Suicidal Erythrocyte Death in Metabolic Syndrome





Abstract
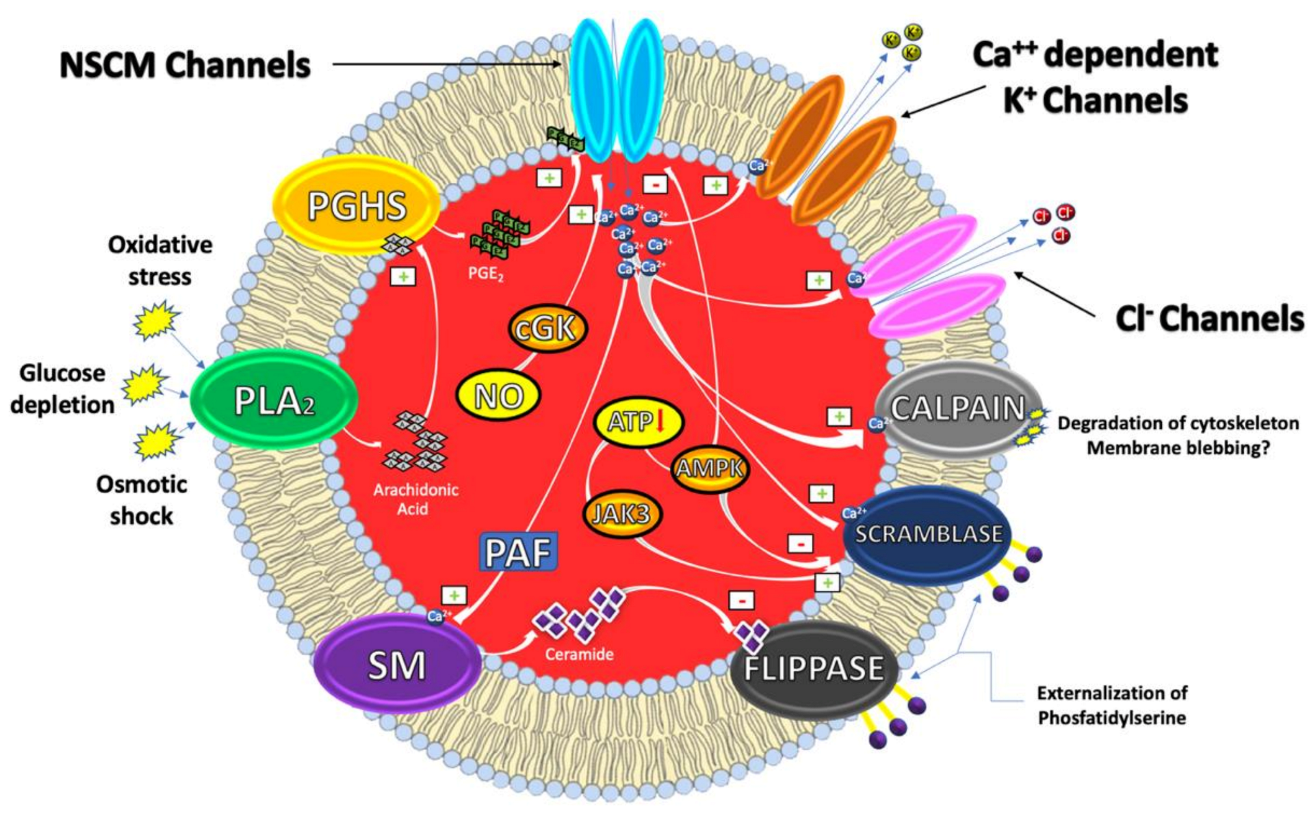
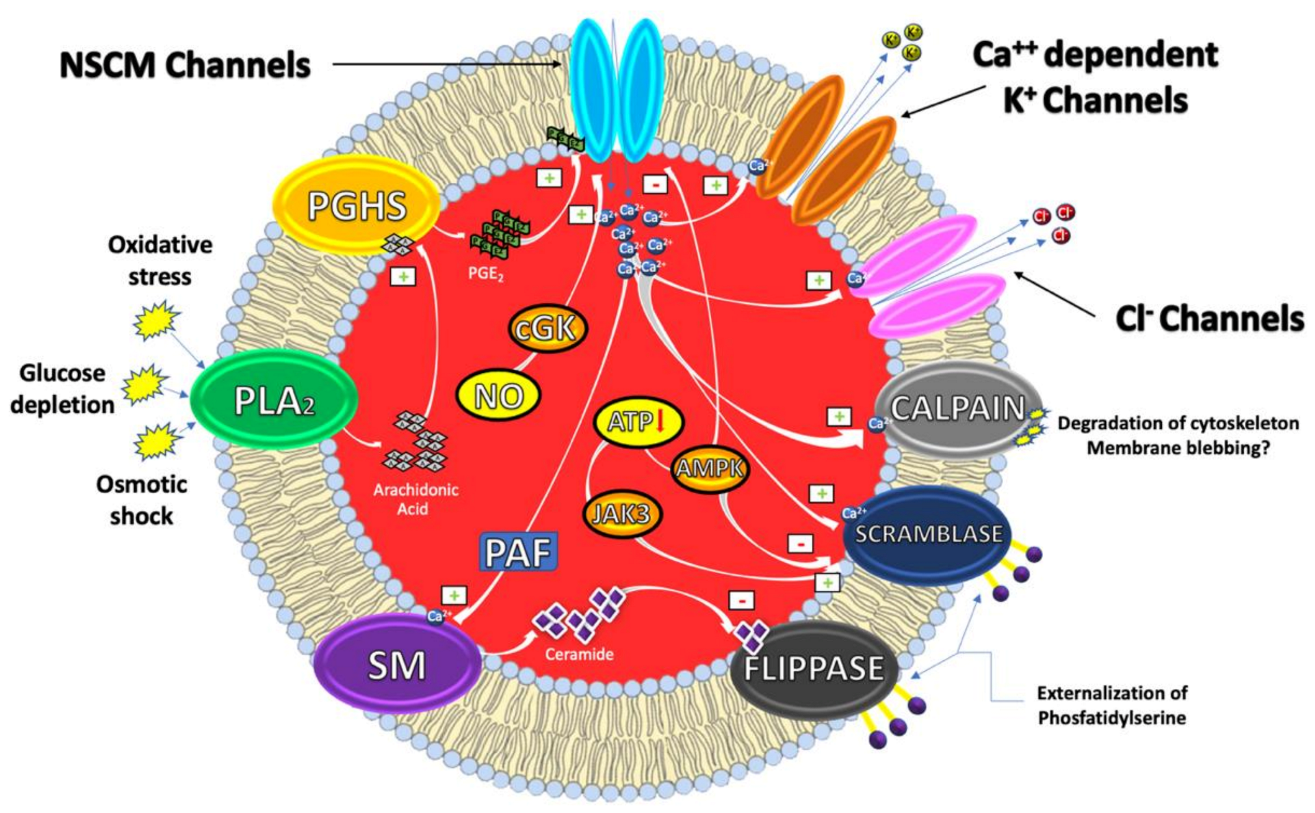
:1. Eryptosis
1.1. Mechanisms
1.2. The Impact of Eryptosis on Endothelial Cells and Thrombocytes
2. The Metabolic Syndrome
3. Eryptosis, Hyperglycemia and Diabetes
4. Eryptosis, Dyslipidemia and Atherosclerosis
5. Eryptosis and Hypertension
6. Eryptosis and Obesity
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bosman, G.J.C.G.M.; Willekens, F.L.A.; Werre, J.M. Erythrocyte aging: A more than superficial resemblance to apoptosis? Cell. Physiol. Biochem. 2005, 16, 1–8. [Google Scholar] [PubMed] [Green Version]
- Arese, P.; Turrini, F.; Schwarzer, E. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell. Physiol. Biochem. 2005, 16, 133–146. [Google Scholar] [PubMed]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [PubMed]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243. [Google Scholar]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Gulbins, E.; Lang, P.A.; Zappulla, D.; Föller, M. Ceramide in suicidal death of erythrocytes. Cell. Physiol. Biochem. 2010, 26, 21–28. [Google Scholar]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide Content Is Increased in Skeletal Muscle from Obese Insulin-Resistant Humans. Diabetes 2004, 53. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Lang, F. Triggers, inhibitors, mechanisms, and significance of eryptosis: The suicidal erythrocyte death. Biomed. Res. Int. 2015. [Google Scholar] [CrossRef] [Green Version]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. Biomed. Res. Int. 2018. [Google Scholar] [CrossRef]
- Pretorius, E.; Du Plooy, J.N.; Bester, J. A Comprehensive Review on Eryptosis. Cell. Physiol. Biochem. 2016, 39, 1977–2000. [Google Scholar]
- Lang, F.; Abed, M.; Lang, E.; Föller, M. Oxidative stress and suicidal erythrocyte death. Antioxid. Redox Signal. 2014, 21. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho) physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Lang, F. Mechanisms and pathophysiological significance of eryptosis, the suicidal erythrocyte death. Semin. Cell Dev. Biol. 2015, 39, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Föller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302. [Google Scholar] [CrossRef] [Green Version]
- Shimaoka, T.; Nakayama, T.; Fukumoto, N.; Kume, N.; Takahashi, S.; Yamaguchi, J.; Minami, M.; Hayashida, K.; Kita, T.; Ohsumi, J.; et al. Cell surface-anchored SR-PSOX/CXC chemokine ligand 16 mediates firm adhesion of CXC chemokine receptor 6-expressing cells. J. Leukoc. Biol. 2004, 75. [Google Scholar] [CrossRef] [Green Version]
- Gayen Betal, S.; Setty, B.N.Y. Phosphatidylserine-positive erythrocytes bind to immobilized and soluble thrombospondin-1 via its heparin-binding domain. Transl. Res. 2008, 152. [Google Scholar] [CrossRef] [Green Version]
- Manodori, A.B.; Barabino, G.A.; Lubin, B.H.; Kuypers, F.A. Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood 2000, 95. [Google Scholar] [CrossRef]
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.B.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405. [Google Scholar] [CrossRef]
- Byrnes, J.R.; Wolberg, A.S. Red blood cells in thrombosis. Blood 2017, 130. [Google Scholar] [CrossRef]
- Walker, B.; Towhid, S.T.; Schmid, E.; Hoffmann, S.M.; Abed, M.; Münzer, P.; Vogel, S.; Neis, F.; Brucker, S.; Gawaz, M.; et al. Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am. J. Physiol. Cell Physiol. 2014, 306. [Google Scholar] [CrossRef]
- Ashraf, M.Z.; Gupta, N. Scavenger receptors: Implications in atherothrombotic disorders. Int. J. Biochem. Cell Biol. 2011, 43, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.M.M.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.T.; Loria, C.M.; Smith, S.C. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; National heart, lung, and blood institute; American heart association; World heart federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed] [Green Version]
- Nilsson, P.M.; Tuomilehto, J.; Rydén, L. The metabolic syndrome—What is it and how should it be managed? Eur. J. Prev. Cardiol. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welty, F.K.; Alfaddagh, A.; Elajami, T.K. Targeting inflammation in metabolic syndrome. Transl. Res. 2016, 167, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.L.; Garber, A.J. Metabolic syndrome. Endocrinol. Metab. Clin. North. Am. 2014, 43, 1–23. [Google Scholar] [CrossRef]
- Samad, F.; Ruf, W. Inflammation, obesity, and thrombosis. Blood 2013, 122, 3415–3422. [Google Scholar] [CrossRef]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vekic, J.; Zeljkovic, A.; Stefanovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V. Obesity and dyslipidemia. Metabolism 2019, 92, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Iikuni, N.; Kwan Lam, Q.; Lu, L.; Matarese, G.; Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4. [Google Scholar] [CrossRef]
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and β-cell dysfunction. Eur. J. Clin. Investig. 2002, 32, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Mollica, M.P.; Lombardi, A.; Cavaliere, G.; Gifuni, G.; Barletta, A. From chronic overnutrition to insulin resistance: The role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 146–152. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Mitron, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93. [Google Scholar] [CrossRef]
- Natarajan, P.; Ray, K.K.; Cannon, C.P. High-Density Lipoprotein and Coronary Heart Disease. Current and Future Therapies. J. Am. Coll. Cardiol. 2010, 55, 1283–1299. [Google Scholar] [CrossRef] [Green Version]
- Landsberg, L.; Aronne, L.J.; Beilin, L.J.; Burke, V.; Igel, L.I.; Lloyd-Jones, D.; Sowers, J. Obesity-Related Hypertension: Pathogenesis, Cardiovascular Risk, and Treatment: A Position Paper of The Obesity Society and the American Society of Hypertension Landsberg et al. Obesity Related Hypertension. J. Clin. Hypertens. 2013, 15. [Google Scholar] [CrossRef]
- Reaven, G.M. Relationships Among Insulin Resistance, Type 2 Diabetes, Essential Hypertension, and Cardiovascular Disease: Similarities and Differences. J. Clin. Hypertens. 2011, 13, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Lam, C.S.Y.; Benzie, I.F.F.; Choi, S.W.; Chan, L.Y.L.; Yeung, V.T.F.; Woo, G.C. Relationships among diabetic retinopathy, antioxidants, and glycemic control. Optom. Vis. Sci. 2011, 88. [Google Scholar] [CrossRef] [PubMed]
- Gorban de Lapertosa, S.; Fereira de Moura, A.; Decroux, C.; Duke, L.; Hammond, L.; Jacobs, E.; Kaundal, A.; Li, J.; Liu, J.; Ohlrogge, A.E.; et al. Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Deray, G.; Heurtier, A.; Grimaldi, A.; Launay Vacher, V.; Isnard Bagnis, C. Anemia and diabetes. Am. J. Nephrol. 2004, 24, 522–526. [Google Scholar] [CrossRef]
- Gauci, R.; Hunter, M.; Bruce, D.G.; Davis, W.A.; Davis, T.M.E. Anemia complicating type 2 diabetes: Prevalence, risk factors and prognosis. J. Diabetes Complic. 2017, 31. [Google Scholar] [CrossRef]
- Singh, D.K.; Winocour, P.; Farrington, K. Erythropoietic stress and anemia in diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 204–210. [Google Scholar] [CrossRef]
- Calderón-Salinas, J.V.; Muñoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodríguez-Morán, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell. Biochem. 2011, 357. [Google Scholar] [CrossRef]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50. [Google Scholar] [CrossRef]
- Vlassopoulos, A.; Lean, M.E.J.; Combet, E. Role of oxidative stress in physiological albumin glycation: A neglected interaction. Free Radic. Biol. Med. 2013, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huebschmann, A.G.; Regensteiner, J.G.; Vlassara, H.; Reusch, J.E.B. Diabetes and advanced glycoxidation end products. Diabetes Care 2006, 29, 1420–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Striker, G.E. Advanced Glycation Endproducts in Diabetes and Diabetic Complications. Endocrinol. Metab. Clin. North. Am. 2013, 42, 697–719. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Gayathiri, S.K.; Ramya, R.; Duraichelvan, R.; Dhason, A.; Saraswathi, N.T. Advanced Glycation-Modified Human Serum Albumin Evokes Alterations in Membrane and Eryptosis in Erythrocytes. Appl. Biochem. Biotechnol. 2015, 177. [Google Scholar] [CrossRef]
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14. [Google Scholar] [CrossRef] [Green Version]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Higashi, T.; Sano, H.; Jinnouchi, Y.; Yoshida, M.; Araki, T.; Ueda, S.; Horiuchi, S. Ne-(carboxymethyl)lysine protein adduct is a major immunological epitope in proteins modified with advanced glycation end products of the maillard reaction. Biochemistry 1996, 35. [Google Scholar] [CrossRef]
- Reddy, S.; Bichler, J.; Wells-Knecht, K.J.; Thorpe, S.R.; Baynes, J.W. Nε-(Carboxymethyl) lysine Is a Dominant Advanced Glycation End Product (AGE) Antigen in Tissue Proteins. Biochemistry 1995, 34. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 2014, 63, 50–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matafome, P.; Sena, C.; Seiça, R. Methylglyoxal, obesity, and diabetes. Endocrine 2013, 43, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48. [Google Scholar] [CrossRef] [PubMed]
- Kilhovd, B.K.; Giardino, I.; Torjesen, P.A.; Birkeland, K.I.; Berg, T.J.; Thornalley, P.J.; Brownlee, M.; Hanssen, K.F. Increased serum levels of the specific AGE-compound methylglyoxal-derived hydroimidazolone in patients with type 2 diabetes. Metabolism 2003, 33. [Google Scholar] [CrossRef]
- Lapolla, A.; Flamini, R.; Dalla Vedova, A.; Senesi, A.; Reitano, R.; Fedele, D.; Basso, E.; Seraglia, R.; Traldi, P. Glyoxal and methylglyoxal levels in diabetic patients: Quantitative determination by a new GC/MS method. Clin. Chem. Lab. Med. 2003, 41. [Google Scholar] [CrossRef]
- Nagai, R.; Deemer, E.K.; Brock, J.W.; Thorpe, S.R.; Baynes, J.W. Effect of glucose concentration on formation of AGEs in erythrocytes In Vitro. Ann. N. Y. Acad. Sci. 2005, 1043, 146–150. [Google Scholar] [CrossRef]
- Nicolay, J.; Schneider, J.; Niemoeller, O.; Artunc, F.; Portero-Otin, M.; Haik, G.; Thornalley, P.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 40. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Jahan, I.; Ng, R. Suppression of the accumulation of triosephosphates and increased formation of methylglyoxal in human red blood cells during hyperglycaemia by thiamine In Vitro. J. Biochem. 2001, 129. [Google Scholar] [CrossRef]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose In Vitro. Biochem. J. 1988, 254. [Google Scholar] [CrossRef] [Green Version]
- Kempe-Teufel, D.S.; Bissinger, R.; Qadri, S.M.; Wagner, R.; Peter, A.; Lang, F. Cellular markers of eryptosis are altered in type 2 diabetes. Clin. Chem. Lab. Med. 2018, 56, e177–e180. [Google Scholar] [CrossRef] [PubMed]
- Wali, R.K.; Jaffe, S.; Kumar, D.; Kalra, V.K. Alterations in organization of phospholipids in erythrocytes as factor in adherence to endothelial cells in diabetes mellitus. Diabetes 1988, 62. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopin, L.; Lowenstein, C. In the Clinic® dyslipidemia. Ann. Intern. Med. 2017, 167. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Baumert, J.; Barbalic, M.; Dupuis, J.; Ellinor, P.T.; Durda, P.; Dehghan, A.; Bis, J.C.; Illig, T.; Morrison, A.C.; et al. Duffy antigen receptor for chemokines (Darc) polymorphism regulates circulating concentrations of monocyte chemoattractant protein-1 and other inflammatory mediators. Blood 2010, 115. [Google Scholar] [CrossRef] [Green Version]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque Hemorrhage and Progression of Coronary Atheroma. N. Engl. J. Med. 2003, 32. [Google Scholar] [CrossRef]
- Virmani, R.; Roberts, W.C. Extravasated erythrocytes, iron, and fibrin in atherosclerotic plaques of coronary arteries in fatal coronary heart disease and their relation to luminal thrombus: Frequency and significance in 57 necropsy patients and in 2958 five mm segments of 224 majo. Am. Heart J. 1983, 105. [Google Scholar] [CrossRef]
- Pinzón-Díaz, C.E.; Calderón-Salinas, J.V.; Rosas-Flores, M.M.; Hernández, G.; López-Betancourt, A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in hypertensive and dyslipidemic patients. Mol. Cell. Biochem. 2018, 11. [Google Scholar] [CrossRef]
- Unruh, D.; Srinivasan, R.; Benson, T.; Haigh, S.; Coyle, D.; Batra, N.; Keil, R.; Sturm, R.; Blanco, V.; Palascak, M.; et al. Red blood cell dysfunction induced by high-fat diet: Potential implications for obesity-related atherosclerosis. Circulation 2015, 132. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis 1999, 142, 1–28. [Google Scholar] [CrossRef]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoeb, M.; Ansari, N.; Srivastava, S.; Ramana, K. 4-Hydroxynonenal in the Pathogenesis and Progression of Human Diseases. Curr. Med. Chem. 2013, 21. [Google Scholar] [CrossRef]
- Leonarduzzi, G.; Chiarpotto, E.; Biasi, F.; Poli, G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1044–1049. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative Stress and Cell Signalling. Curr. Med. Chem. 2012, 11. [Google Scholar] [CrossRef]
- Poli, G.; Sottero, B.; Gargiulo, S.; Leonarduzzi, G. Cholesterol oxidation products in the vascular remodeling due to atherosclerosis. Mol. Aspects Med. 2009, 30, 180–189. [Google Scholar] [CrossRef]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34. [Google Scholar] [CrossRef] [Green Version]
- Attanzio, A.; Frazzitta, A.; Cilla, A.; Livrea, M.A.; Tesoriere, L.; Allegra, M. 7-keto-cholesterol and cholestan-3beta, 5alpha, 6beta-Triol induce eryptosis through distinct pathways leading to NADPH oxidase and nitric oxide synthase activation. Cell. Physiol. Biochem. 2019, 53. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Allegra, M.; Restivo, I.; Fucarino, A.; Pitruzzella, A.; Vasto, S.; Livrea, M.A.; Tesoriere, L.; Attanzio, A. Proeryptotic activity of 4-hydroxynonenal: A new potential physiopathological role for lipid peroxidation products. Biomolecules 2020, 770. [Google Scholar] [CrossRef] [PubMed]
- Davy, K.P.; Halle, J.E. Obesity and hypertension: Two epidemics or one? Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2004, 286. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from Coronary Heart Disease in Subjects with Type 2 Diabetes and in Nondiabetic Subjects with and without Prior Myocardial Infarction. N. Engl. J. Med. 1998, 339. [Google Scholar] [CrossRef]
- World Health Organization. A Global Brief on Hypertension: World Silent Killer, Global Health Crisis; WHO: Geneva, Switzerland, 2013. [Google Scholar]
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picone, P.; Di Carlo, M.; Nuzzo, D. Obesity and Alzheimer’s disease: Molecular bases. Eur. J. Neurosci. 2020, 52, 3944–3950. [Google Scholar] [CrossRef]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I. Morbidity and mortality risk associated with an overweight BMI in older men and women. Obesity 2007, 15. [Google Scholar] [CrossRef] [Green Version]
- Solá, E.; Vayá, A.; Martínez, M.; Moscardó, A.; Corella, D.; Santaolaria, M.L.; Espãa, F.; Hernández-Mijares, A. Erythrocyte membrane phosphatidylserine exposure in obesity. Obesity 2009, 17. [Google Scholar] [CrossRef]
- Pernow, J.; Mahdi, A.; Yang, J.; Zhou, Z. Red blood cell dysfunction: A new player in cardiovascular disease. Cardiovasc. Res. 2019, 115. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Parameters | Cut-Points |
|---|---|
| Waist Circumference | Population- and Country-specific Definitions |
| Triacylglycerols | ≥150 mg/dL |
| HDL Cholesterol | Males: ≤40 mg/dL Females: ≤50 mg/dL |
| Blood Pressure | Systolic: ≥130 and/or Diastolic: ≥85 mmHg |
| Fasting Plasma Glucose | ≥100 mg/dL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Restivo, I.; Attanzio, A.; Tesoriere, L.; Allegra, M. Suicidal Erythrocyte Death in Metabolic Syndrome. Antioxidants 2021, 10, 154. https://doi.org/10.3390/antiox10020154
Restivo I, Attanzio A, Tesoriere L, Allegra M. Suicidal Erythrocyte Death in Metabolic Syndrome. Antioxidants. 2021; 10(2):154. https://doi.org/10.3390/antiox10020154
Chicago/Turabian StyleRestivo, Ignazio, Alessandro Attanzio, Luisa Tesoriere, and Mario Allegra. 2021. "Suicidal Erythrocyte Death in Metabolic Syndrome" Antioxidants 10, no. 2: 154. https://doi.org/10.3390/antiox10020154
APA StyleRestivo, I., Attanzio, A., Tesoriere, L., & Allegra, M. (2021). Suicidal Erythrocyte Death in Metabolic Syndrome. Antioxidants, 10(2), 154. https://doi.org/10.3390/antiox10020154