
Platycodon grandiflorum Root Protects against Aβ-Induced Cognitive Dysfunction and Pathology in Female Models of Alzheimer’s Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Analysis of Platycoside E and Platycodin D Using HPLC-ELSD
2.3. Animals and PGE Administration
2.4. Behavioral Test
2.5. Preparation of Brain Tissue
2.6. Immunofluorescence Labeling
2.7. Immunoperoxidase Labeling
2.8. Cell Viability Assay in HT22 Cells
2.9. Intracellular ROS Generation in HT22 Cells
2.10. Image Acquisition and Analysis
2.11. Statistical Analysis
3. Results
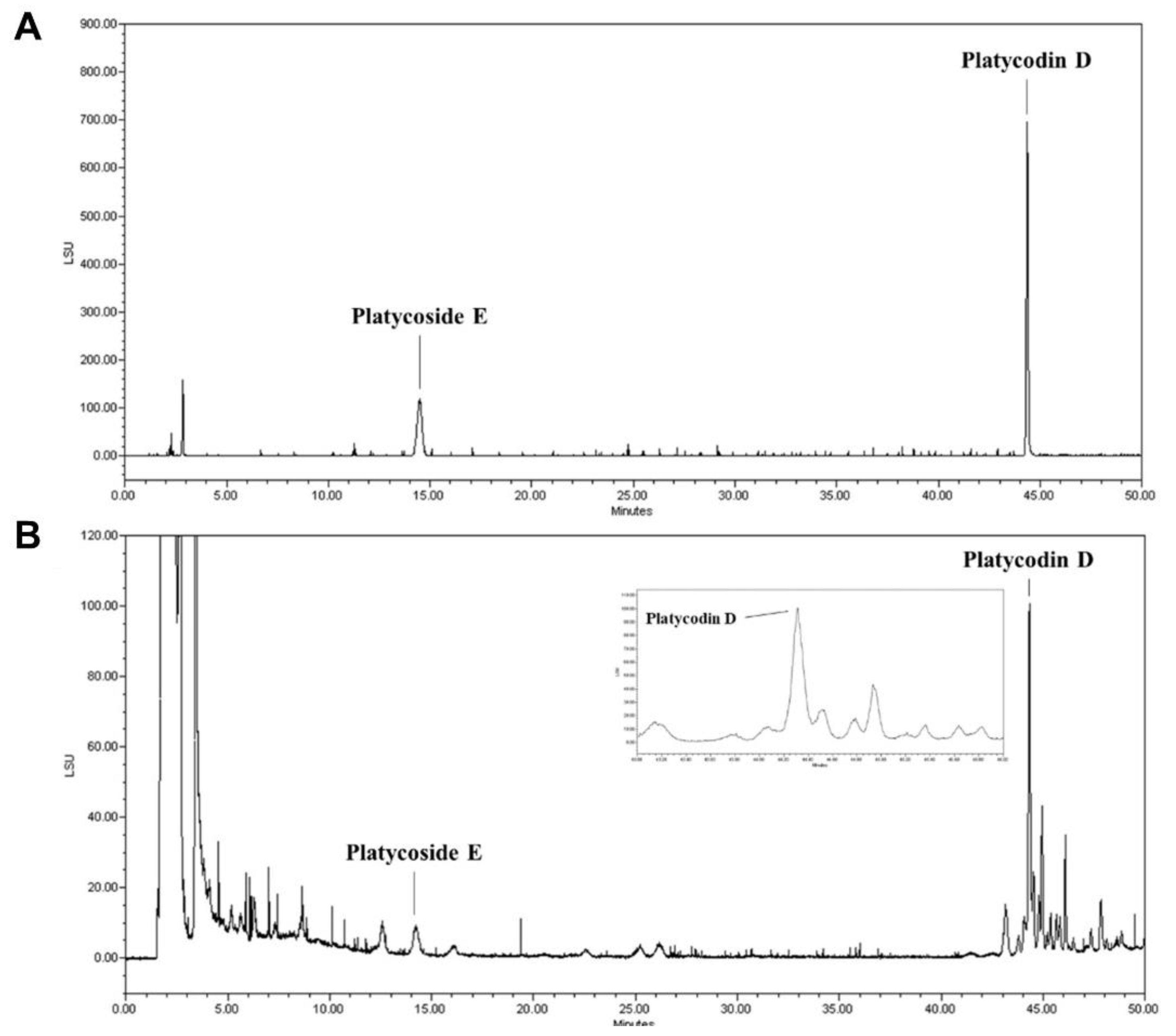
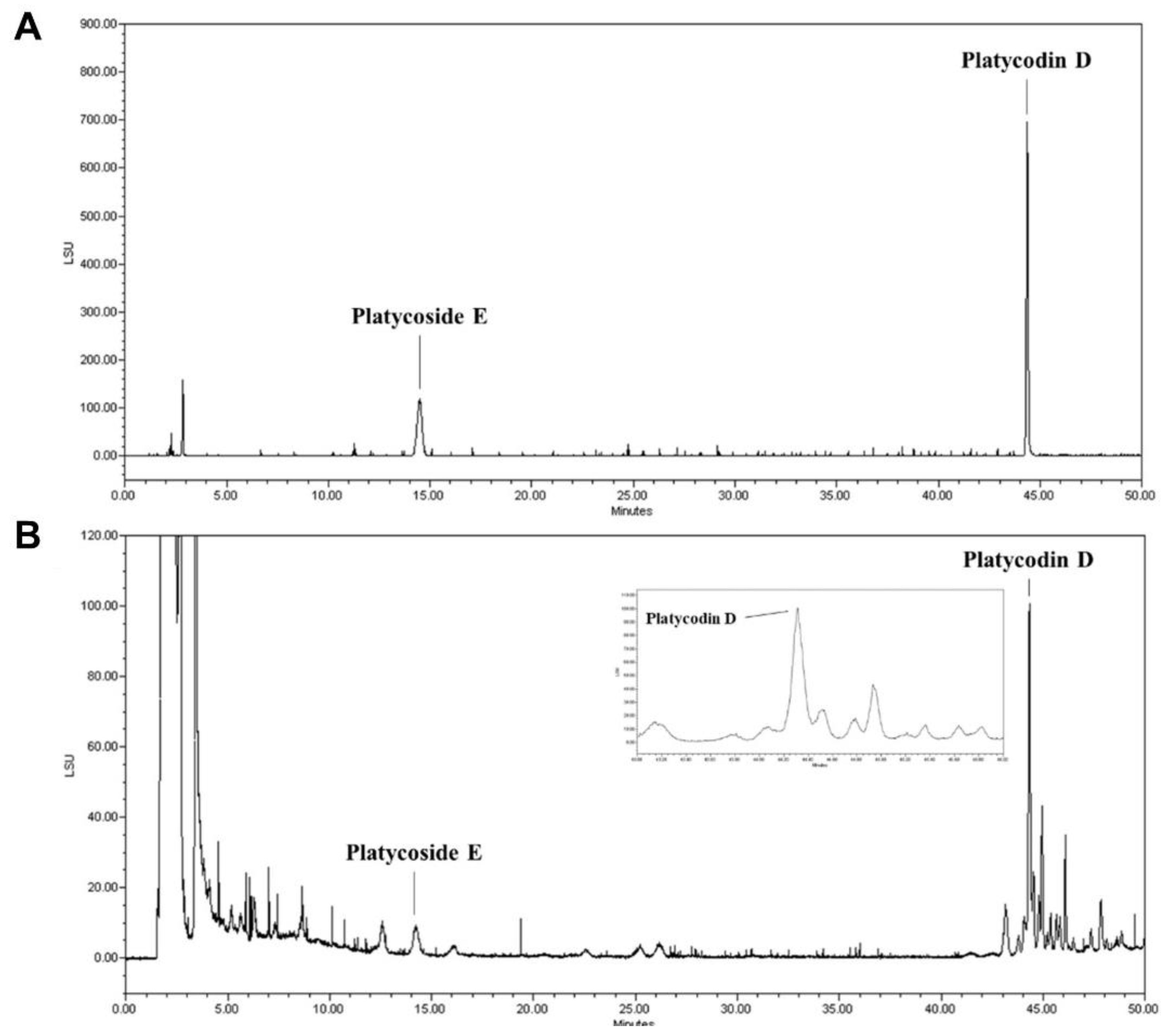
3.1. Standardization of Indicator Components in PGE Using HPLC
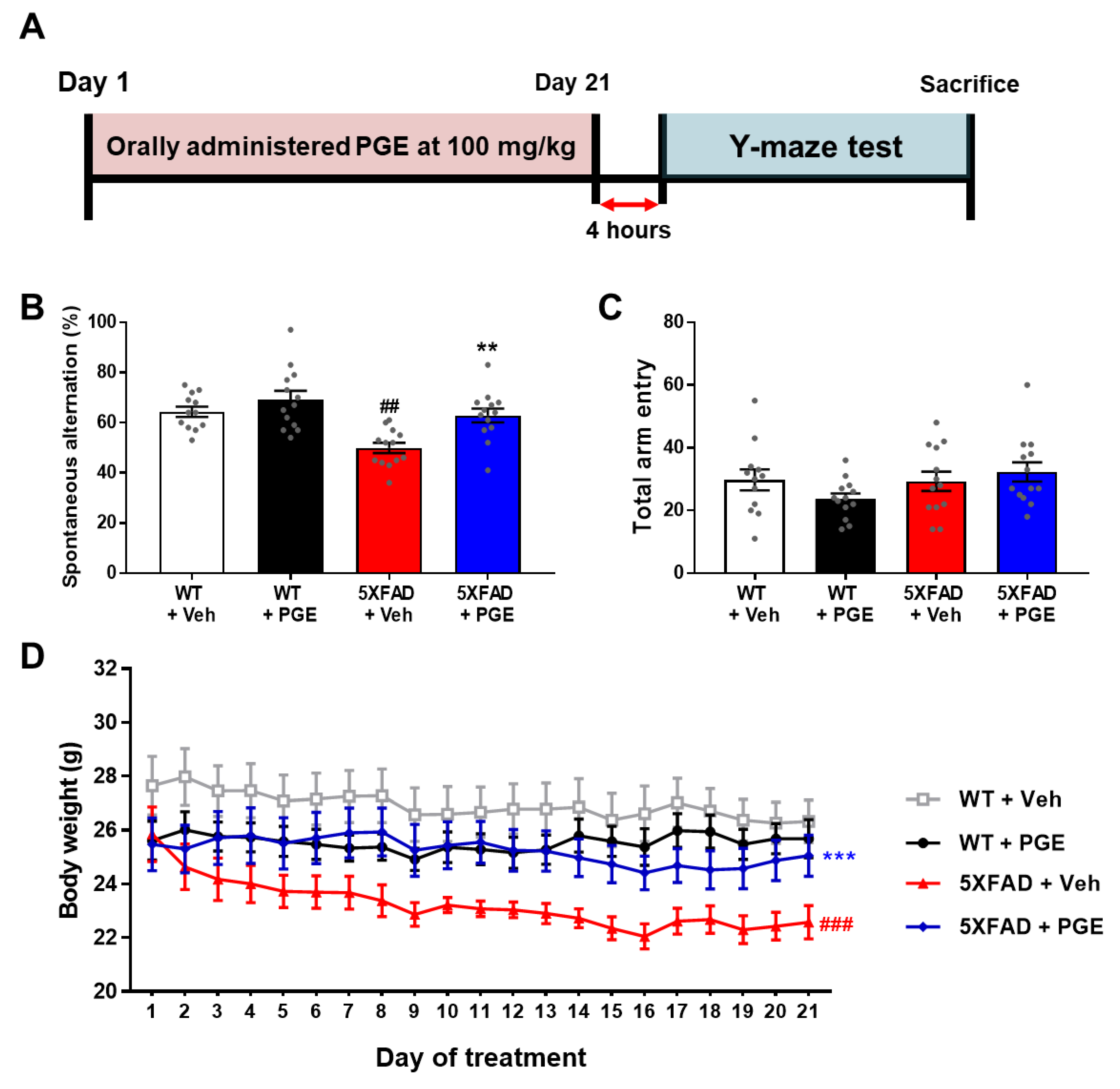
3.2. Effect of PGE on Cognitive and Physiological Dysfunction in an Animal Model of AD
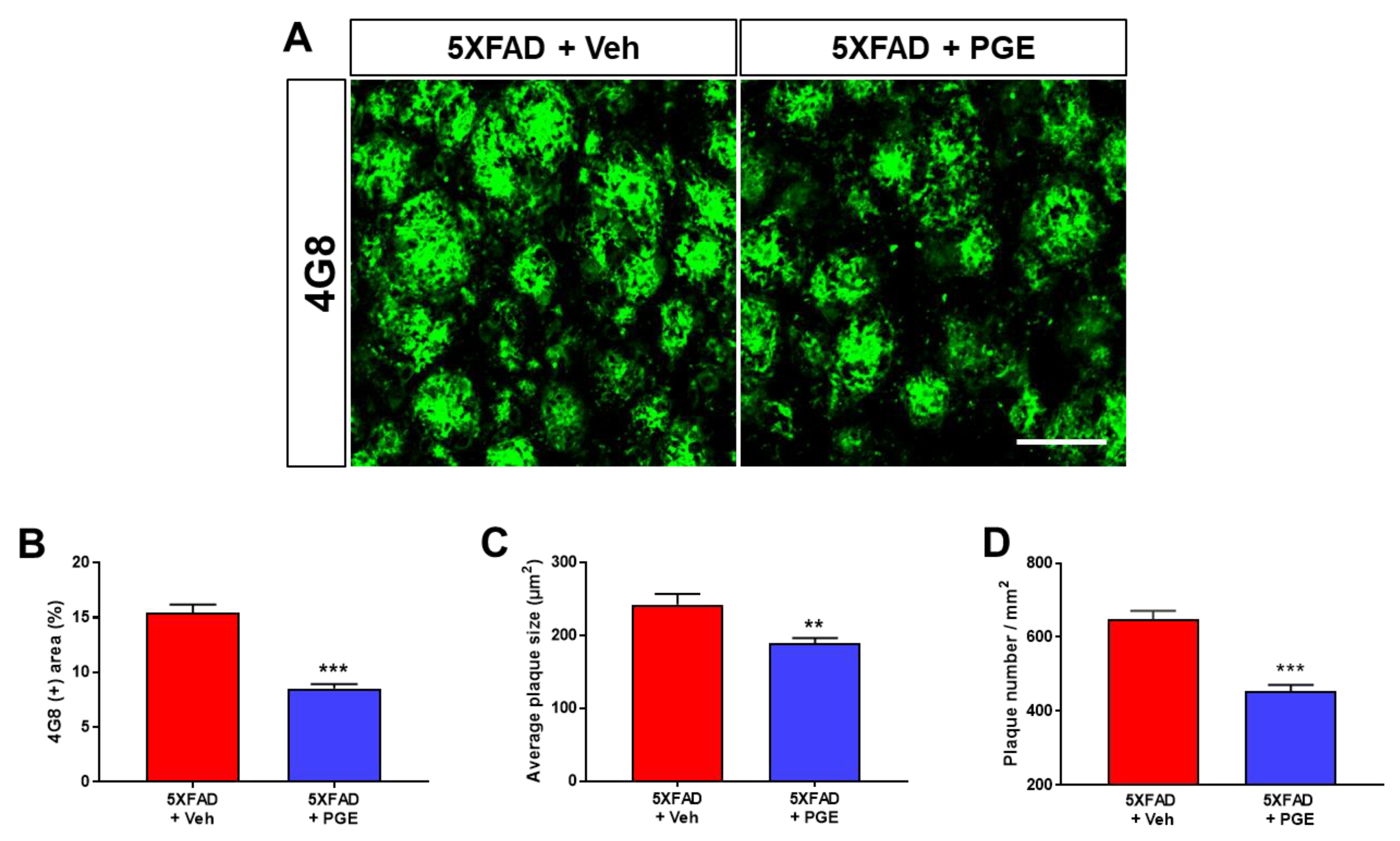
3.3. Effect of PGE on Aβ Accumulation in the Aβ-Overexpressing Brain
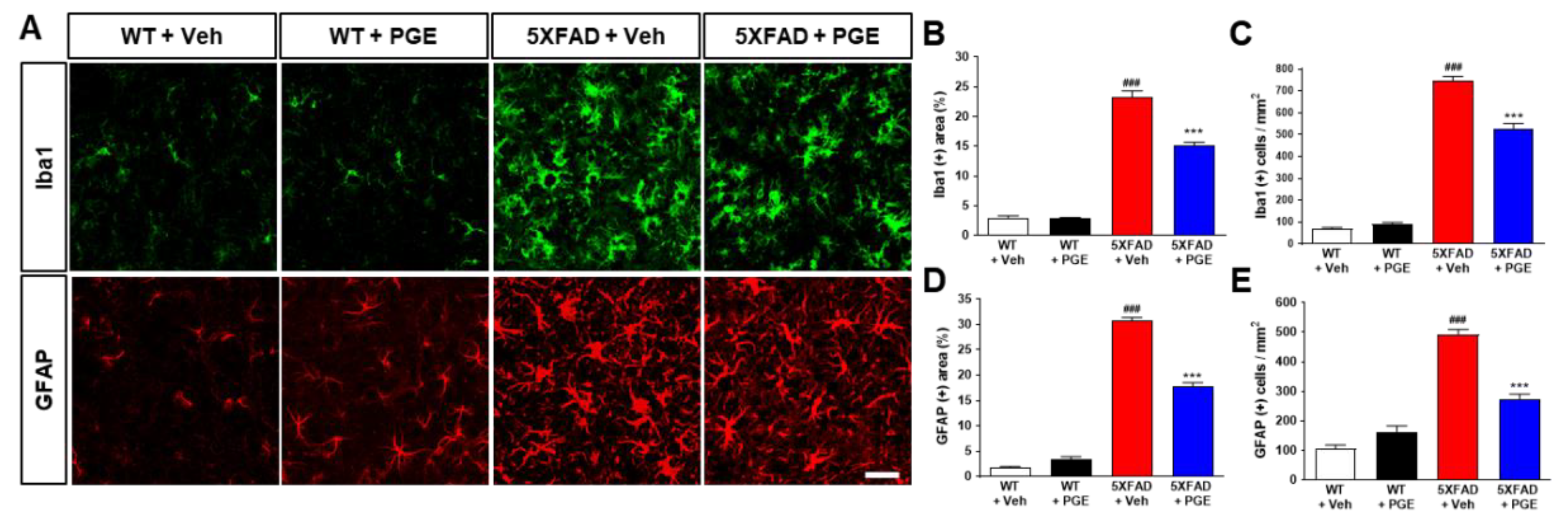
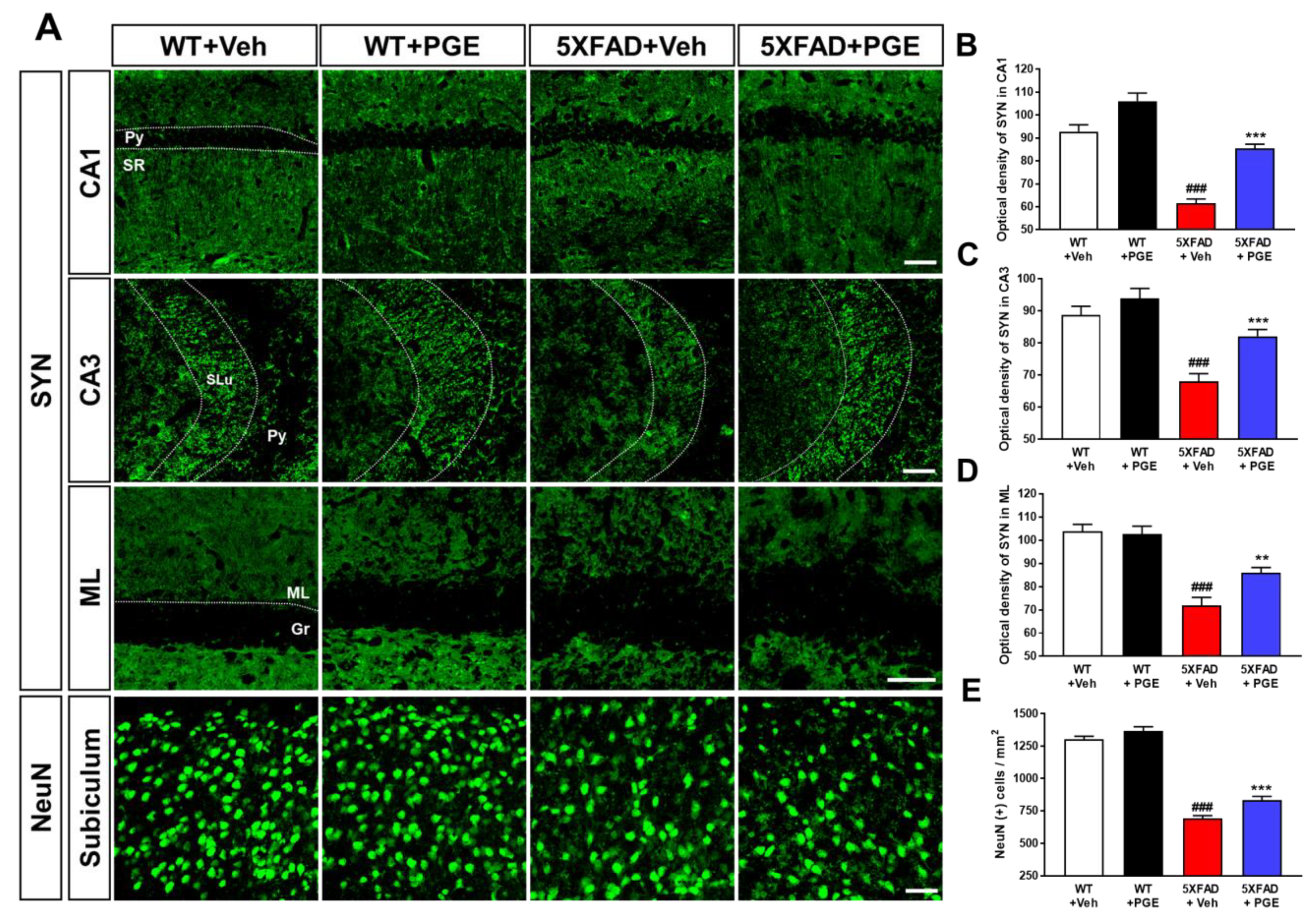
3.4. Effect of PGE on Neurodegeneration in the Brain of 5XFAD Mice
3.5. Effect of PGE on Oxidative Stress in the Animal Models of AD
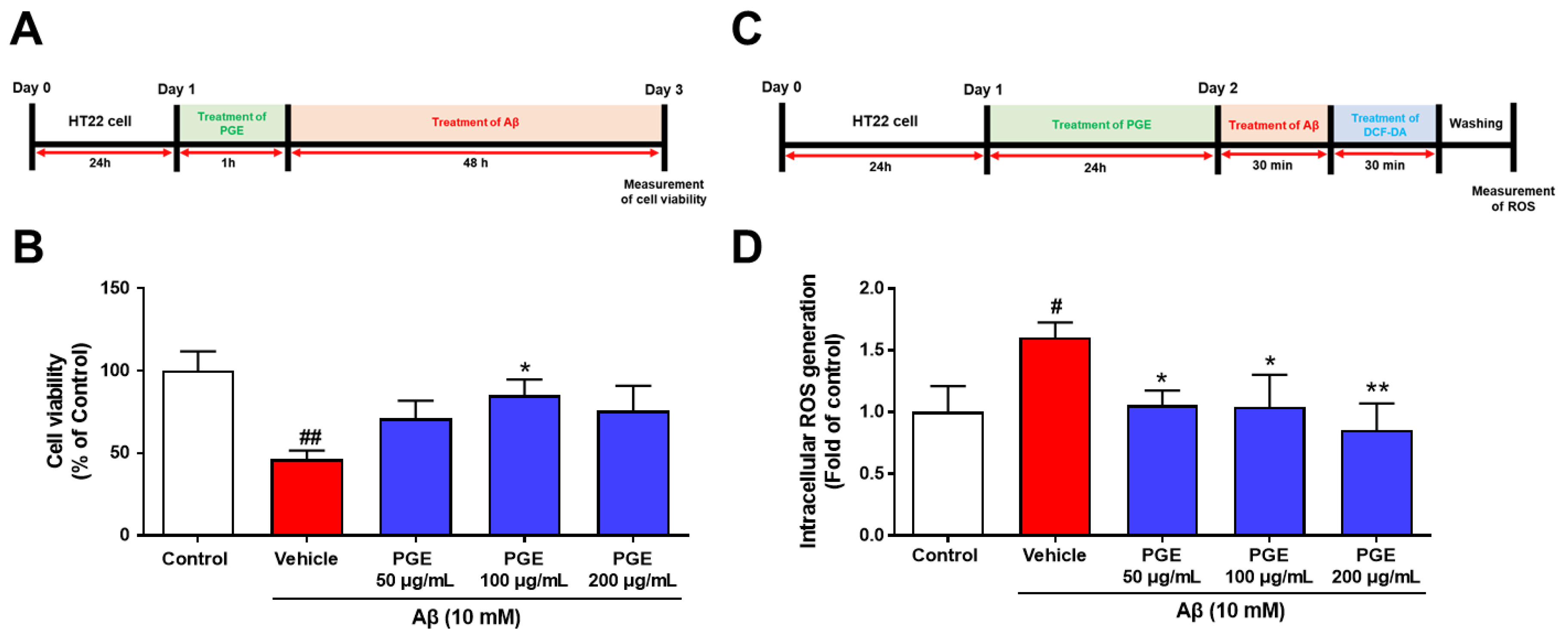
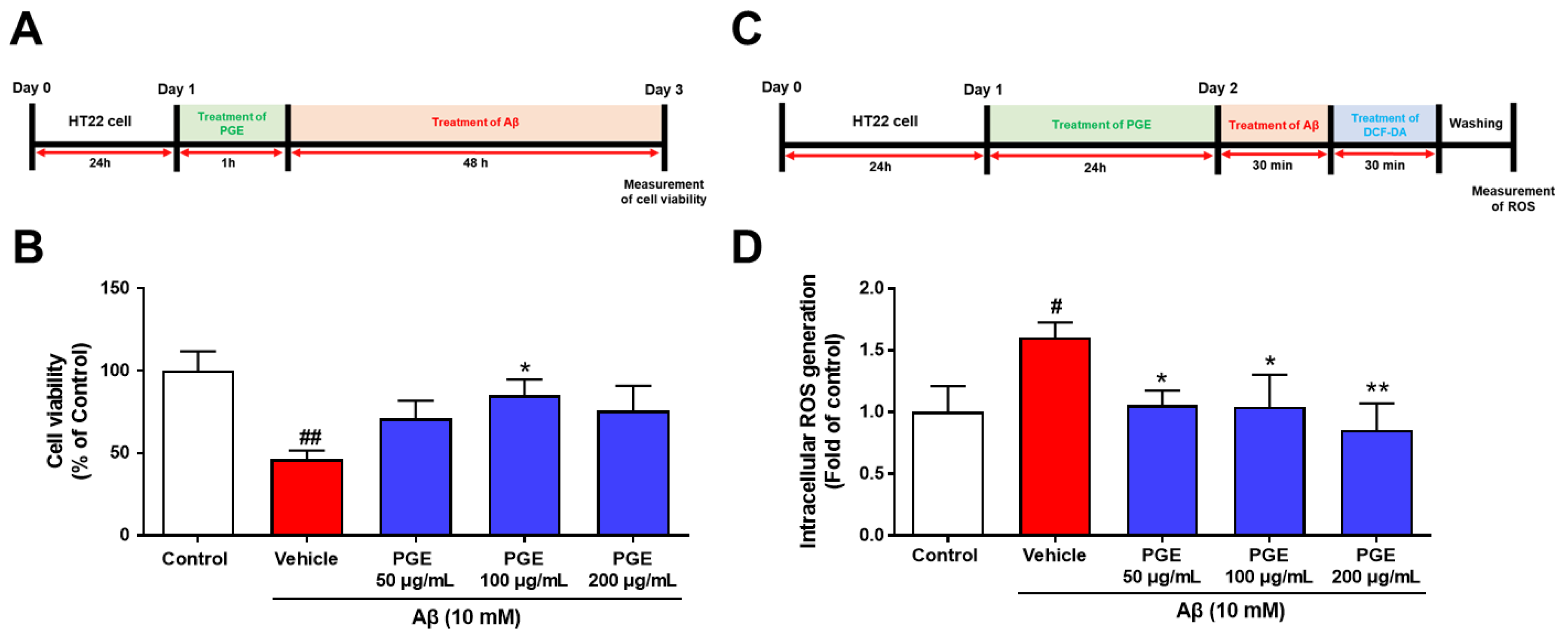
3.6. Effect of PGE on Aβ-Induced Cell Death and Oxidative Stress in Cultured Hippocampal Neurons
3.7. Effect of PGE on Neuroinflammation in the Brain of AD Transgenic Mice
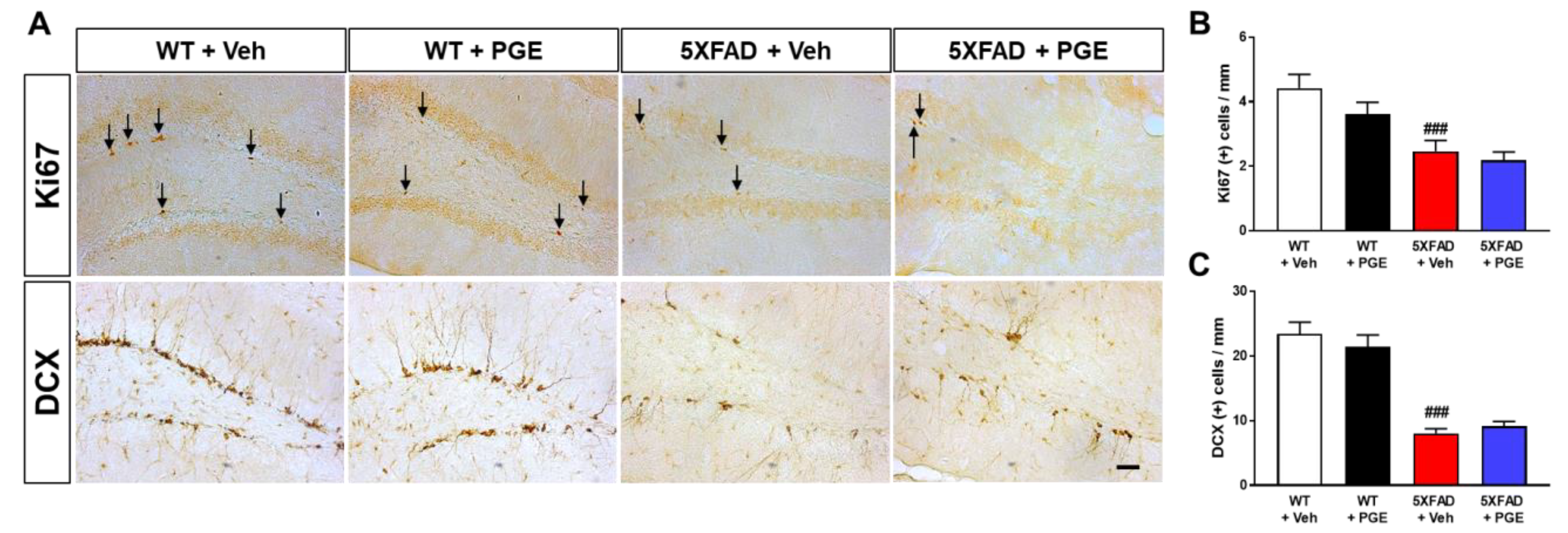
3.8. Effect of PGE on Adult Hippocampal Neurogenesis in the Hippocampus of 5XFAD Mice
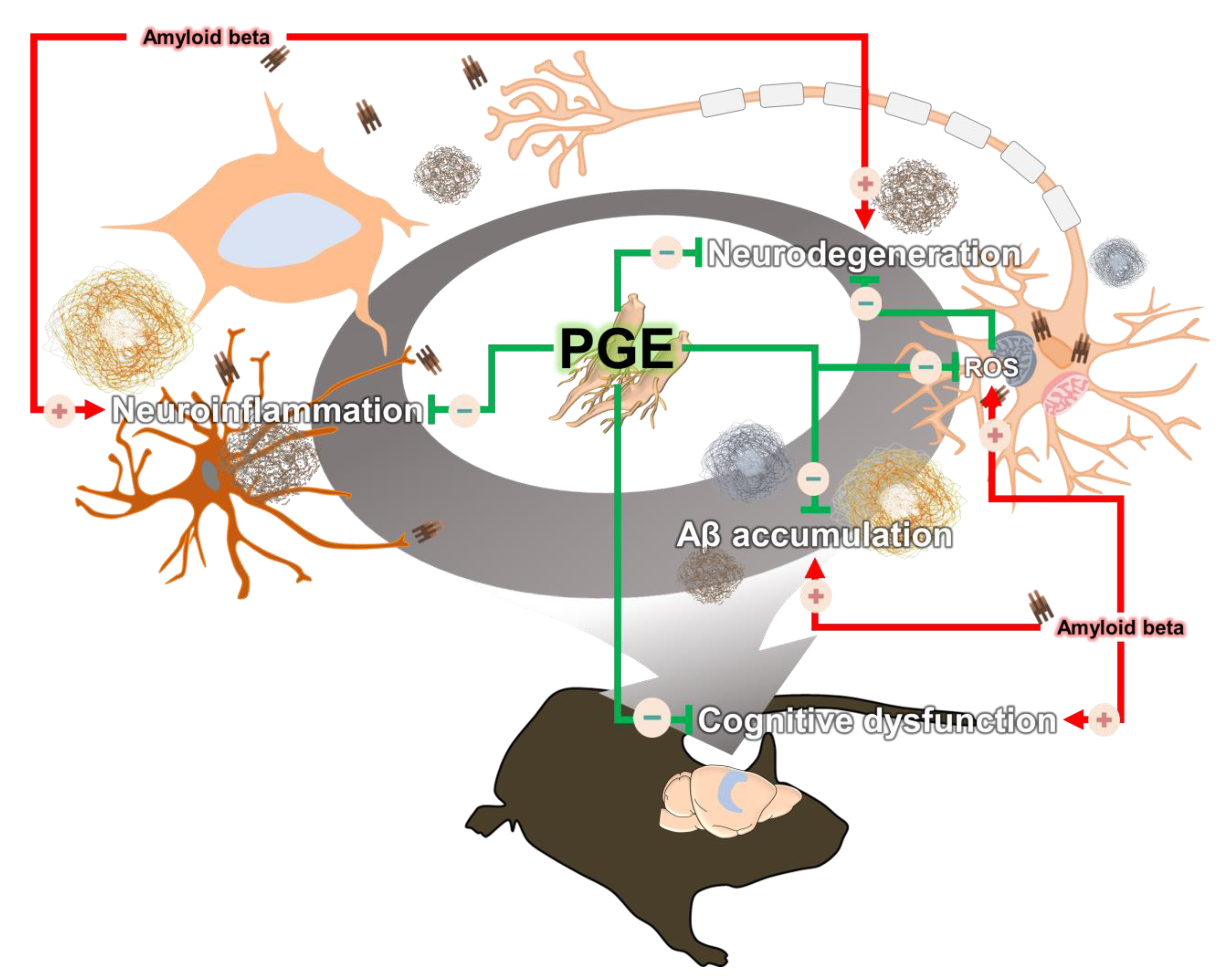
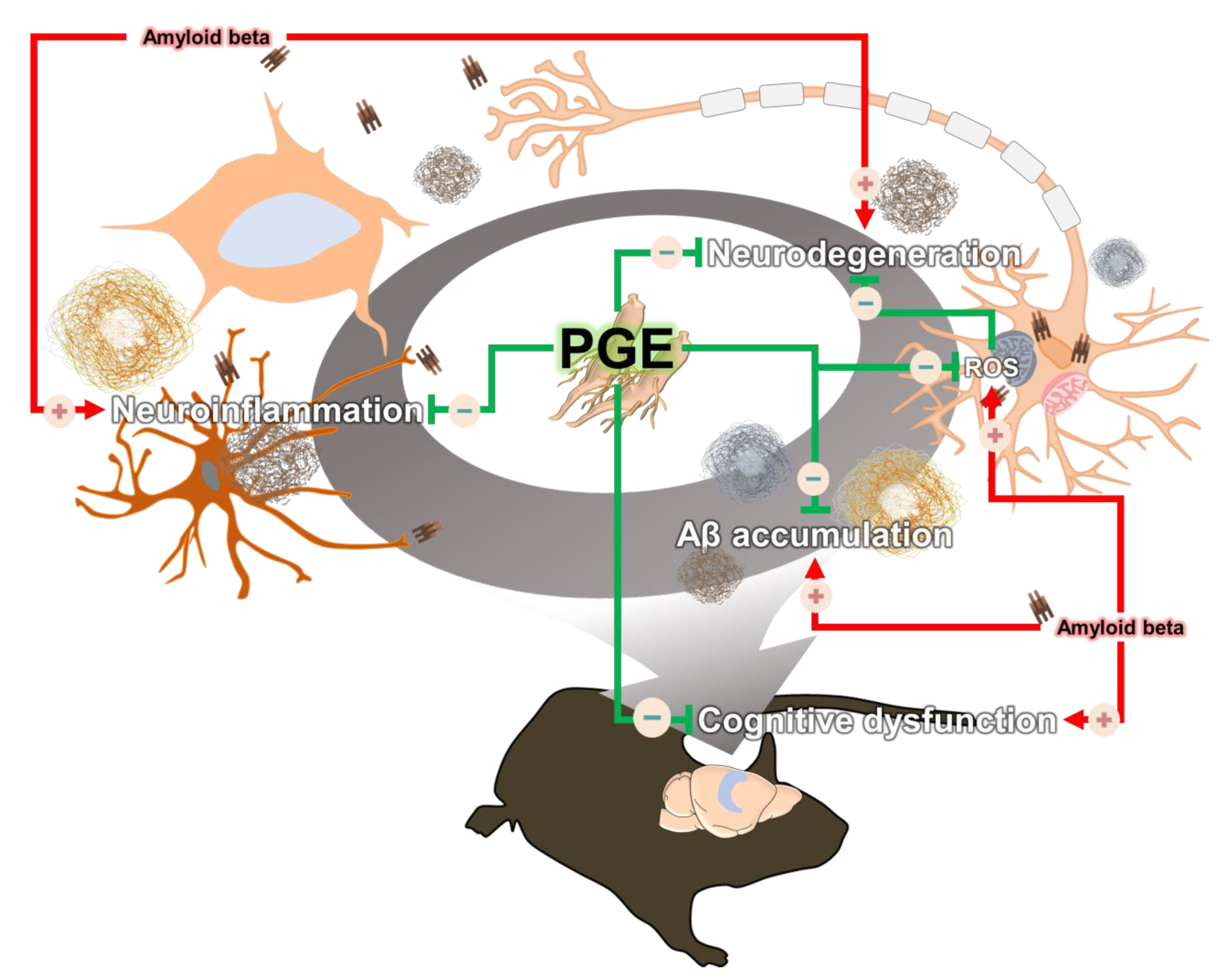
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.G.; Hong, S.B.; Nam, Y.; Tae, J.; Yoo, A.; Song, E.J.; Kim, K.I.; Lee, D.; Park, J.; Lee, S.M.; et al. Ghrelin in Alzheimer’s disease: Pathologic roles and therapeutic implications. Ageing Res. Rev. 2019, 55, 100945. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2004, 12, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Halabisky, B.; Zhou, Y.; Palop, J.J.; Yu, G.; Mucke, L.; Gan, L. Imbalance between GABAergic and Glutamatergic Transmission Impairs Adult Neurogenesis in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2009, 5, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, Y.; Yang, D.; Zhang, C.; Zhang, N.; Li, M.; Liu, Y. Platycodon grandiflorus—An ethnopharmacological, phytochemical and pharmacological review. J. Ethnopharmacol. 2015, 164, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Han, S.B.; Park, S.H.; Lee, K.H.; Lee, C.W.; Lee, S.H.; Kim, H.C.; Kim, Y.S.; Lee, H.S.; Kim, H.M. Polysaccharide isolated from the radix of Platycodon grandiflorum selectively activates B cells and macrophages but not T cells. Int. Immunopharmacol. 2001, 1, 1969–1978. [Google Scholar] [CrossRef]
- Kim, Y.P.; Lee, E.B.; Kim, S.Y.; Li, D.; Ban, H.S.; Lim, S.S.; Shin, K.H.; Ohuchi, K. Inhibition of prostaglandin E2 production by platycodin D isolated from the root of Platycodon grandiflorum. Planta Med. 2001, 67, 362–364. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, J.S.; Choi, S.U.; Kim, J.S.; Lee, H.S.; Roh, S.H.; Jeong, Y.C.; Kim, Y.K.; Ryu, S.Y. Isolation of a new saponin and cytotoxic effect of saponins from the root of Platycodon grandiflorum on human tumor cell lines. Planta Med. 2005, 71, 566–568. [Google Scholar] [CrossRef]
- Lee, K.J.; Choi, C.Y.; Chung, Y.C.; Kim, Y.S.; Ryu, S.Y.; Roh, S.H.; Jeong, H.G. Protective effect of saponins derived from roots of Platycodon grandiflorum on tert-butyl hydroperoxide-induced oxidative hepatotoxicity. Toxicol. Lett. 2004, 147, 271–282. [Google Scholar] [CrossRef]
- Kim, T.; Yoon, E.; Lee, D.; Imm, J.-Y. Antioxidant and anti-inflammatory activities of Platycodon grandiflorum seeds extract. CyTA J. Food 2020, 18, 435–444. [Google Scholar] [CrossRef]
- Oh, H.-G.; Kim, J.-H.; Shin, E.-H.; Kang, Y.-R.; Lee, B.-G.; Park, S.-H.; Moon, D.-I.; Kwon, L.-S.; Kim, Y.-P.; Choi, M.-H. Improving effects of platycodon extracts jelly on β-amyloid-induced cytotoxicity and scopolamine-induced cognitive impairment animal models. Korean J. Plant Resour. 2013, 26, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Um, M.Y.; Ha, T.Y.; Seong, K.S.; Kim, Y.S. In vitro screening of the acetylcholinesterase inhibition, antioxidant activity, and neuronal cell protective effect of medicinal plant extracts. Korean J. Food Preserv. 2013, 20, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Liu, G.; Wang, M.; Lv, Z.; Du, P. A selenium polysaccharide from Platycodon grandiflorum rescues PC12 cell death caused by H2O2 via inhibiting oxidative stress. Int. J. Biol. Macromol. 2017, 104, 393–399. [Google Scholar] [CrossRef]
- Son, I.H.; Park, Y.H.; Lee, S.I.; Yang, H.D.; Moon, H.I. Neuroprotective activity of triterpenoid saponins from Platycodi radix against glutamate-induced toxicity in primary cultured rat cortical cells. Molecules 2007, 12, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Xin, Z.; Liu, B.; Wang, J.; Wang, J.; Zhang, X.; Wang, Y.; Li, F. Platycodin D Inhibits Inflammatory Response in LPS-Stimulated Primary Rat Microglia Cells through Activating LXRalpha-ABCA1 Signaling Pathway. Front. Immunol. 2017, 8, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.H.; Kim, C.J.; Kim, E.H.; Kim, M.G.; Leem, K.H.; Kim, J. Effects of Platycodon grandiflorum on lipopolysaccharide-stimulated production of prostaglandin E2, nitric oxide, and interleukin-8 in mouse microglial BV2 cells. J. Med.Food 2006, 9, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Kim, Y.S.; Yeo, S.J.; Roh, S.H.; Jeong, Y.C.; Kang, J.S.; Ryu, S.Y. Ameliorating effect of balloon flower saponin on the ethanol-induced memory impairment in mice. Phytother. Res. 2008, 22, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.K.; Ahn, J.Y.; Kim, S.; Ryu, S.Y.; Kim, Y.S.; Ha, T.Y. Ethanol extract and saponin of Platycodon grandiflorum ameliorate scopolamine-induced amnesia in mice. J. Med. Food 2010, 13, 584–588. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Haertel, C.; Maelicke, A.; Montag, D. Galantamine slows down plaque formation and behavioral decline in the 5XFAD mouse model of Alzheimer’s disease. PLoS ONE 2014, 9, e89454. [Google Scholar] [CrossRef]
- Kimura, R.; Devi, L.; Ohno, M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J. Neurochem. 2010, 113, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Kimura, R.; Ohno, M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 2009, 33, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.I.; Jeon, S.G.; Kim, K.A.; Kim, J.J.; Song, E.J.; Jeon, Y.; Kim, E.; Lee, K.B.; Kwak, J.H.; Moon, M. Platycodon grandiflorus Root Extract Improves Learning and Memory by Enhancing Synaptogenesis in Mice Hippocampus. Nutrients 2017, 9, 794. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.; Choi, J.G.; Nam, D.W.; Hong, H.S.; Choi, Y.J.; Oh, M.S.; Mook-Jung, I. Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1-42 oligomer-injected mice. J. Alzheimers Dis. 2011, 23, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, M.; Kim, S.; Hwang, L.; Park, S. Ghrelin regulates hippocampal neurogenesis in adult mice. Endocr. J. 2009, 56, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.J.; You, H.J.; Park, M.S.; Johnston, T.V.; Ku, S.; Ji, G.E. Biocatalysis of Platycoside E and Platycodin D3 Using Fungal Extracellular beta-Glucosidase Responsible for Rapid Platycodin D Production. Int. J. Mol. Sci. 2018, 19, 2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendron, W.H. Age-Related Weight Loss in the 5XFAD Mouse Model of Alzheimer S Disease: A Behavioural and Hormonal Analysis. Ph.D. Thesis, Dalhousie University, Halifax, NS, Canada, 2015. [Google Scholar]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Cuestas Torres, D.M.; Cardenas, F.P. Synaptic plasticity in Alzheimer’s disease and healthy aging. Rev. Neurosci. 2020, 31, 245–268. [Google Scholar] [CrossRef]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Passamonti, L.; Tsvetanov, K.A.; Jones, P.S.; Bevan-Jones, W.R.; Arnold, R.; Borchert, R.J.; Mak, E.; Su, L.; O’Brien, J.T.; Rowe, J.B. Neuroinflammation and Functional Connectivity in Alzheimer’s Disease: Interactive Influences on Cognitive Performance. J. Neurosci. 2019, 39, 7218–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oomen, C.A.; Bekinschtein, P.; Kent, B.A.; Saksida, L.M.; Bussey, T.J. Adult hippocampal neurogenesis and its role in cognition. Wiley Interdiscip. Rev. Cogn. Sci. 2014, 5, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Jimenez, E.P.; Flor-Garcia, M.; Terreros-Roncal, J.; Rabano, A.; Cafini, F.; Pallas-Bazarra, N.; Avila, J.; Llorens-Martin, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. MolNeurodegener 2011, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, X.H.; Guan, J.; Ge, S.; Wu, M.B.; Lin, J.P.; Yang, L.R. Advancement of multi-target drug discoveries and promising applications in the field of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 169, 200–223. [Google Scholar] [CrossRef]
- Yoo, K.Y.; Park, O.K.; Hwang, I.K.; Li, H.; Ryu, S.Y.; Kang, I.J.; Yi, J.S.; Bae, Y.S.; Park, J.; Kim, Y.S.; et al. Induction of cell proliferation and neuroblasts in the subgranular zone of the dentate gyrus by aqueous extract from Platycodon grandiflorum in middle-aged mice. Neurosci. Lett. 2008, 444, 97–101. [Google Scholar] [CrossRef]
- Moon, M.; Cha, M.Y.; Mook-Jung, I. Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimers Dis. 2014, 41, 233–241. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Morphology of Alzheimer disease. Fortschr. Med. 1990, 108, 621–624. [Google Scholar]
- Rattan, S.I.; Demirovic, D. Hormesis can and does work in humans. Dose Response 2009, 8, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaviani, E.; Franceschi, C. A new theory on the common evolutionary origin of natural immunity, inflammation and stress response: The invertebrate phagocytic immunocyte as an eye-witness. Domest. Anim. Endocrinol. 1998, 15, 291–296. [Google Scholar] [CrossRef]
- Joseph, J.A.; Shukitt-Hale, B.; Casadesus, G. Reversing the deleterious effects of aging on neuronal communication and behavior: Beneficial properties of fruit polyphenolic compounds. Am. J. Clin. Nutr. 2005, 81, 313S–316S. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormesis and Ginseng: Ginseng Mixtures and Individual Constituents Commonly Display Hormesis Dose Responses, Especially for Neuroprotective Effects. Molecules 2020, 25, 2719. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, G.; Di Rosa, G.; Scuto, M.; Leri, M.; Stefani, M.; Schmitz-Linneweber, C.; Calabrese, V.; Saul, N. Healthspan Maintenance and Prevention of Parkinson’s-like Phenotypes with Hydroxytyrosol and Oleuropein Aglycone in C. elegans. Int. J. Mol. Sci. 2020, 21, 2588. [Google Scholar] [CrossRef] [Green Version]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Dumont, M.; Beal, M.F. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic. Biol. Med. 2011, 51, 1014–1026. [Google Scholar] [CrossRef] [Green Version]
- Tillement, L.; Lecanu, L.; Papadopoulos, V. Alzheimer’s disease: Effects of beta-amyloid on mitochondria. Mitochondrion 2011, 11, 13–21. [Google Scholar] [CrossRef]
- Umeno, A.; Biju, V.; Yoshida, Y. In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic. Res. 2017, 51, 413–427. [Google Scholar] [CrossRef]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/Antioxidant Imbalance in Alzheimer’s Disease: Therapeutic and Diagnostic Prospects. Oxid. Med. Cell Longev. 2018, 2018, 6435861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminzadeh, M.; Roghani, M.; Sarfallah, A.; Riazi, G.H. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int. Immunopharmacol. 2018, 54, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, C.; Zheng, M.; Liu, S.; Shi, R. Amentoflavone suppresses amyloid beta1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci 2019, 239, 117043. [Google Scholar] [CrossRef] [PubMed]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid. Med. Cell Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chausse, B.; Lewen, A.; Poschet, G.; Kann, O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain Behav. Immun. 2020, 88, 802–814. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef] [Green Version]
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Jeong, C.-H.; Choi, G.N.; Kim, J.H.; Kwak, J.H.; Kim, D.O.; Kim, Y.J.; Heo, H.J. Antioxidant activities from the aerial parts of Platycodon grandiflorum. Food Chem. 2010, 118, 278–282. [Google Scholar] [CrossRef]
- Ryu, C.S.; Kim, C.H.; Lee, S.Y.; Lee, K.S.; Choung, K.J.; Song, G.Y.; Kim, B.H.; Ryu, S.Y.; Lee, H.S.; Kim, S.K. Evaluation of the total oxidant scavenging capacity of saponins isolated from Platycodon grandiflorum. Food Chem. 2012, 132, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Konnerth, A. Impairments of neural circuit function in Alzheimer’s disease. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Flores Guzman, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, P.T. The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectr. 2005, 10, 6–9. [Google Scholar] [CrossRef]
- Martorana, A.; Di Lorenzo, F.; Belli, L.; Sancesario, G.; Toniolo, S.; Sallustio, F.; Sancesario, G.M.; Koch, G. Cerebrospinal Fluid Abeta42 Levels: When Physiological Become Pathological State. CNS Neurosci. Ther. 2015, 21, 921–925. [Google Scholar] [CrossRef]
- Ji, M.Y.; Bo, A.; Yang, M.; Xu, J.F.; Jiang, L.L.; Zhou, B.C.; Li, M.H. The Pharmacological Effects and Health Benefits of Platycodon grandiflorus-A Medicine Food Homology Species. Foods 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Martorana, A.; Di Lorenzo, F.; Manenti, G.; Semprini, R.; Koch, G. Homotaurine induces measurable changes of short latency afferent inhibition in a group of mild cognitive impairment individuals. Front. Aging Neurosci. 2014, 6, 254. [Google Scholar] [CrossRef]
- Choi, J.H.; Yoo, K.Y.; Park, O.K.; Lee, C.H.; Won, M.H.; Hwang, I.K.; Ryu, S.Y.; Kim, Y.S.; Yi, J.S.; Bae, Y.S.; et al. Platycodin D and 2″-O-acetyl-polygalacin D2 isolated from Platycodon grandiflorum protect ischemia/reperfusion injury in the gerbil hippocampus. Brain Res. 2009, 1279, 197–208. [Google Scholar] [CrossRef]
- Shi, C.; Li, Q.; Zhang, X. Platycodin D Protects Human Fibroblast Cells from Premature Senescence Induced by H2O2 through Improving Mitochondrial Biogenesis. Pharmacology 2020, 105, 598–608. [Google Scholar] [CrossRef]
- Wang, G.; Guo, H.; Wang, X. Platycodin D protects cortical neurons against oxygen-glucose deprivation/reperfusion in neonatal hypoxic-ischemic encephalopathy. J. Cell Biochem. 2019, 120, 14028–14034. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Jin, S.W.; Kim, H.G.; Choi, C.Y.; Lee, H.S.; Ryu, S.Y.; Chung, Y.C.; Hwang, Y.J.; Um, Y.J.; Jeong, T.C.; et al. Saponins, especially platyconic acid A, from Platycodon grandiflorum reduce airway inflammation in ovalbumin-induced mice and PMA-exposed A549 cells. J. Agric. Food Chem. 2015, 63, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Ramassamy, C.; Dore, S.; Christen, Y.; Poirier, J.; Quirion, R. The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by beta-amyloid. Eur. J. Neurosci. 2000, 12, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimers Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.B.; Spangler, E.L.; Devan, B.D.; Guo, Z.; Bowker, J.L.; Janas, A.M.; Hagepanos, A.; Minor, R.K.; DeCabo, R.; Mouton, P.R.; et al. A blueberry-enriched diet provides cellular protection against oxidative stress and reduces a kainate-induced learning impairment in rats. Neurobiol. Aging 2008, 29, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.M.; Hashimoto, M.; Katakura, M.; Hara, Y.; Shido, O. Green tea catechins prevent cognitive deficits caused by Abeta1-40 in rats. J. Nutr. Biochem. 2008, 19, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Hoi, C.P.; Ho, Y.P.; Baum, L.; Chow, A.H. Neuroprotective effect of honokiol and magnolol, compounds from Magnolia officinalis, on beta-amyloid-induced toxicity in PC12 cells. Phytother. Res. 2010, 24, 1538–1542. [Google Scholar] [CrossRef]
- Joseph, J.A.; Denisova, N.A.; Arendash, G.; Gordon, M.; Diamond, D.; Shukitt-Hale, B.; Morgan, D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr. Neurosci. 2003, 6, 153–162. [Google Scholar] [CrossRef]
- Maitra, I.; Marcocci, L.; Droy-Lefaix, M.T.; Packer, L. Peroxyl radical scavenging activity of Ginkgo biloba extract EGb 761. Biochem. Pharmacol. 1995, 49, 1649–1655. [Google Scholar] [CrossRef]
- Rigacci, S. Olive Oil Phenols as Promising Multi-targeting Agents Against Alzheimer’s Disease. Adv. Exp. Med. Biol. 2015, 863, 1–20. [Google Scholar] [CrossRef]
- Wang, J.; Santa-Maria, I.; Ho, L.; Ksiezak-Reding, H.; Ono, K.; Teplow, D.B.; Pasinetti, G.M. Grape derived polyphenols attenuate tau neuropathology in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Thomas, P.; Zhong, J.H.; Bi, F.F.; Kosaraju, S.; Pollard, A.; Fenech, M.; Zhou, X.F. Consumption of grape seed extract prevents amyloid-beta deposition and attenuates inflammation in brain of an Alzheimer’s disease mouse. Neurotox. Res. 2009, 15, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, F.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Duroux, J.L.; Olivier, Y.; Trouillas, P. Free radical scavenging by natural polyphenols: Atom versus electron transfer. J. Phys. Chem. A 2013, 117, 2082–2092. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.K.; Surh, Y.J. Modulation of Nrf2-mediated antioxidant and detoxifying enzyme induction by the green tea polyphenol EGCG. Food Chem. Toxicol. 2008, 46, 1271–1278. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.I.; Lee, Y.K.; Park, S.G.; Choi, I.S.; Ban, J.O.; Park, H.K.; Nam, S.Y.; Yun, Y.W.; Han, S.B.; Oh, K.W.; et al. l-Theanine, an amino acid in green tea, attenuates beta-amyloid-induced cognitive dysfunction and neurotoxicity: Reduction in oxidative damage and inactivation of ERK/p38 kinase and NF-κB pathways. Free Radic. Biol. Med. 2009, 47, 1601–1610. [Google Scholar] [CrossRef]
- Longpre, F.; Garneau, P.; Christen, Y.; Ramassamy, C. Protection by EGb 761 against beta-amyloid-induced neurotoxicity: Involvement of NF-kappaB, SIRT1, and MAPKs pathways and inhibition of amyloid fibril formation. Free Radic. Biol. Med. 2006, 41, 1781–1794. [Google Scholar] [CrossRef]
- Oh, J.H.; Kang, L.L.; Ban, J.O.; Kim, Y.H.; Kim, K.H.; Han, S.B.; Hong, J.T. Anti-inflammatory effect of 4-O-methylhonokiol, compound isolated from Magnolia officinalis through inhibition of NF-κB [corrected]. Chem. Biol. Interact. 2009, 180, 506–514. [Google Scholar] [CrossRef]
- Zuo, J.; Yin, B.; Hu, X. Research Progress in the Chemical Constituents and Modern Pharmacology of Platycodon. J. Liaoning Univ. TCM 2019, 21, 113–116. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, Y.; Shin, S.J.; Park, Y.H.; Kim, M.-J.; Jeon, S.G.; Lee, H.; Choi, Y.; Kim, T.-J.; Shin, S.M.; Kim, J.-J.; et al. Platycodon grandiflorum Root Protects against Aβ-Induced Cognitive Dysfunction and Pathology in Female Models of Alzheimer’s Disease. Antioxidants 2021, 10, 207. https://doi.org/10.3390/antiox10020207
Nam Y, Shin SJ, Park YH, Kim M-J, Jeon SG, Lee H, Choi Y, Kim T-J, Shin SM, Kim J-J, et al. Platycodon grandiflorum Root Protects against Aβ-Induced Cognitive Dysfunction and Pathology in Female Models of Alzheimer’s Disease. Antioxidants. 2021; 10(2):207. https://doi.org/10.3390/antiox10020207
Chicago/Turabian StyleNam, Yunkwon, Soo Jung Shin, Yong Ho Park, Min-Jeong Kim, Seong Gak Jeon, Hyewon Lee, Yeji Choi, Tae-Jin Kim, Seong Min Shin, Jwa-Jin Kim, and et al. 2021. "Platycodon grandiflorum Root Protects against Aβ-Induced Cognitive Dysfunction and Pathology in Female Models of Alzheimer’s Disease" Antioxidants 10, no. 2: 207. https://doi.org/10.3390/antiox10020207
APA StyleNam, Y., Shin, S. J., Park, Y. H., Kim, M.-J., Jeon, S. G., Lee, H., Choi, Y., Kim, T.-J., Shin, S. M., Kim, J.-J., Yoo, D.-H., Kim, H. D., Kim, S., & Moon, M. (2021). Platycodon grandiflorum Root Protects against Aβ-Induced Cognitive Dysfunction and Pathology in Female Models of Alzheimer’s Disease. Antioxidants, 10(2), 207. https://doi.org/10.3390/antiox10020207