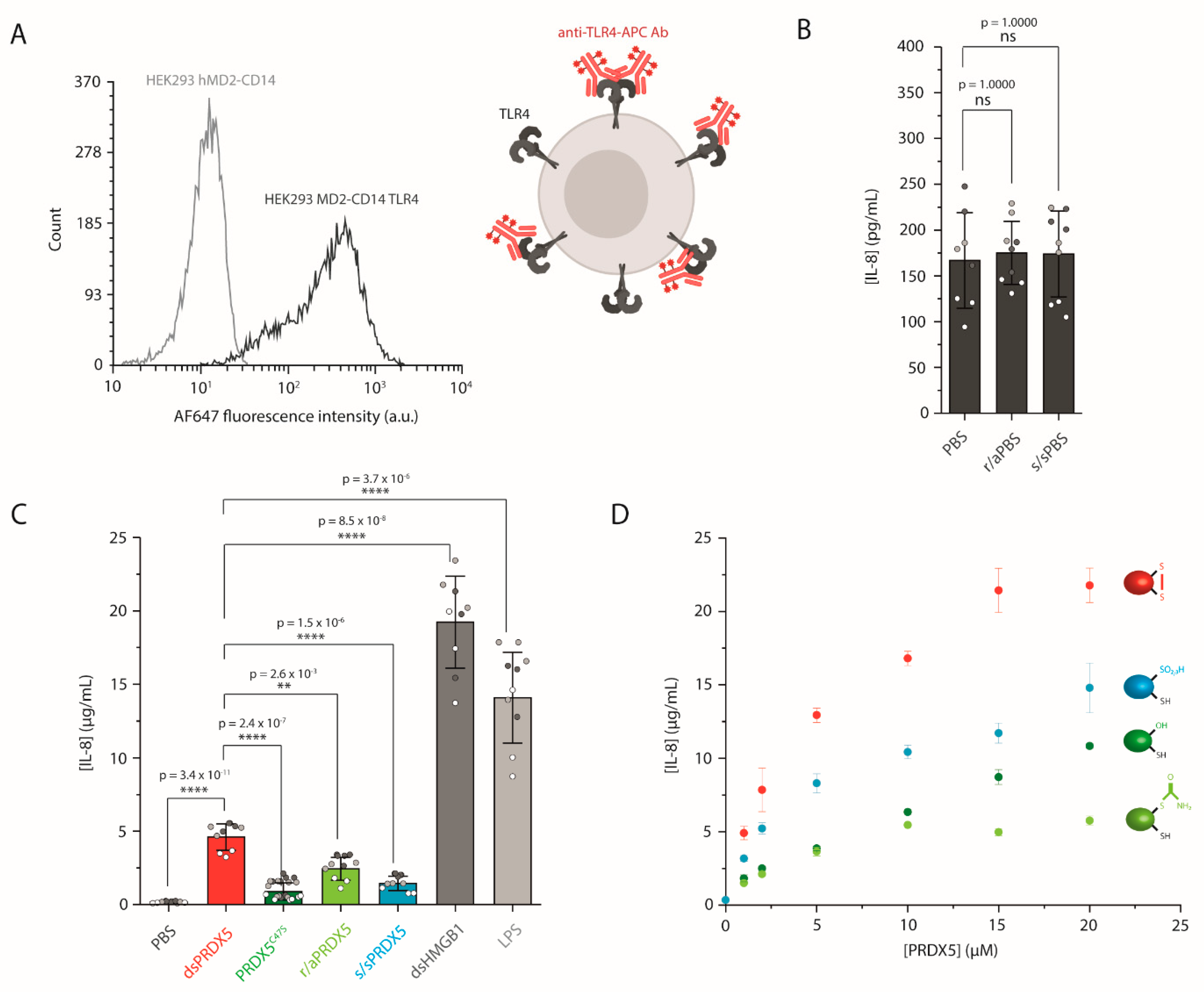
3.2. The PRDX5 Disulfide Bond Allows a Stronger Cytokine-Stimulating Activity
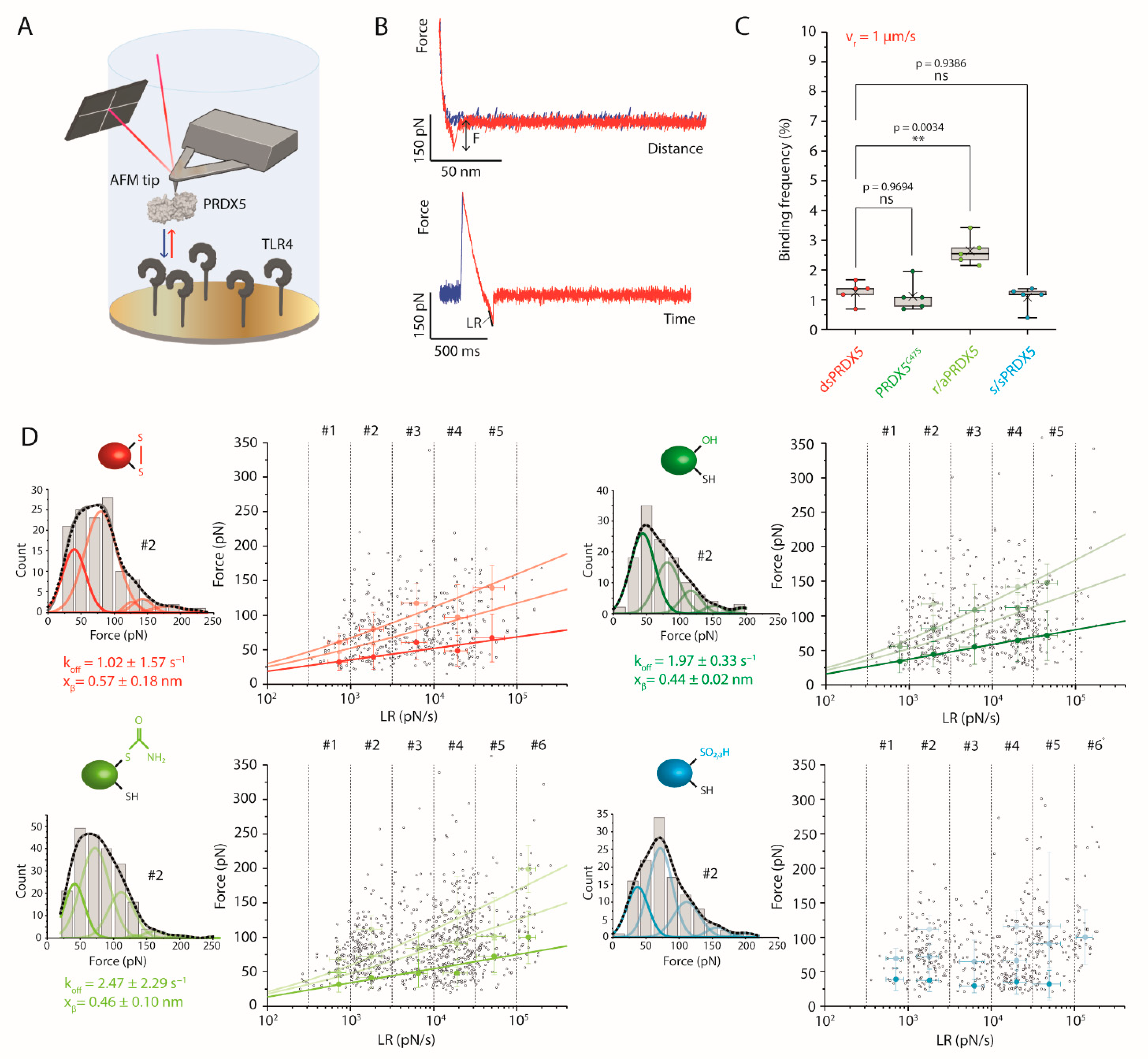
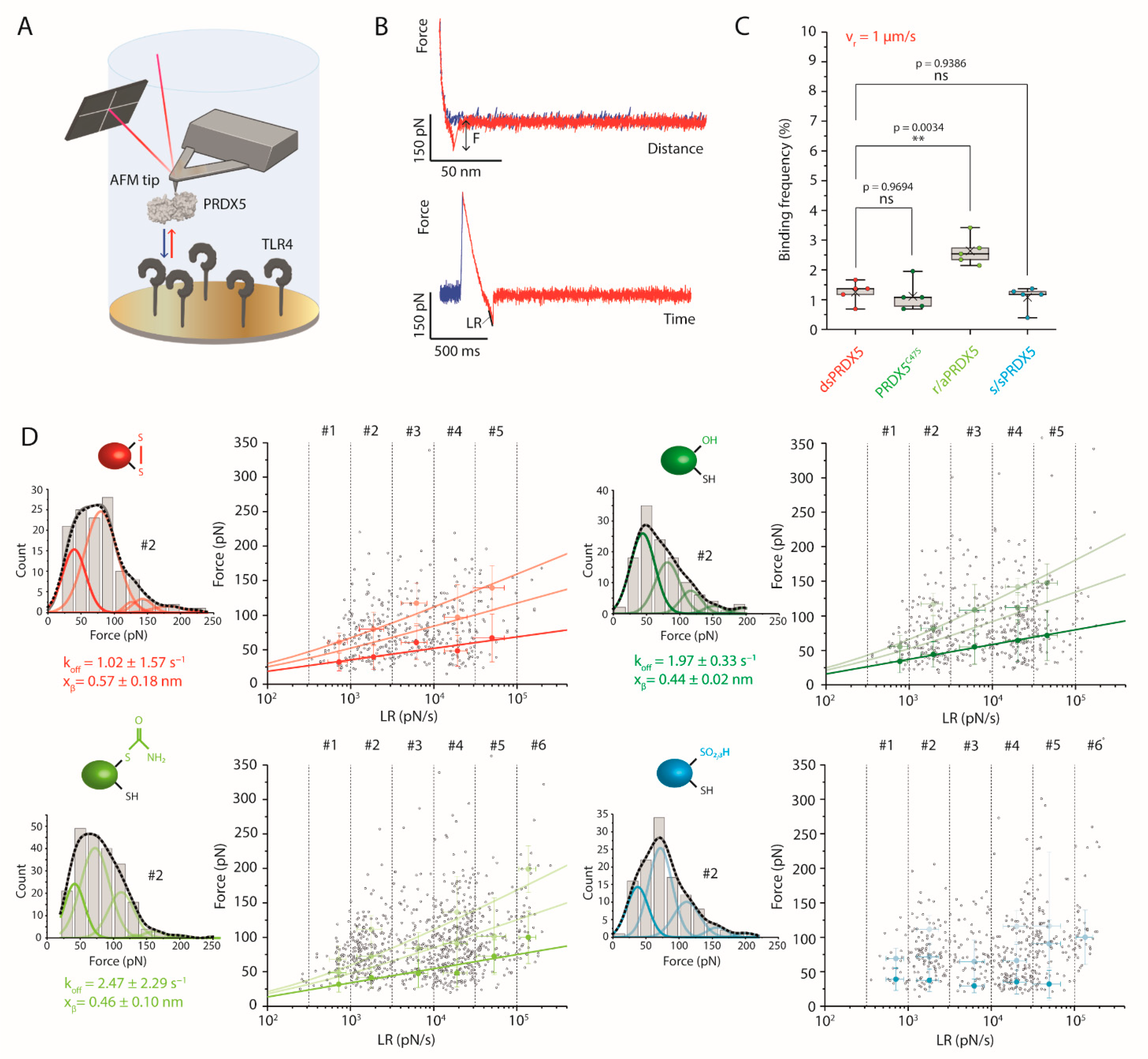
3.3. Disulfide, Mutant and Reduced PRDX5 Bind to TLR4 with Similar Kinetic Off-Rates
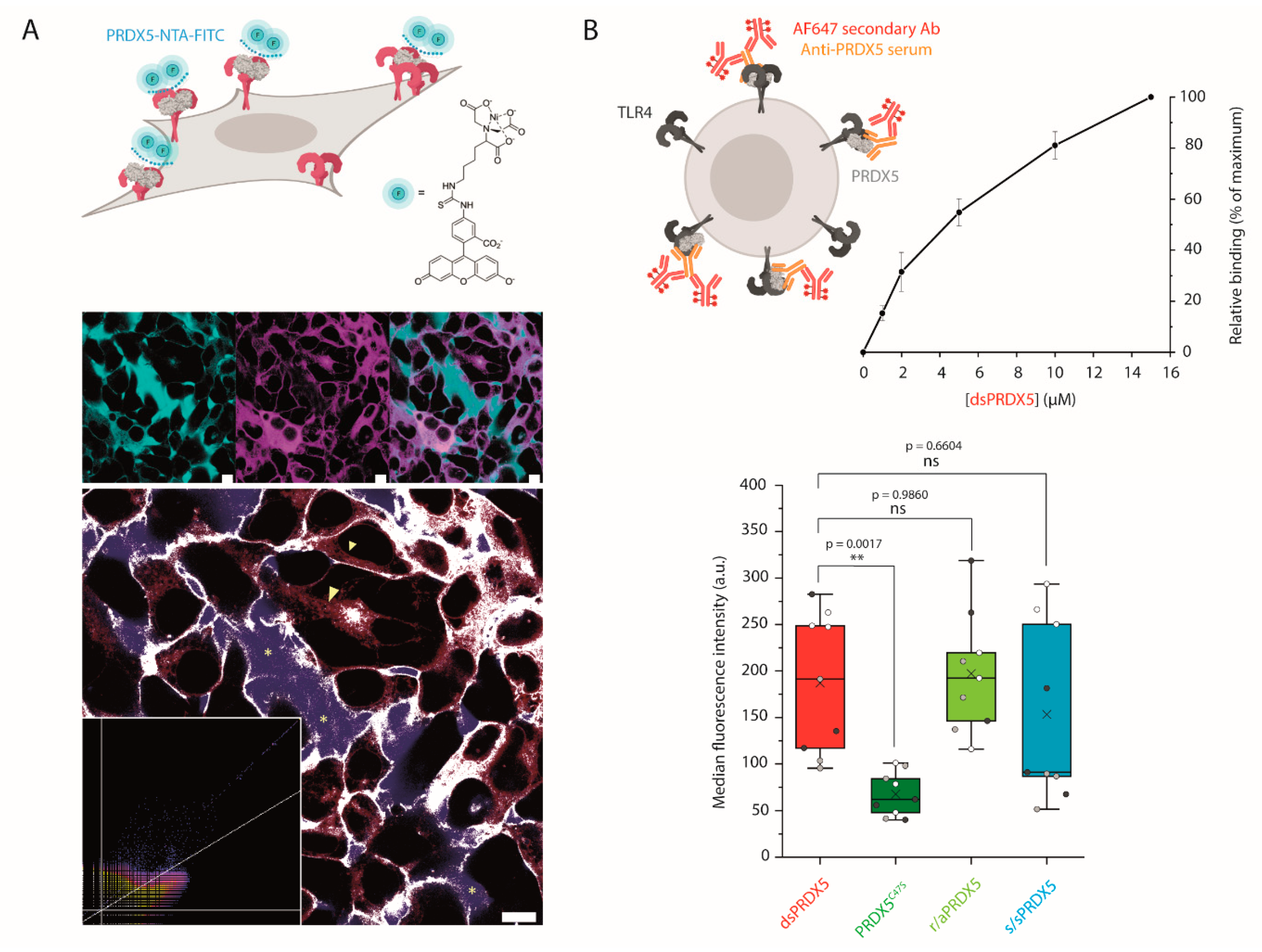
3.4. PRDX5C47S Shows a Lower Propensity to Bind to the Cell Surface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shau, H.; Gupta, R.K.; Golub, S.H. Identification of a Natural Killer Enhancing Factor (NKEF) from Human Erythroid Cells. Cell. Immunol. 1993, 147, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative Protein Folding by an Endoplasmic Reticulum Localized Peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.C.; Hah, Y.-S.; Kim, W.Y.; Jung, B.G.; Jang, H.H.; Lee, J.R.; Kim, S.Y.; Lee, Y.M.; Jeon, M.G.; Kim, C.W.; et al. Oxidative Stress-Dependent Structural and Functional Switching of a Human 2-Cys Peroxiredoxin Isotype II That Enhances HeLa Cell Resistance to H2O2-Induced Cell Death. J. Biol. Chem. 2005, 280, 28775–28784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.-J.; Kang, S.W.; Yang, C.-H.; Rhee, S.G.; Ryu, S.-E. Crystal Structure of a Novel Human Peroxidase Enzyme at 2.0 Å Resolution. Nat. Struct. Biol. 1998, 5, 400–406. [Google Scholar] [CrossRef]
- Hirotsu, S.; Abe, Y.; Okada, K.; Nagahara, N.; Hori, H.; Nishino, T.; Hakoshima, T. Crystal Structure of a Multifunctional 2-Cys Peroxiredoxin Heme-Binding Protein 23 KDa/Proliferation-Associated Gene Product. Proc. Natl. Acad. Sci. USA 1999, 96, 12333–12338. [Google Scholar] [CrossRef] [Green Version]
- Declercq, J.P.; Evrard, C.; Clippe, A.; Stricht, D.V.; Bernard, A.; Knoops, B. Crystal Structure of Human Peroxiredoxin 5, a Novel Type of Mammalian Peroxiredoxin at 1.5 A Resolution. J. Mol. Biol. 2001, 311, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the Antioxidant Enzyme 1-CYS Peroxiredoxin Requires Glutathionylation Mediated by Heterodimerization with Pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a New Type of Mammalian Peroxiredoxin That Forms an Intramolecular Disulfide as a Reaction Intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef] [Green Version]
- Peshenko, I.V.; Novoselov, V.I.; Evdokimov, V.A.; Nikolaev, T.M.; Shuvaeva, V.M.; Lipkin, V.M.; Fesenko, E.E. Novel 28-KDa Secretory Protein from Rat Olfactory Epithelium. FEBS Lett. 1996, 381, 12–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.-S.; Kang, S.W.; Woo, H.A.; Hwang, S.C.; Chae, H.Z.; Kim, K.; Rhee, S.G. Inactivation of Human Peroxiredoxin I during Catalysis as the Result of the Oxidation of the Catalytic Site Cysteine to Cysteine-Sulfinic Acid. J. Biol. Chem. 2002, 277, 38029–38036. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.A.; Chae, H.Z.; Hwang, S.C.; Yang, K.-S.; Kang, S.W.; Kim, K.; Rhee, S.G. Reversing the Inactivation of Peroxiredoxins Caused by Cysteine Sulfinic Acid Formation. Science 2003, 300, 653–656. [Google Scholar] [CrossRef]
- Lim, J.C.; Choi, H.-I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.-S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible Oxidation of the Active-Site Cysteine of Peroxiredoxin to Cysteine Sulfonic Acid for Enhanced Molecular Chaperone Activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [Green Version]
- Riddell, J.R.; Wang, X.-Y.; Minderman, H.; Gollnick, S.O. Peroxiredoxin 1 Stimulates Secretion of Pro-Inflammatory Cytokines by Binding to Toll-like Receptor 4. J. Immunol. Baltim. Md 1950 2010, 184, 1022–1030. [Google Scholar] [CrossRef]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin Family Proteins Are Key Initiators of Post-Ischemic Inflammation in the Brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Rafikova, O.; Lamoke, F.; Gutsaeva, D.; Jahng, W.J.; Repossi, A.; Facchiano, F.; Bartoli, M. Modulation of Toll-like Receptor 4 Signaling in Human Diabetic Retina by Peroxiredoxin 1. Invest. Ophthalmol. Vis. Sci. 2015, 56, 4288. [Google Scholar]
- Lu, Y.; Zhang, X.-S.; Zhang, Z.-H.; Zhou, X.-M.; Gao, Y.-Y.; Liu, G.-J.; Wang, H.; Wu, L.-Y.; Li, W.; Hang, C.-H. Peroxiredoxin 2 Activates Microglia by Interacting with Toll-like Receptor 4 after Subarachnoid Hemorrhage. J. Neuroinflammation 2018, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, S.; Tang, D.; Peng, Y.; Meng, J.; Peng, S.; Deng, Z.; Qiu, S.; Liao, X.; Chen, H.; et al. Circulating Peroxiredoxin-1 Is a Novel Damage-Associated Molecular Pattern and Aggravates Acute Liver Injury via Promoting Inflammation. Free Radic. Biol. Med. 2019, 137, 24–36. [Google Scholar] [CrossRef]
- Nakamura, K.; Shichita, T. Cellular and Molecular Mechanisms of Sterile Inflammation in Ischaemic Stroke. J. Biochem. (Tokyo) 2019, 165, 459–464. [Google Scholar] [CrossRef] [Green Version]
- Knoops, B.; Becker, S.; Poncin, M.A.; Glibert, J.; Derclaye, S.; Clippe, A.; Alsteens, D. Specific Interactions Measured by AFM on Living Cells between Peroxiredoxin-5 and TLR4: Relevance for Mechanisms of Innate Immunity. Cell Chem. Biol. 2018, 25, 550–559. [Google Scholar] [CrossRef]
- Plaisant, F.; Clippe, A.; Vander Stricht, D.; Knoops, B.; Gressens, P. Recombinant Peroxiredoxin 5 Protects against Excitotoxic Brain Lesions in Newborn Mice. Free Radic. Biol. Med. 2003, 34, 862–872. [Google Scholar] [CrossRef]
- Woo, H.A.; Rhee, S.G. Immunoblot Detection of Proteins That Contain Cysteine Sulfinic or Sulfonic Acids with Antibodies Specific for the Hyperoxidized Cysteine-Containing Sequence. In Methods Redox Signal; Mary Ann Liebert: New Rochelle, NY, 2010; pp. 19–23. ISBN 978-1-934857-06-8. [Google Scholar]
- Goemaere, J.; Knoops, B. Peroxiredoxin Distribution in the Mouse Brain with Emphasis on Neuronal Populations Affected in Neurodegenerative Disorders. J. Comp. Neurol. 2012, 520, 258–280. [Google Scholar] [CrossRef]
- Denoncin, K.; Vertommen, D.; Arts, I.S.; Goemans, C.V.; Rahuel-Clermont, S.; Messens, J.; Collet, J.-F. A New Role for Escherichia Coli DsbC Protein in Protection against Oxidative Stress. J. Biol. Chem. 2014, 289, 12356–12364. [Google Scholar] [CrossRef] [Green Version]
- Arts, I.S.; Vertommen, D.; Baldin, F.; Laloux, G.; Collet, J.-F. Comprehensively Characterizing the Thioredoxin Interactome In Vivo Highlights the Central Role Played by This Ubiquitous Oxidoreductase in Redox Control. Mol. Cell. Proteomics MCP 2016, 15, 2125–2140. [Google Scholar] [CrossRef] [Green Version]
- Butt, H.-J.; Jaschke, M. Calculation of Thermal Noise in Atomic Force Microscopy. Nanotechnology 1995, 6, 1. [Google Scholar] [CrossRef]
- Evans, E.; Ritchie, K. Dynamic Strength of Molecular Adhesion Bonds. Biophys. J. 1997, 72, 1541–1555. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.M. Analytical Descriptions of Dynamic Force Spectroscopy: Behaviour of Multiple Connections. Anal. Chim. Acta 2003, 479, 107–115. [Google Scholar] [CrossRef]
- Smeets, A.; Marchand, C.; Linard, D.; Knoops, B.; Declercq, J.-P. The Crystal Structures of Oxidized Forms of Human Peroxiredoxin 5 with an Intramolecular Disulfide Bond Confirm the Proposed Enzymatic Mechanism for Atypical 2-Cys Peroxiredoxins. Arch. Biochem. Biophys. 2008, 477, 98–104. [Google Scholar] [CrossRef]
- Dubuisson, M.; Vander Stricht, D.; Clippe, A.; Etienne, F.; Nauser, T.; Kissner, R.; Koppenol, W.H.; Rees, J.-F.; Knoops, B. Human Peroxiredoxin 5 Is a Peroxynitrite Reductase. FEBS Lett. 2004, 571, 161–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banmeyer, I.; Marchand, C.; Clippe, A.; Knoops, B. Human Mitochondrial Peroxiredoxin 5 Protects from Mitochondrial DNA Damages Induced by Hydrogen Peroxide. FEBS Lett. 2005, 579, 2327–2333. [Google Scholar] [CrossRef] [Green Version]
- Barelier, S.; Linard, D.; Pons, J.; Clippe, A.; Knoops, B.; Lancelin, J.-M.; Krimm, I. Discovery of Fragment Molecules That Bind the Human Peroxiredoxin 5 Active Site. PLOS ONE 2010, 5, e9744. [Google Scholar] [CrossRef] [PubMed]
- Sardi, F.; Manta, B.; Portillo-Ledesma, S.; Knoops, B.; Comini, M.A.; Ferrer-Sueta, G. Determination of Acidity and Nucleophilicity in Thiols by Reaction with Monobromobimane and Fluorescence Detection. Anal. Biochem. 2013, 435, 74–82. [Google Scholar] [CrossRef]
- Portillo-Ledesma, S.; Sardi, F.; Manta, B.; Tourn, M.V.; Clippe, A.; Knoops, B.; Alvarez, B.; Coitiño, E.L.; Ferrer-Sueta, G. Deconstructing the Catalytic Efficiency of Peroxiredoxin-5 Peroxidatic Cysteine. Biochemistry 2014, 53, 6113–6125. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Ledesma, S.; Randall, L.M.; Parsonage, D.; Dalla Rizza, J.; Karplus, P.A.; Poole, L.B.; Denicola, A.; Ferrer-Sueta, G. Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry 2018, 57, 3416–3424. [Google Scholar] [CrossRef] [PubMed]
- Baković, J.; Yu, B.Y.K.; Silva, D.; Chew, S.P.; Kim, S.; Ahn, S.-H.; Palmer, L.; Aloum, L.; Stanzani, G.; Malanchuk, O.; et al. A Key Metabolic Integrator, Coenzyme A, Modulates the Activity of Peroxiredoxin 5 via Covalent Modification. Mol. Cell. Biochem. 2019, 461, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Mann, M.; Meng, C.K.; Fenn, J.B. Interpreting Mass Spectra of Multiply Charged Ions. Anal. Chem. 1989, 61, 1702–1708. [Google Scholar] [CrossRef]
- Reinhold, B.B.; Reinhold, V.N. Electrospray Ionization Mass Spectrometry: Deconvolution by an Entropy-Based Algorithm. J. Am. Soc. Mass Spectrom. 1992, 3, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Ferrige, A.; Seddon, M.J.; Green, B.; Jarvis, S.; Skilling, J.; Staunton, J. Disentangling Electrospray Spectra with Maximum Entropy. Rapid Commun. Mass Spectrom. 1992, 6, 707–711. [Google Scholar] [CrossRef]
- Apostol, I.; Aitken, J.; Levine, J.; Lippincott, J.; Davidson, J.S.; Abbott-Brown, D. Recombinant Protein Sequences Can Trigger Methylation of N-Terminal Amino Acids in Escherichia Coli. Protein Sci. Publ. Protein Soc. 1995, 4, 2616–2618. [Google Scholar] [CrossRef] [Green Version]
- Schwark, D.G.; Schmitt, M.A.; Fisk, J.D. Directed Evolution of the Methanosarcina Barkeri Pyrrolysyl TRNA/Aminoacyl TRNA Synthetase Pair for Rapid Evaluation of Sense Codon Reassignment Potential. Int. J. Mol. Sci. 2021, 22, E895. [Google Scholar] [CrossRef]
- Barranco-Medina, S.; Lázaro, J.-J.; Dietz, K.-J. The Oligomeric Conformation of Peroxiredoxins Links Redox State to Function. FEBS Lett. 2009, 583, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, Mechanism and Regulation of Peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.-P. Peroxiredoxin 5: Structure, Mechanism, and Function of the Mammalian Atypical 2-Cys Peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Evrard, C.; Capron, A.; Marchand, C.; Clippe, A.; Wattiez, R.; Soumillion, P.; Knoops, B.; Declercq, J.-P. Crystal Structure of a Dimeric Oxidized Form of Human Peroxiredoxin 5. J. Mol. Biol. 2004, 337, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a Receptor for Complexes of Lipopolysaccharide (LPS) and LPS Binding Protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a Molecule That Confers Lipopolysaccharide Responsiveness on Toll-like Receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.M. Dynamic Lipopolysaccharide Transfer Cascade to TLR4/MD2 Complex via LBP and CD14. BMB Rep. 2017, 50, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 Signals through Toll-like Receptor (TLR) 4 and TLR2. Shock Augusta Ga 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A Critical Cysteine Is Required for HMGB1 Binding to Toll-like Receptor 4 and Activation of Macrophage Cytokine Release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually Exclusive Redox Forms of HMGB1 Promote Cell Recruitment or Proinflammatory Cytokine Release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Matsuguchi, T.; Tsuboi, N.; Yajima, T.; Yoshikai, Y. Differences in Expression of Toll-like Receptors and Their Reactivities in Dendritic Cells in BALB/c and C57BL/6 Mice. Infect. Immun. 2002, 70, 6638–6645. [Google Scholar] [CrossRef] [Green Version]
- López, L.; Chiribao, M.L.; Girard, M.C.; Gómez, K.A.; Carasi, P.; Fernandez, M.; Hernandez, Y.; Robello, C.; Freire, T.; Piñeyro, M.D. The Cytosolic Tryparedoxin Peroxidase from Trypanosoma Cruzi Induces a Pro-Inflammatory Th1 Immune Response in a Peroxidatic Cysteine-Dependent Manner. Immunology 2021, 163, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, A.R.; Cannistraro, S. Dynamic Force Spectroscopy and Biomolecular Recognition; CRC Press: Boca Raton, FL, USA, 2012; ISBN 978-0-429-06733-4. [Google Scholar]
- Alsteens, D.; Newton, R.; Schubert, R.; Martinez-Martin, D.; Delguste, M.; Roska, B.; Müller, D.J. Nanomechanical Mapping of First Binding Steps of a Virus to Animal Cells. Nat. Nanotechnol. 2017, 12, 177–183. [Google Scholar] [CrossRef]
- Delguste, M.; Zeippen, C.; Machiels, B.; Mast, J.; Gillet, L.; Alsteens, D. Multivalent Binding of Herpesvirus to Living Cells Is Tightly Regulated during Infection. Sci. Adv. 2018, 4, eaat1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, E.A.; Calderwood, D.A. Forces and Bond Dynamics in Cell Adhesion. Science 2007, 316, 1148–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, E.; Williams, P. Dynamic Force Spectroscopy. Physics of Bio-Molecules and Cells. Physique des Biomolécules et des Cellules; Flyvbjerg, F., Jülicher, F., Ormos, P., David, F., Eds.; Springer: Berlin, Heidelberg, 2002; pp. 145–204. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poncin, M.A.; Van Meerbeeck, P.; Simpson, J.D.; Clippe, A.; Tyckaert, F.; Bouillenne, F.; Degand, H.; Matagne, A.; Morsomme, P.; Knoops, B.; et al. Role of the Redox State of Human Peroxiredoxin-5 on Its TLR4-Activating DAMP Function. Antioxidants 2021, 10, 1902. https://doi.org/10.3390/antiox10121902
Poncin MA, Van Meerbeeck P, Simpson JD, Clippe A, Tyckaert F, Bouillenne F, Degand H, Matagne A, Morsomme P, Knoops B, et al. Role of the Redox State of Human Peroxiredoxin-5 on Its TLR4-Activating DAMP Function. Antioxidants. 2021; 10(12):1902. https://doi.org/10.3390/antiox10121902
Chicago/Turabian StylePoncin, Mégane A., Pierre Van Meerbeeck, Joshua D. Simpson, André Clippe, François Tyckaert, Fabrice Bouillenne, Hervé Degand, André Matagne, Pierre Morsomme, Bernard Knoops, and et al. 2021. "Role of the Redox State of Human Peroxiredoxin-5 on Its TLR4-Activating DAMP Function" Antioxidants 10, no. 12: 1902. https://doi.org/10.3390/antiox10121902
APA StylePoncin, M. A., Van Meerbeeck, P., Simpson, J. D., Clippe, A., Tyckaert, F., Bouillenne, F., Degand, H., Matagne, A., Morsomme, P., Knoops, B., & Alsteens, D. (2021). Role of the Redox State of Human Peroxiredoxin-5 on Its TLR4-Activating DAMP Function. Antioxidants, 10(12), 1902. https://doi.org/10.3390/antiox10121902