
Oxidative Stress-Related Mechanisms in Melanoma and in the Acquired Resistance to Targeted Therapies

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Melanoma and Targeted Therapies
2. Oxidative Stress in Physiopathology and in Cancer
3. Oxidative Stress in Melanoma
3.1. ROS/RNS in Melanoma
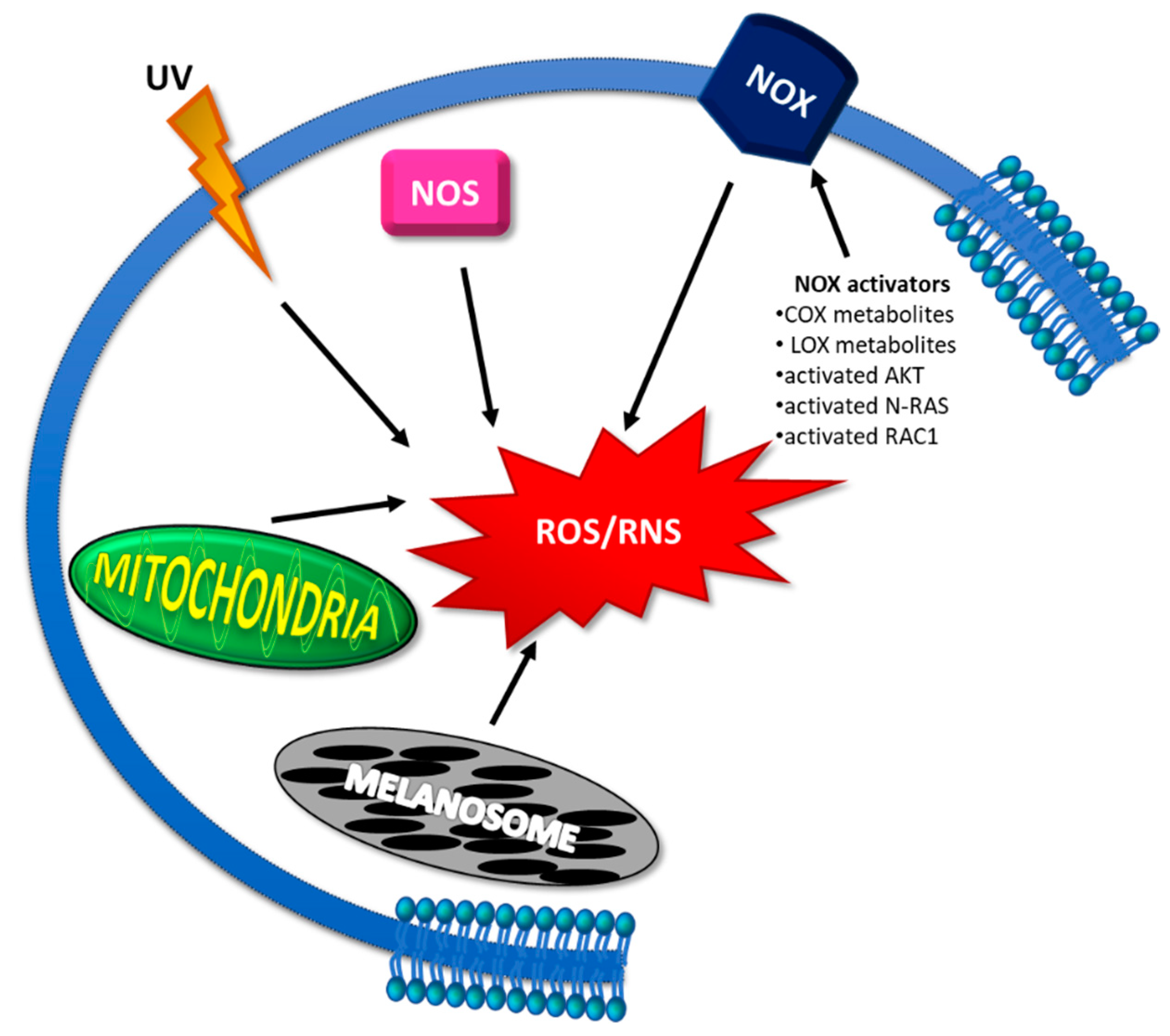
3.1.1. Reasons for ROS/RNS Increase
3.1.2. Consequences of ROS/RNS Increase
3.1.3. Enhancing ROS Production as Melanoma Anticancer Therapy
3.2. Antioxidant Systems in Melanoma
3.2.1. Nfr2 and Its Signaling Pathway
3.2.2. Other Antioxidant Enzymes and Molecules
3.2.3. Targeting Redox Homeostasis as Melanoma Anticancer Therapy
3.3. Lipid Peroxidation in Melanoma
3.4. Enzymatic Systems Detoxifying LPO Products in Melanoma
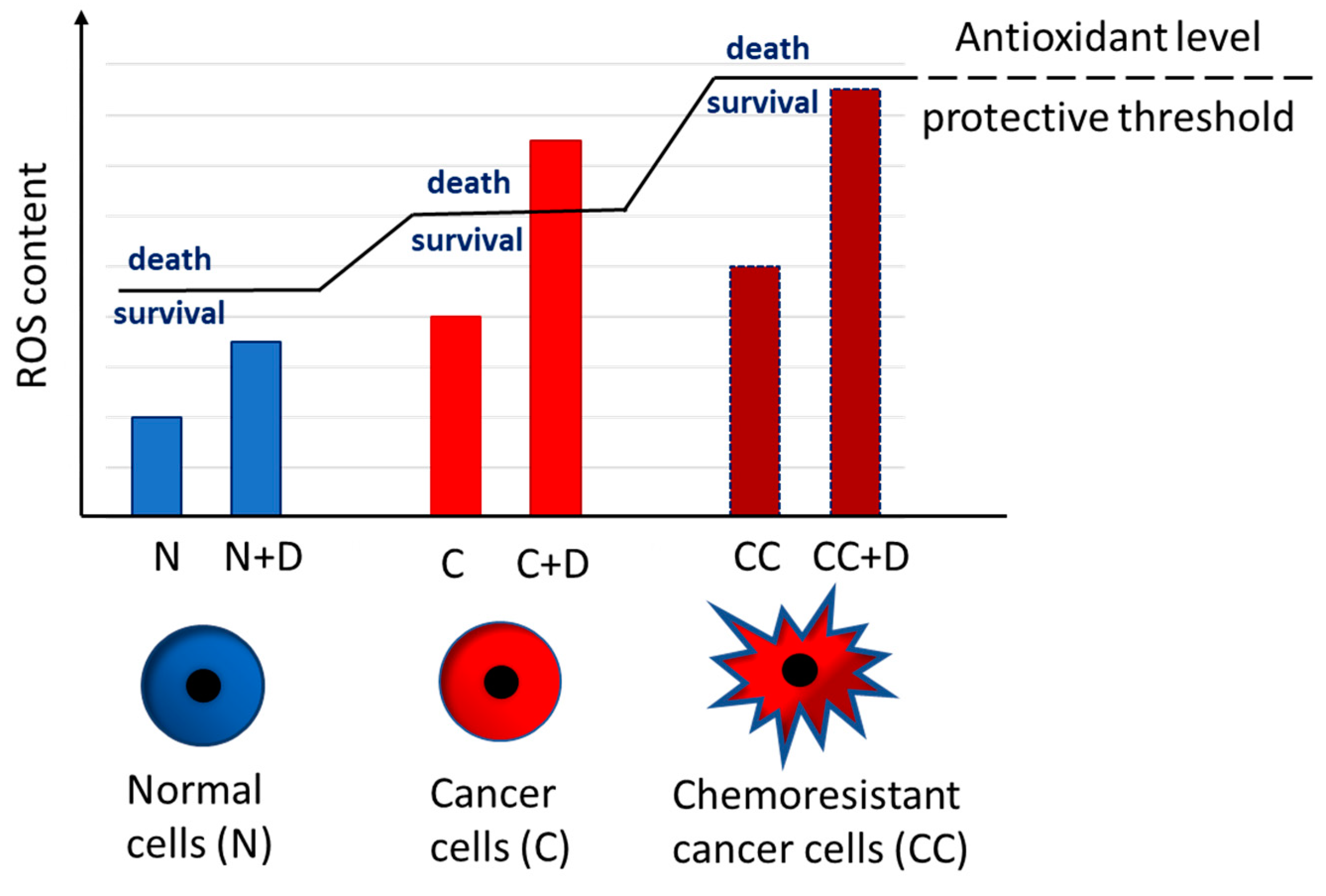
4. Oxidative Stress in Resistance to Targeted Therapies in Melanoma
4.1. Therapeutic Strategies to Overcome MAPKi Resistance
5. Microbiota, Oxidative Stress, and Melanoma
6. Conclusions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984, Erratum in Lancet 2019, 393, 746. [Google Scholar] [CrossRef]
- Tarver, T. Cancer Facts and Figures; America Cancer Society: Atlanta, GA, USA, 2012. [Google Scholar]
- Ribero, S.; Longo, C.; Glass, D.; Nathan, P.; Bataille, V. What Is New in Melanoma Genetics and Treatment? Dermatology 2016, 232, 259–264. [Google Scholar] [CrossRef]
- Quaglino, P.; Fava, P.; Tonella, L.; Rubatto, M.; Ribero, S.; Fierro, M.T. Treatment of Advanced Metastatic Melanoma. Dermatol. Pract. Concept. 2021, 11 (Suppl. S1), e2021164S. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.; Bradley, W.D.; Wang, Q.; Yang, H.; Xu, L.; Higgins, B.; Kolinsky, K.; Packman, K.; Kim, M.J.; Trunzer, K.; et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012, 72, 969–978. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Grob, J.J.; Amonkar, M.M.; Karaszewska, B.; Schachter, J.; Dummer, R.; Mackiewicz, A.; Stroyakovskiy, D.; Drucis, K.; Grange, F.; Chiarion-Sileni, V.; et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): Results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015, 16, 1389–1398. [Google Scholar] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Flaherty, K.; Goff, S. Emerging Strategies in Systemic Therapy for the Treatment of Melanoma. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 751–758. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.V.; Basile, K.J.; Kugel, C.H., 3rd; Witkiewicz, A.K.; Le, K.; Amaravadi, R.K.; Karakousis, G.C.; Xu, X.; Xu, W.; Schuchter, L.M.; et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Investig. 2013, 123, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glitza, I.C.; Davies, M.A. Genotyping of cutaneous melanoma. Chin. Clin. Oncol. 2014, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. On Behalf Of Scito Youth. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Moerke, N.J.; Niepel, M.; Zhang, T.; Gray, N.S.; Sorger, P.K. Systematic analysis of BRAF(V600E) melanomas reveals a role for JNK/c-Jun pathway in adaptive resistance to drug-induced apoptosis. Mol. Syst. Biol. 2015, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M.; et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signal. 2018, ra82. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Abel, E.V.; Koomen, J.M.; Fang, B.; Wood, E.R.; Chen, Y.A.; Fisher, K.J.; Iyengar, S.; Dahlman, K.B.; Wargo, J.A.; et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene 2016, 35, 1225–1235. [Google Scholar] [CrossRef] [Green Version]
- Titz, B.; Lomova, A.; Le, A.; Hugo, W.; Kong, X.; Ten Hoeve, J.; Friedman, M.; Shi, H.; Moriceau, G.; Song, C.; et al. JUN dependency in distinct early and late BRAF inhibition adaptation states of melanoma. Cell Discov. 2016, 2, 16028. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Cadenas, E. Oxidative stress: Damage to intact cells and organs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1985, 311, 617–631. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [Green Version]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, G. In Memoriam: Mario Umberto Dianzani’s scientific legacy. Free Radic. Biol. Med. 2016, 92, 165–166. [Google Scholar] [CrossRef]
- Comporti, M.; Saccocci, C.; Dianzani, M.U. Effect of CCl-4 in vitro and in vivo on lipid peroxidation of rat liver homogenates and subcellular fractions. Enzymologia 1965, 29, 185–204. [Google Scholar]
- Dianzani, M.U. Hermann Esterbauer. Free Radic. Biol. Med. 1997, 23, 835–837. [Google Scholar]
- Benedetti, A.; Comporti, M.; Esterbauer, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta 1980, 620, 281–296. [Google Scholar] [CrossRef]
- Dianzani, M.U. 4-Hydroxynonenal from pathology to physiology. Mol. Asp. Med. 2003, 24, 263–272. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Dianzani, M.U. Lipid peroxidation: Control of cell proliferation, cell differentiation and cell death. Mol. Asp. Med. 2008, 29, 1–8. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G.; Pizzimenti, S.; Ciamporcero, E.S.; Daga, M.; Ullio, C.; Arcaro, A.; Cetrangolo, G.P.; Ferretti, C.; Dianzani, C.; Lepore, A.; et al. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid. Redox Signal. 2015, 22, 1681–1702. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G.; Gentile, F.; Pizzimenti, S.; Canuto, R.A.; Daga, M.; Arcaro, A.; Cetrangolo, G.P.; Lepore, A.; Ferretti, C.; Dianzani, C.; et al. Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products. Antioxidants 2016, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Zarkovic, N. Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 2020, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Muzio, G.; Salvo, R.A.; Taniguchi, N.; Maggiora, M.; Canuto, R.A. 4-Hydroxynonenal metabolism by aldo/keto reductase in hepatoma cells. Adv. Exp. Med. Biol. 1999, 463, 445–452. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [Green Version]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell. Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Kim, E.K.; Jang, M.; Song, M.J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Mijatović, S.; Savić-Radojević, A.; Plješa-Ercegovac, M.; Simić, T.; Nicoletti, F.; Maksimović-Ivanić, D. The Double-Faced Role of Nitric Oxide and Reactive Oxygen Species in Solid Tumors. Antioxidants 2020, 9, 374. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L., Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meierjohann, S. Oxidative stress in melanocyte senescence and melanoma transformation. Eur. J. Cell Biol. 2014, 93, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [Green Version]
- Xian, D.; Lai, R.; Song, J.; Xiong, X.; Zhong, J. Emerging Perspective: Role of Increased ROS and Redox Imbalance in Skin Carcinogenesis. Oxid. Med. Cell. Longev. 2019, 2019, 8127362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic. Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Salimian Rizi, B.; Achreja, A.; Nagrath, D. Nitric Oxide: The Forgotten Child of Tumor Metabolism. Trends Cancer 2017, 3, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, P.; Pani, G.; Giannoni, E.; Taddei, L.; Colavitti, R.; Raugei, G.; Symons, M.; Borrello, S.; Galeotti, T.; Ramponi, G. Reactive oxygen species as essential mediators of cell adhesion: The oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell. Biol. 2003, 161, 933–944. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Bittinger, F.; González-García, J.L.; Klein, C.L.; Brochhausen, C.; Offner, F.; Kirkpatrick, C.J. Production of superox-ide by human malignant melanoma cells. Melanoma Res. 1998, 8, 381–387. [Google Scholar] [CrossRef]
- Wittgen, H.G.M.; van Kempen, L.C.L.T. Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res. 2007, 17, 400–409. [Google Scholar] [CrossRef]
- Terra, V.A.; Souza-Neto, F.P.; Pereira, R.C.; Silva, T.N.; Costa, A.C.; Luiz, R.C.; Cecchini, R.; Cecchini, A.L. Time-dependent reactive species formation and oxidative stress damage in the skin after UVB irradiation. J. Photochem. Photobiol. B 2012, 109, 34–41. [Google Scholar] [CrossRef]
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to cellular DNA by UVB and UVA radiation. Photochem. Photobiol. 2015, 91, 140–155. [Google Scholar] [CrossRef]
- Valencia, A.; Kochevar, I.E. Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes. J. Investig. Dermatol. 2008, 128, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef] [Green Version]
- Nasti, T.H.; Timares, L. MC1R, eumelanin and pheomelanin: Their role in determining the susceptibility to skin cancer. Photochem. Photobiol. 2015, 91, 188–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer. 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19, 2667. [Google Scholar] [CrossRef] [Green Version]
- Kadekaro, A.L.; Kavanagh, R.; Kanto, H.; Terzieva, S.; Hauser, J.; Kobayashi, N.; Schwemberger, S.; Cornelius, J.; Babcock, G.; Shertzer, H.G.; et al. alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res. 2005, 65, 4292–4299. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Brar, S.S.; Kennedy, T.P.; Sturrock, A.B.; Huecksteadt, T.P.; Quinn, M.T.; Whorton, A.R.; Hoidal, J.R. An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am. J. Physiol. Cell. Physiol. 2002, 282, C1212–C1224. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gomez Garcia, A.M.; Meyskens, F.L., Jr. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. J. Investig. Dermatol. 2012, 132, 2033–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyano, K.; Ueno, N.; Takeya, R.; Sumimoto, H. Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J. Biol. Chem. 2006, 28, 21857–21868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Kaibuchi, K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell. Biol. 2001, 2, 887–897. [Google Scholar] [CrossRef]
- Stalin, J.; Garrido-Urbani, S.; Heitz, F.; Szyndralewiez, C.; Jemelin, S.; Coquoz, O.; Ruegg, C.; Imhof, B.A. Inhibition of host NOX1 blocks tumor growth and enhances checkpoint inhibitor-based immunotherapy. Life Sci Alliance 2019, 2, e201800265. [Google Scholar] [CrossRef] [Green Version]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation pheno-type of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [Green Version]
- Kircher, D.A.; Arave, R.A.; Cho, J.H.; Holmen, S.L. Melanoma metastases caught in the AKT. Mol. Cell. Oncol. 2016, 3, e1128516. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Pereira, C.; Moraes, J.A.; Souza, M.d.J.; Laurindo, F.R.; Arruda, M.A.; Barja-Fidalgo, C. Redox modulation of FAK controls melanoma survival—Role of NOX4. PLoS ONE 2014, 9, e99481. [Google Scholar] [CrossRef]
- Antony, S.; Jiang, G.; Wu, Y.; Meitzler, J.L.; Makhlouf, H.R.; Haines, D.C.; Butcher, D.; Hoon, D.S.; Ji, J.; Zhang, Y.; et al. NADPH oxidase 5 (NOX5)-induced reactive oxygen signaling modulates normoxic HIF-1α and p27Kip1 expression in malignant melanoma and other human tumors. Mol. Carcinog. 2017, 56, 2643–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef]
- Yang, Z.; Misner, B.; Ji, H.; Poulos, T.L.; Silverman, R.B.; Meyskens, F.L.; Yang, S. Targeting Nitric Oxide Signaling with nNOS Inhibitors As a Novel Strategy for the Therapy and Prevention of Human Melanoma. Antioxid. Redox Signal. 2013, 19, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rivera, E.; Jayaraman, P.; Parikh, F.; Davies, M.A.; Ekmekcioglu, S.; Izadmehr, S.; Milton, D.R.; Chipuk, J.E.; Grimm, E.A.; Estrada, Y.; et al. Inducible Nitric Oxide Synthase Drives mTOR Pathway Activation and Proliferation of Human Melanoma by Reversible Nitrosylation of TSC2. Cancer Res. 2014, 74, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Sikora, A.G.; Gelbard, A.; Davies, M.A.; Sano, D.; Ekmekcioglu, S.; Kwon, J.; Hailemichael, Y.; Jayaraman, P.; Myers, J.N.; Grimm, E.A.; et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1834–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekmekcioglu, S.; Ellerhorst, J.; Smid, C.M.; Prieto, V.G.; Munsell, M.; Buzaid, A.C.; Grimm, E.A. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 4768–4775. [Google Scholar]
- Lahdenranta, J.; Hagendoorn, J.; Padera, T.P.; Hoshida, T.; Nelson, G.; Kashiwagi, S.; Jain, R.K.; Fukumura, D. Endothelial nitric oxide synthase mediates lymphangiogenesis and lymphatic metastasis. Cancer Res. 2009, 69, 2801–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhart, V.; Reiß, K.; Kollau, A.; Mayer, B.; Gorren, A.C.F. Site and mechanism of uncoupling of nitric-oxide synthase: Uncoupling by monomerization and other misconceptions. Nitric Oxide 2019, 89, 14–21. [Google Scholar] [CrossRef]
- Li, H.; Förstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 61–67. [Google Scholar] [CrossRef]
- Melo, F.H.; Molognoni, F.; Morais, A.S.; Toricelli, M.; Mouro, M.G.; Higa, E.M.; Lopes, J.D.; Jasiulionis, M.G. Endothelial nitric oxide synthase uncoupling as a key mediator of melanocyte malignant transformation associated with sustained stress conditions. Free Radic. Biol. Med. 2011, 50, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef]
- Reich, R.; Martin, G.R. Identification of arachidonic acid pathways required for the invasive and metastatic activity of malignant tumor cells. Prostaglandins 1996, 51, 1–17. [Google Scholar] [CrossRef]
- Zhang, H.J.; Zhao, W.; Venkataraman, S.; Robbins, M.E.; Buettner, G.R.; Kregel, K.C.; Oberley, L.W. Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J. Biol. Chem. 2002, 277, 20919–20926. [Google Scholar] [CrossRef] [Green Version]
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 2001, 63, 88–102. [Google Scholar] [CrossRef]
- Gorini, F.; Scala, G.; Cooke, M.S.; Majello, B.; Amente, S. Towards a comprehensive view of 8-oxo-7,8-dihydro-2′-deoxyguanosine: Highlighting the intertwined roles of DNA damage and epigenetics in genomic instability. DNA Repair 2021, 97, 103027. [Google Scholar] [CrossRef] [PubMed]
- Murtas, D.; Piras, F.; Minerba, L.; Ugalde, J.; Floris, C.; Maxia, C.; Demurtas, P.; Perra, M.T.; Sirigu, P. Nuclear 8-hydroxy-2′-deoxyguanosine as survival biomarker in patients with cutaneous melanoma. Oncol. Rep. 2010, 23, 329–335. [Google Scholar]
- Farhood, B.; Najafi, M.; Salehi, E.; Hashemi Goradel, N.; Nashtaei, M.S.; Khanlarkhani, N.; Mortezaee, K. Disruption of the redox balance with either oxidative or anti-oxidative overloading as a promising target for cancer therapy. J. Cell. Biochem. 2019, 120, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer. Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, C.; Kruger, C.A.; Abrahamse, H. Photodynamic Therapy for Metastatic Melanoma Treatment: A Review. Technol. Cancer Res. Treat. 2018, 17, 1533033818791795. [Google Scholar] [CrossRef] [Green Version]
- Jabir, N.R.; Tabrez, S.; Ashraf, G.M.; Shakil, S.; Damanhouri, G.A.; Kamal, M.A. Nanotechnology-based approaches in anticancer research. Int. J. Nanomed. 2012, 7, 4391–4408. [Google Scholar] [CrossRef] [Green Version]
- Dianzani, C.; Zara, G.P.; Maina, G.; Pettazzoni, P.; Pizzimenti, S.; Rossi, F.; Gigliotti, C.L.; Ciamporcero, E.S.; Daga, M.; Barrera, G. Drug delivery nanoparticles in skin cancers. Biomed. Res. Int. 2014, 2014, 895986. [Google Scholar] [CrossRef]
- El-Kenawy, A.E.M.; Constantin, C.; Hassan, S.M.A.; Mostafa, A.M.; Neves, A.F.; de Araújo, T.G.; Neagu, M. Nanomedicine in Melanoma: Current Trends and Future Perspectives. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017; Volume 10. [Google Scholar] [CrossRef]
- Beiu, C.; Giurcaneanu, C.; Grumezescu, A.M.; Holban, A.M.; Popa, L.G.; Mihai, M.M. Nanosystems for Improved Targeted Therapies in Melanoma. J. Clin. Med. 2020, 9, 318. [Google Scholar] [CrossRef] [Green Version]
- Clemente, N.; Argenziano, M.; Gigliotti, C.L.; Ferrara, B.; Boggio, E.; Chiocchetti, A.; Caldera, F.; Trotta, F.; Benetti, E.; Annaratone, L.; et al. Paclitaxel-Loaded Nanosponges Inhibit Growth and Angiogenesis in Melanoma Cell Models. Front. Pharmacol. 2019, 10, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudlowitz, D.; Muggia, F. Nanoparticle albumin-bound paclitaxel (nab-paclitaxel): Extending its indications. Expert Opin. Drug Saf. 2014, 13, 681–685. [Google Scholar] [CrossRef]
- Specenier, P. Efficacy of nab-paclitaxel in treating metastatic melanoma. Expert Opin. Pharmacother. 2019, 20, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.N.; Suman, V.J.; Javed, A.; Reid, J.M.; Wall, D.J.; Erickson, L.A.; Ernstoff, M.; Anderson, D.M. Sequencing Ipilimumab Immunotherapy Before or After Chemotherapy (Nab-Paclitaxel and Bevacizumab) for the Treatment of BRAFwt (BRAF Wild-Type) Metastatic Malignant Melanoma: Results of a Study of Academic and Community Cancer Research United (ACCRU) RU261206I. Am. J. Clin. Oncol. 2020, 43, 115–121. [Google Scholar] [CrossRef]
- Mayola, E.; Gallerne, C.; Esposti, D.D.; Martel, C.; Pervaiz, S.; Larue, L.; Debuire, B.; Lemoine, A.; Brenner, C.; Lemaire, C. Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2. Apoptosis 2011, 16, 1014–1027. [Google Scholar] [CrossRef]
- Hsiao, Y.P.; Tsai, C.H.; Wu, P.P.; Hsu, S.C.; Liu, H.C.; Huang, Y.P.; Yang, J.H.; Chung, J.G. Cantharidin induces G2/M phase arrest by inhibition of Cdc25c and Cyclin A and triggers apoptosis through reactive oxygen species and the mitochondria-dependent pathways of A375.S2 human melanoma cells. Int. J. Oncol. 2014, 45, 2393–2402. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Ye, W.; Zhang, M.; Liang, J. The effects of nrf2 on tumor angiogenesis: A review of the possible mechanisms of action. Crit. Rev. Eukaryot Gene Expr. 2012, 22, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Choi, B.H.; Kwak, M.K. Shadows of NRF2 in cancer: Resistance to chemotherapy. Curr. Opin. Toxicol. 2016, 1, 20–28. [Google Scholar] [CrossRef]
- Hintsala, H.R.; Jokinen, E.; Haapasaari, K.M.; Moza, M.; Ristimäki, A.; Soini, Y.; Koivunen, J.; Karihtala, P. Nrf2/Keap1 Pathway and Expression of Oxidative Stress Lesions 8-hydroxy-2′-deoxyguanosine and Nitrotyrosine in Melanoma. Anticancer Res. 2016, 36, 1497–1506. [Google Scholar] [PubMed]
- Hintsala, H.R.; Haapasaari, K.M.; Soini, Y.; Karihtala, P. An immunohistochemical study of NFE2L2, KEAP1 and 8-hydroxy-2′-deoxyguanosine and the EMT markers SNAI2, ZEB1 and TWIST1 in metastatic melanoma. Histol. Histopathol. 2017, 32, 129–136. [Google Scholar] [CrossRef]
- Hämäläinen, M.; Teppo, H.R.; Skarp, S.; Haapasaari, K.M.; Porvari, K.; Vuopala, K.; Kietzmann, T.; Karihtala, P. NRF1 and NRF2 mRNA and Protein Expression Decrease Early during Melanoma Carcinogenesis: An Insight into Survival and MicroRNAs. Oxid. Med. Cell. Longev. 2019, 2019, 2647068. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhao, Z.; Meng, X.; Chen, H.; Fu, G. Migration and invasion in B16-F10 mouse melanoma cells are regulated by Nrf2 inhibition during treatment with ionizing radiation. Oncol. Lett. 2018, 16, 1959–1966. [Google Scholar] [CrossRef] [Green Version]
- Jessen, C.; Kreß, J.K.C.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef]
- Miura, S.; Shibazaki, M.; Kasai, S.; Yasuhira, S.; Watanabe, A.; Inoue, T.; Kageshita, Y.; Tsunoda, K.; Takahashi, K.; Akasaka, T.; et al. A somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J. Investig. Dermatol. 2014, 134, 553–556. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Jia, Z.; Trush, M.A.; Li, Y.R. Nrf2 Deficiency Promotes Melanoma Growth and Lung Metastasis. React. Oxyg. Species 2016, 2, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep. 2018, 8, 12846. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [CrossRef] [Green Version]
- Schmidlin, C.J.; Tian, W.; Dodson, M.; Chapman, E.; Zhang, D.D. FAM129B-dependent activation of NRF2 promotes an invasive phenotype in BRAF mutant melanoma cells. Mol. Carcinog. 2021, 60, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef] [Green Version]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jang, H.H. The Role of Peroxiredoxin Family in Cancer Signaling. J. Cancer Prev. 2019, 24, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Manevich, Y.; Chen, J.W.; Feinstein, S.I. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J. Biol. Chem. 1999, 274, 21326–21334. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.N.; Lee, S.B.; Lee, S.S.; Yoon, S.H.; Kang, G.Y.; Hwang, S.G.; Um, H.D. Phospholipase A2 activity of peroxiredoxin 6 promotes invasion and metastasis of lung cancer cells. Mol. Cancer Ther. 2010, 9, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, D.Y.; Ahn, S.J.; Lee, R.A.; Kim, S.W.; Park, I.A.; Chae, H.Z. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 2001, 21, 2085–2090. [Google Scholar]
- Quan, C.; Cha, E.J.; Lee, H.L.; Han, K.H.; Lee, K.M.; Kim, W.J. Enhanced expression of peroxiredoxin I and VI correlates with development, recurrence and progression of human bladder cancer. J. Urol. 2006, 175, 1512–1516. [Google Scholar] [CrossRef]
- Chang, X.Z.; Li, D.Q.; Hou, Y.F.; Wu, J.; Lu, J.S.; Di, G.H.; Jin, W.; Ou, Z.L.; Shen, Z.Z.; Shao, Z.M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, R76. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.M.; Park, K.R.; Lee, H.P.; Lee, D.H.; Jo, M.; Shin, D.H.; Yoon, D.Y.; Han, S.B.; Hong, J.T. PRDX6 promotes lung tumor progression via its GPx and iPLA2 activities. Free Radic. Biol. Med. 2014, 69, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.E.; Park, D.S.; Kim, Y.S.; Kim, B.R.; Park, S.N.; Lee, M.K.; Park, S.H.; Yoon, K.H.; Jeong, E.T.; Kim, H.R. Prx1 modulates the chemosensitivity of lung cancer to docetaxel through suppression of FOXO1-induced apoptosis. Int. J. Oncol. 2013, 43, 72–78. [Google Scholar] [CrossRef]
- Schmitt, A.; Schmitz, W.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Peroxiredoxin 6 triggers melanoma cell growth by increasing arachidonic acid-dependent lipid signalling. Biochem. J. 2015, 471, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Lokaj, K.; Meierjohann, S.; Schütz, C.; Teutschbein, J.; Schartl, M.; Sickmann, A. Quantitative differential proteome analysis in an animal model for human melanoma. J. Proteome Res. 2009, 8, 1818–1827. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola-Villava, M.; Martin-Gonzalez, M.; Lazaro, P.; Pizarro, A.; Lluch, A.; Ribas, G. Role of glutathione S-transferases in melanoma susceptibility: Association with GSTP1 rs1695 polymorphism. Br. J. Dermatol. 2012, 166, 1176–1183. [Google Scholar] [CrossRef]
- Wang, X.; Dong, H.; Li, Q.; Li, Y.; Hong, A. Thioredoxin induces Tregs to generate an immunotolerant tumor microenvironment in metastatic melanoma. Oncoimmunology 2015, 4, e1027471. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zheng, Z.; Kim, K.Y.; Jin, X.; Roh, M.R.; Jin, Z. Hypermethylation and downregulation of glutathione peroxidase 3 are related to pathogenesis of melanoma. Oncol. Rep. 2016, 36, 2737–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef]
- Papa, L.M.; Manfredi, G.; Germain, D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer 2014, 5, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Griess, B.M.; Tom, E.; Domann, F.; Teoh-Fitzgerald, M. Extracellular superoxide dismutase and its role in cancer. Free Radic. Biol. Med. 2017, 112, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Longo, D.L.; Oppenheim, J.J.; Matsushima, K. Overexpression of mitochondrial manganese superoxide dismutase promotes the survival of tumor cells exposed to interleukin-1, tumor necrosis factor, selected anticancer drugs, and ionizing radiation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1993, 7, 361–368. [Google Scholar] [CrossRef]
- Suresh, A.; Guedez, L.; Moreb, J.; Zucali, J. Overexpres-sion of manganese superoxide dismutase promotes survival in cell lines after doxorubicin treatment. Br. J. Haematol. 2003, 120, 457–463. [Google Scholar] [CrossRef]
- Bisevac, J.P.; Djukic, M.; Stanojevic, I.; Stevanovic, I.; Mijuskovic, Z.; Djuric, A.; Gobeljic, B.; Banovic, T.; Vojvodic, D. Association Between Oxidative Stress and Melanoma Progression. J. Med. Biochem. 2018, 37, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Church, S.L.; Grant, J.W.; Ridnour, L.A.; Oberley, L.W.; Swanson, P.E.; Meltzer, P.S.; Trent, J.M. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc. Natl. Acad. Sci. USA 1993, 90, 3113–3117. [Google Scholar] [CrossRef] [Green Version]
- Bracalente, C.; Ibañez, I.L.; Berenstein, A.; Notcovich, C.; Cerda, M.B.; Klamt, F.; Chernomoretz, A.; Durán, H. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Upregulation of antioxidant genes correlates with regression of melanoma malignancy and with malignant progression when downregulated. Oncotarget 2016, 7, 41154–41171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, T.D.; Diaconeasa, Z. Recent Advances in Phenolic Metabolites and Skin Cancer. Int. J. Mol. Sci. 2021, 22, 9707. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Afaq, F.; Maddodi, N.; Johnson, J.J.; Sarfaraz, S.; Ahmad, A.; Setaluri, V.; Mukhtar, H. Inhibition of human melanoma cell growth by the dietary flavonoid fisetin is associ-ated with disruption of Wnt/β-catenin signaling and decreased Mitf levels. J. Investig. Dermatol. 2011, 131, 1291–1299. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Kappes, J.C.; Katiyar, S.K. Inhibition of NADPH oxidase 1 activity and blocking the binding of cytosolic and membrane-bound proteins by honokiol inhibit migratory potential of melanoma cells. Oncotarget 2016, 7, 7899–7912. [Google Scholar] [CrossRef] [Green Version]
- Diaconeasa, Z.; Ayvaz, H.; Ruginǎ, D.; Leopold, L.; Stǎnilǎ, A.; Socaciu, C.; Tăbăran, F.; Luput, L.; Mada, D.C.; Pintea, A.; et al. Melanoma Inhibition by Anthocyanins Is Associated with the Reduction of Oxidative Stress Biomarkers and Changes in Mitochondrial Membrane Potential. Plant Foods Hum. Nutr. 2017, 72, 404–410. [Google Scholar] [CrossRef]
- Enaru, B.; Socaci, S.; Farcas, A.; Socaciu, C.; Danciu, C.; Stanila, A.; Diaconeasa, Z. Novel Delivery Systems of Polyphenols and Their Potential Health Benefits. Pharmaceuticals 2021, 14, 946. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Maddodi, N.; Abu Zaid, M.; Khan, N.; bin Hafeez, B.; Asim, M.; Suh, Y.; Yun, J.-M.; Setaluri, V.; Mukhtar, H. Lupeol inhibits growth of highly aggressive human metastatic melanoma cells in vitro and in vivo by inducing apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Cotter, M.A.; Thomas, J.; Cassidy, P.; Robinette, K.; Jenkins, N.; Florell, S.R.; Leachman, S.; Samlowski, W.E.; Grossman, D. N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet-induced melanoma in mice. Clin. Cancer Res. 2007, 13, 5952–5958. [Google Scholar] [CrossRef] [Green Version]
- Goodson, A.G.; Cotter, M.A.; Cassidy, P.; Wade, M.; Florell, S.R.; Liu, T.; Boucher, K.M.; Grossman, D. Use of oral N-acetylcysteine for protection of melanocytic nevi against UV-induced oxidative stress: Towards a novel paradigm for melanoma chemoprevention. Clin. Cancer Res. 2009, 15, 7434–7440. [Google Scholar] [CrossRef] [Green Version]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Green, A.C. Dietary Antioxidants and Melanoma: Evidence from Cohort and Intervention Studies. Nutr. Cancer 2015, 67, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddles-tun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Sznarkowska, A.; Kostecka, A.; Meller, K.; Bielawski, K.P. Inhibition of cancer antioxidant defense by natural compounds. Oncotarget 2017, 8, 15996–16016. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Shi, G.; Bian, C.; Nisar, M.F.; Guo, Y.; Wu, Y.; Li, W.; Huang, X.; Jiang, X.; Bartsch, J.W.; et al. UVA Irradiation Enhances Brusatol-Mediated Inhibition of Melanoma Growth by Downregulation of the Nrf2-Mediated Antioxidant Response. Oxid. Med. Cell. Longev. 2018, 9742154. [Google Scholar] [CrossRef] [PubMed]
- Daga, M.; Pizzimenti, S.; Dianzani, C.; Cucci, M.A.; Cavalli, R.; Grattarola, M.; Ferrara, B.; Scariot, V.; Trotta, F.; Barrera, G. Ailanthone inhibits cell growth and migration of cisplatin resistant bladder cancer cells through down-regulation of Nrf2, YAP, and c-Myc expression. Phytomedicine 2019, 56, 156–164. [Google Scholar] [CrossRef]
- Cucci, M.A.; Grattarola, M.; Dianzani, C.; Damia, G.; Ricci, F.; Roetto, A.; Trotta, F.; Barrera, G.; Pizzimenti, S. Ailanthone increases oxidative stress in CDDP-resistant ovarian and bladder cancer cells by inhibiting of Nrf2 and YAP expression through a post-translational mechanism. Free Radic. Biol. Med. 2020, 150, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Grattarola, M.; Cucci, M.A.; Roetto, A.; Dianzani, C.; Barrera, G.; Pizzimenti, S. Post-translational down-regulation of Nrf2 and YAP proteins, by targeting deubiquitinases, reduces growth and chemoresistance in pancreatic cancer cells. Free Radic. Biol. Med. 2021, 174, 202–210. [Google Scholar] [CrossRef]
- He, Y.; Peng, S.; Wang, J.; Chen, H.; Cong, X.; Chen, A.; Hu, M.; Qin, M.; Wu, H.; Gao, S.; et al. Ailanthone targets p23 to overcome MDV3100 resistance in castration-resistant prostate cancer. Nat. Commun. 2016, 7, 13122. [Google Scholar] [CrossRef]
- Yang, P.; Sun, D.; Jiang, F. Ailanthone promotes human vestibular schwannoma cell apoptosis and autophagy by downregulation of miR-21. Oncol. Res. 2018, 26, 941–948. [Google Scholar] [CrossRef]
- Gao, W.; Ge, S.; Sun, J. Ailanthone exerts anticancer effect by up-regulating miR-148a expression in MDA-MB-231 breast cancer cells and inhibiting proliferation, migration and invasion. Biomed. Pharmacother. 2019, 109, 1062–1069. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Pan, Z.; Wang, D.; Li, M.; Chen, X.; Zhou, L.; Xu, M.; Li, D.; Zheng, Q. Ailanthone induces cell cycle arrest and apoptosis in melanoma B16 and A375 cells. Biomolecules 2019, 9, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.R.; Kim, S.M.; Hwang, K.A.; Kang, J.H.; Choi, K.C. Resveratrol induced reactive oxygen species and endoplasmic reticulum stress-mediated apoptosis, and cell cycle arrest in the A375SM malignant melanoma cell line. Int. J. Mol. Med. 2018, 42, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Balyan, R.; Kudugunti, S.K.; Hamad, H.A.; Yousef, M.S.; Moridani, M.Y. Bioactivation of luteolin by tyrosinase selectively inhibits glutathione S-transferase. Chem. Biol. Interact. 2015, 240, 208–218. [Google Scholar] [CrossRef]
- De Luca, A.; Rotili, D.; Carpanese, D.; Lenoci, A.; Calderan, L.; Scimeca, M.; Mai, A.; Bonanno, E.; Rosato, A.; Geroni, C.; et al. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget 2015, 6, 4126–4143. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Auge, V.; Basaga, H.; Boada, J.; Brenke, R.; Chapple, S.; Cohen, G.; Feher, J.; Grune, T.; Lengyel, G. Pathological aspects of lipid peroxidation. Free Radic. Res. 2010, 44, 1125–1171. [Google Scholar] [CrossRef]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteom. 2013, 92, 239–247. [Google Scholar] [CrossRef]
- Rossi, M.A.; Cecchini, G. Lipid peroxidation in hepatomas of different degrees of deviation. Cell. Biochem. Funct. 1983, 1, 49–54. [Google Scholar] [CrossRef]
- Dianzani, M.U.; Canuto, R.A.; Rossi, M.A.; Poli, G.; Garcea, R.; Biocca, M.E.; Cecchini, G.; Biasi, F.; Ferro, M.; Bassi, A.M. Further experiments on lipid peroxidation in transplanted and experimental hepatomas. Toxicol. Pathol. 1984, 12, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberley, T.D.; Toyokuni, S.; Szweda, L.I. Localization of hydroxynonenal protein adducts in normal human kidney and selected human kidney cancers. Free Radic. Biol. Med. 1999, 27, 695–703. [Google Scholar] [CrossRef]
- Zanetti, D.; Poli, G.; Vizio, B.; Zingaro, B.; Chiarpotto, E.; Biasi, F. 4-hydroxynonenal and transforming growth factor-beta1 expression in colon cancer. Mol. Asp. Med. 2003, 24, 273–280. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Stankiewicz, A.; Sulkowska, M.; Sulkowski, S.; Kasacka, I. Antioxidant status and lipid peroxidation in colorectal cancer. J. Toxicol. Environ. Health A 2001, 64, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Young, O.; Crotty, T.; O’Connell, R.; O’Sullivan, J.; Curran, A.J. Levels of oxidative damage and lipid peroxidation in thyroid neoplasia. Head Neck. 2010, 32, 750–756. [Google Scholar] [CrossRef]
- Karihtala, P.; Kauppila, S.; Puistola, U.; Jukkola-Vuorinen, A. Divergent behaviour of oxidative stress markers 8-hydroxydeoxyguanosine (8-OHdG) and 4-hydroxy-2-nonenal (HNE) in breast carcinogenesis. Histopathology 2011, 58, 854–862. [Google Scholar] [CrossRef]
- Juric-Sekhar, G.; Zarkovic, K.; Waeg, G.; Cipak, A.; Zarkovic, N. Distribution of 4-hydroxynonenal-protein conjugates as a marker of lipid peroxidation and parameter of malignancy in astrocytic and ependymal tumors of the brain. Tumori 2009, 95, 762–768. [Google Scholar] [CrossRef]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol. 2003, 148, 913–922. [Google Scholar] [CrossRef]
- Woźniak, A.; Drewa, G.; Woźniak, B.; Schachtschabel, D.O. Activity of antioxidant enzymes and concentration of lipid peroxidation products in selected tissues of mice of different ages, both healthy and melanoma-bearing. Z. Gerontol. Geriatr. 2004, 37, 184–189. [Google Scholar] [CrossRef]
- Blendea, A.; Şerban, I.L.; Brănişteanu, D.C.; Brănişteanu, D. Evaluation of Immunostaining for 4-Hydroxy-2-Nonenal Receptors in Cutaneous Malignant Melanoma Immunohistochemical Study of 55 Cases. J. Mol. Biomark. Diagn. 2017, 8, 1–7. [Google Scholar]
- Pizzimenti, S.; Ferracin, M.; Sabbioni, S.; Toaldo, C.; Pettazzoni, P.; Dianzani, M.U.; Negrini, M.; Barrera, G. MicroRNA expression changes during human leukemic HL-60 cell differentiation induced by 4-hydroxynonenal, a product of lipid peroxidation. Free Radic. Biol. Med. 2009, 46, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Cucci, M.A.; Compagnone, A.; Daga, M.; Grattarola, M.; Ullio, C.; Roetto, A.; Palmieri, A.; Rosa, A.C.; Argenziano, M.; Cavalli, R.; et al. Post-translational inhibition of YAP oncogene expression by 4-hydroxynonenal in bladder cancer cells. Free Radic. Biol. Med. 2019, 141, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Martinotti, S.; Fazio, V.; Manzari, V.; Paradisi, L.; Parola, M.; Frati, L.; Dianzani, M.U. Effect of 4-hydroxynonenal on c-myc expression. Toxicol. Pathol. 1987, 15, 238–240. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Serra, A.; Ferretti, C.; Fazio, V.M.; Saglio, G.; Dianzani, M.U. 4-hydroxynonenal specifically inhibits c-myb but does not affect c-fos expressions in HL-60 cells. Biochem. Biophys. Res. Commun. 1996, 227, 589–593. [Google Scholar] [CrossRef]
- Kreuzer, T.; Zarković, N.; Grube, R.; Schaur, R.J. Inhibition of HeLa cell proliferation by 4-hydroxynonenal is associated with enhanced expression of the c-fos oncogene. Cancer Biother. Radiopharm. 1997, 12, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, T.; Grube, R.; Wutte, A.; Zarkovic, N.; Schaur, R.J. 4-Hydroxynonenal modifies the effects of serum growth factors on the expression of the c-fos proto-oncogene and the proliferation of HeLa carcinoma cells. Free Radic. Biol. Med. 1998, 25, 42–49. [Google Scholar] [CrossRef]
- Zarkovic, N.; Tillian, M.H.; Schaur, J.; Waeg, G.; Jurin, M.; Esterbauer, H. Inhibition of melanoma B16-F10 growth by lipid peroxidation product 4-hydroxynonenal. Cancer Biother. 1995, 10, 153–156. [Google Scholar] [CrossRef]
- Zarkovic, N.; Schaur, R.J.; Puhl, H.; Jurin, M.; Esterbauer, H. Mutual dependence of growth modifying effects of 4-hydroxynonenal and fetal calf serum in vitro. Free Radic. Biol. Med. 1994, 16, 877–884. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Pettazzoni, P.; Osella-Abate, S.; Novelli, M.; Toaldo, C.; Husse, M.; Daga, M.; Minelli, R.; Bisazza, A.; et al. The inclusion complex of 4-hydroxynonenal with a polymeric derivative of β-cyclodextrin enhances the antitumoral efficacy of the aldehyde in several tumor cell lines and in a three-dimensional human melanoma model. Free Radic. Biol. Med. 2013, 65, 765–777. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Daga, M.; Ciamporcero, E.; Toaldo, C.; Pettazzoni, P.; Osella-Abate, S.; Novelli, M.; Minelli, R.; Bisazza, A.; Gamba, P.; et al. Improved Anti-Tumoral Therapeutic Efficacy of 4-Hydroxynonenal Incorporated in Novel Lipid Nanocapsules in 2D and 3D Models. J. Biomed. Nanotechnol. 2015, 11, 2169–2185. [Google Scholar] [CrossRef]
- Muzio, G.; Maggiora, M.; Paiuzzi, E.; Oraldi, M.; Canuto, R.A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012, 52, 735–746. [Google Scholar] [CrossRef]
- Canuto, R.A.; Muzio, G.; Biocca, M.E.; Dianzani, M.U. Oxidative metabolism of 4-hydroxy-2,3-nonenal during diethyl-nitrosamine-induced carcinogenesis in rat liver. Cancer Lett. 1989, 46, 7–13. [Google Scholar] [CrossRef]
- Canuto, R.A.; Muzio, G.; Maggiora, M.; Poli, G.; Biasi, F.; Dianzani, M.U.; Ferro, M.; Bassi, A.M.; Penco, S.; Marinari, U.M. Ability of different hepatoma cells to metabolize 4-hydroxynonenal. Cell Biochem. Funct. 1993, 11, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lv, D.L.; Duan, J.J.; Xu, S.L.; Zhang, J.F.; Yang, X.J.; Zhang, X.Y.; Cui, H.; Bian, X.W.; Yu, S.C. ALDH1A1 expression correlates with clinicopathologic features and poor prognosis of breast cancer patients: A systematic review and meta-analysis. BMC Cancer 2014, 14, 444. [Google Scholar] [CrossRef] [Green Version]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem. Cells 2011, 29, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Parikh, H.; Xiao, W.; Hoskins, J.W.; Pflicke, H.; Liu, X.; Collins, I.; Zhou, W.; Wang, Z.; Powell, J.; et al. An integrated transcriptome and epigenome analysis identifies a novel candidate gene for pancreatic cancer. BMC Med. Genom. 2013, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Poturnajova, M.; Kozovska, Z.; Matuskova, M. Aldehyde dehydrogenase 1A1 and 1A3 isoforms—Mechanism of activation and regulation in cancer. Cell Signal. 2021, 87, 110120. [Google Scholar] [CrossRef]
- Sladek, N.E.; Dockham, P.A.; Lee, M.O. Human and mouse hepatic aldehyde dehydrogenases important in the biotransformation of cyclophosphamide and the retinoids. Adv. Exp. Med. Biol. 1991, 284, 97–104. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Suwala, A.K.; Koch, K.; Rios, D.H.; Aretz, P.; Uhlmann, C.; Ogorek, I.; Felsberg, J.; Reifenberger, G.; Köhrer, K.; Deenen, R.; et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget 2018, 9, 22703–22716. [Google Scholar] [CrossRef] [Green Version]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem. Cell. 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassalli, G. Aldehyde Dehydrogenases: Not just markers, but functional regulators of stem cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.; Luo, Y.; Fujita, M. Aldehyde dehydrogenase isozymes: Markers of cancer stem cells in human melanoma. Expert Rev. Dermatol. 2013, 8, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasmickaite, L.; Engesaeter, B.O.; Skrbo, N.; Hellenes, T.; Kristian, A.; Oliver, N.K.; Suo, Z.; Maelandsmo, G.M. Aldehyde dehydrogenase (ALDH) activity does not select for cells with enhanced aggressive properties in malignant melanoma. PLoS ONE 2010, 5, e10731. [Google Scholar] [CrossRef]
- Pérez-Alea, M.; McGrail, K.; Sánchez-Redondo, S.; Ferrer, B.; Fournet, G.; Cortés, J.; Muñoz, E.; Hernandez-Losa, J.; Tenbaum, S.; Martin, G.; et al. ALDH1A3 is epigenetically regulated during melanocyte transformation and is a target for melanoma treatment. Oncogene 2017, 36, 5695–5708. [Google Scholar] [CrossRef]
- Yue, L.; Huang, Z.M.; Fong, S.; Leong, S.; Jakowatz, J.G.; Charruyer-Reinwald, A.; Wei, M.; Ghadially, R. Targeting ALDH1 to decrease tumorigenicity, growth and metastasis of human melanoma. Melanoma Res. 2015, 25, 138–148. [Google Scholar] [CrossRef]
- Jin, N.; Zhu, X.; Cheng, F.; Zhang, L. Disulfiram/copper targets stem cell-like ALDH+ population of multiple myeloma by inhibition of ALDH1A1 and Hedgehog pathway. J. Cell. Biochem. 2018, 119, 6882–6893. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, W.; Xia, J.; Gu, Z.; Wendlandt, E.; Zhan, X.; Janz, S.; Tricot, G.; Zhan, F. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget 2014, 5, 11986–11997. [Google Scholar] [CrossRef] [Green Version]
- Dinavahi, S.S.; Gowda, R.; Gowda, K.; Bazewicz, C.G.; Chirasani, V.R.; Battu, M.B.; Berg, A.; Dokholyan, N.V.; Amin, S.; Robertson, G.P. Development of a novel multi-isoform ALDH inhibitor effective as an antimelanoma agent. Mol. Cancer Ther. 2020, 19, 447–459. [Google Scholar] [CrossRef]
- Sarvi, S.; Crispin, R.; Lu, Y.; Zeng, L.; Hurley, T.D.; Houston, D.R.; von Kriegsheim, A.; Chen, C.H.; Mochly-Rosen, D.; Ranzani, M.; et al. ALDH1 bio-activates Nifuroxazide to eradicate ALDHHigh Melanoma-Initiating Cells. Cell. Chem. Biol. 2018, 25, 1456–1469.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, J.M.; Ravindran Menon, D.; Smith, D.E.; Baird, E.; Kitano, T.; Gao, D.; Tan, A.C.; Fujita, M. Clinical implications of ALDH1A1 and ALDH1A3 mRNA expression in melanoma subtypes. Chem. Biol. Interact. 2019, 314, 108822. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic flexibility in melanoma: A potential therapeutic target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef]
- Audrito, V.; Managò, A.; Gaudino, F.; Deaglio, S. Targeting metabolic reprogramming in metastatic melanoma: The key role of nicotinamide phosphoribosyltransferase (NAMPT). Semin. Cell. Dev. Biol. 2019, 98, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [Green Version]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Mishra, R.; Patel, H.; Alanazi, S.; Wei, X.; Ma, Z.; Garrett, J.T. BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers 2020, 12, 1661. [Google Scholar] [CrossRef]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef] [Green Version]
- Pecinova, A.; Drahota, Z.; Kovalcikova, J.; Kovarova, N.; Pecina, P.; Alan, L.; Zima, M.; Houstek, J.; Mracek, T. Pleiotropic Effects of Biguanides on Mitochondrial Reactive Oxygen Species Production. Oxid. Med. Cell Longev. 2017, 2017, 7038603. [Google Scholar] [CrossRef] [Green Version]
- Schöckel, L.; Glasauer, A.; Basit, F.; Bitschar, K.; Truong, H.; Erdmann, G.; Algire, C.; Hägebarth, A.; Willems, P.H.; Kopitz, C.; et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Li, X.; Ding, W.; Zeng, X.; Kong, H.; Wang, H.; Xie, W. Deguelin induces the apoptosis of lung cancer cells through regulating a ROS driven Akt pathway. Cancer Cell Int. 2015, 15, 25. [Google Scholar] [CrossRef] [Green Version]
- Basit, F.; van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.L.; Chagani, S.; Nelson, D.; Cassidy, P.B.; Laws, M.; Ganguli-Indra, G.; Indra, A.K. Mitochondrial complex I inhibitor deguelin induces metabolic reprogramming and sensitizes vemurafenib-resistant BRAFV600E mutation bearing metastatic melanoma cells. Mol. Carcinog. 2019, 58, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Kumari, V.; Dyba, M.A.; Holland, R.J.; Liang, Y.H.; Singh, S.V.; Ji, X. Irreversible inhibition of glutathione S-transferase by phenethyl isothiocyanate (PEITC), a dietary cancer chemopreventive phytochemical. PLoS ONE 2016, 11, e0163821. [Google Scholar] [CrossRef] [PubMed]
- Queirolo, P.; Spagnolo, F.; Picasso, V.; Spano, L.; Tanda, E.; Fontana, V.; Giorello, L.; Merlo, D.F.; Simeone, E.; Grimaldi, A.M.; et al. Combined vemurafenib and fotemustine in patients with BRAF V600 melanoma progressing on vemurafenib. Oncotarget 2018, 9, 12408–12417. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.A.; Luong, M.K.; Shaw, H.; Nathan, P.; Bataille, V.; Spector, T.D. The gut microbiome: What the oncologist ought to know. Br. J. Cancer 2021, 125, 1197–1209. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, X.; Zhu, Y.; Gamallat, Y.; Meyiah, A.; Ma, S.; Xin, Y. Intestinal Dysbiosis Increases the Incidence of Malignant Melanoma in Mice Model. Genet. Mol. Res. 2017, 16, gmr16039840. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.V.; Robeson, M.S., 2nd; Griffin, R.J.; Quick, C.M.; Siegel, E.R.; Cannon, M.J.; Vang, K.B.; Dings, R.P.M. Gastrointestinal Tract Dysbiosis Enhances Distal Tumor Progression through Suppression of Leukocyte Trafficking. Cancer Res. 2019, 79, 5999–6009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Elmén, L.; Segota, I.; Xian, Y.; Tinoco, R.; Feng, Y.; Fujita, Y.; Segura Muñoz, R.R.; Schmaltz, R.; Bradley, L.M.; et al. Prebiotic-Induced Anti-tumor Immunity Attenuates Tumor Growth. Cell Rep. 2020, 30, 1753–1766.e6. [Google Scholar] [CrossRef] [Green Version]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Squarzanti, D.F.; Zavattaro, E.; Pizzimenti, S.; Amoruso, A.; Savoia, P.; Azzimonti, B. Non-Melanoma Skin Cancer: News from microbiota research. Crit. Rev. Microbiol. 2020, 46, 433–449. [Google Scholar] [CrossRef]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Mrázek, J.; Mekadim, C.; Kučerová, P.; Švejstil, R.; Salmonová, H.; Vlasáková, J.; Tarasová, R.; Čížková, J.; Červinková, M. Melanoma-related changes in skin microbiome. Folia Microbiol. 2019, 64, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Marciano, F.; Vajro, P. Oxidative Stress and Gut Microbiota. In Gastrointestinal Tissue Oxidative Stress and Dietary Antioxidant; Gracia-Sancho, J., Salvadó, J., Eds.; Academic Press: Cambridge, MA, USA, 2017; Chapter 8; pp. 113–123. [Google Scholar]
- Vamanu, E. Polyphenolic Nutraceuticals to Combat Oxidative Stress through Microbiota Modulation. Front. Pharmacol. 2019, 10, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, A.; Bhatia, D.; Bishayee, A. Anti-Inflammatory Mechanism Involved in Pomegranate-Mediated Prevention of Breast Cancer: The Role of NF-κB and Nrf2 Signaling Pathways. Nutrients 2017, 9, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Olejar, K.J.; On, S.L.W.; Chelikani, V. The Potential of Lactobacillus spp. for Modulating Oxidative Stress in the Gastrointestinal Tract. Antioxidants 2020, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Yokokura, T.; Azuma, I. Antimetastatic effect of Lactobacillus casei YIT9018 (LC 9018) on a highly metastatic variant of B16 melanoma in C57BL/6J mice. Cancer Immunol. Immunother. 1987, 24, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Hu, M.; Feng, X.; Xiao Li, W.; Dong, D.; Wang, W. Preventive effect of Lactobacillus reuteri on melanoma. Biomed. Pharmacother. 2020, 126, 109929. [Google Scholar] [CrossRef] [PubMed]
- Le Noci, V.; Guglielmetti, S.; Arioli, S.; Camisaschi, C.; Bianchi, F.; Sommariva, M.; Storti, C.; Triulzi, T.; Castelli, C.; Balsari, A.; et al. Modulation of Pulmonary Microbiota by Antibiotic or Probiotic Aerosol Therapy: A Strategy to Promote Immunosurveillance against Lung Metastases. Cell Rep. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizzimenti, S.; Ribero, S.; Cucci, M.A.; Grattarola, M.; Monge, C.; Dianzani, C.; Barrera, G.; Muzio, G. Oxidative Stress-Related Mechanisms in Melanoma and in the Acquired Resistance to Targeted Therapies. Antioxidants 2021, 10, 1942. https://doi.org/10.3390/antiox10121942
Pizzimenti S, Ribero S, Cucci MA, Grattarola M, Monge C, Dianzani C, Barrera G, Muzio G. Oxidative Stress-Related Mechanisms in Melanoma and in the Acquired Resistance to Targeted Therapies. Antioxidants. 2021; 10(12):1942. https://doi.org/10.3390/antiox10121942
Chicago/Turabian StylePizzimenti, Stefania, Simone Ribero, Marie Angele Cucci, Margherita Grattarola, Chiara Monge, Chiara Dianzani, Giuseppina Barrera, and Giuliana Muzio. 2021. "Oxidative Stress-Related Mechanisms in Melanoma and in the Acquired Resistance to Targeted Therapies" Antioxidants 10, no. 12: 1942. https://doi.org/10.3390/antiox10121942