
Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress in Cancer Cells
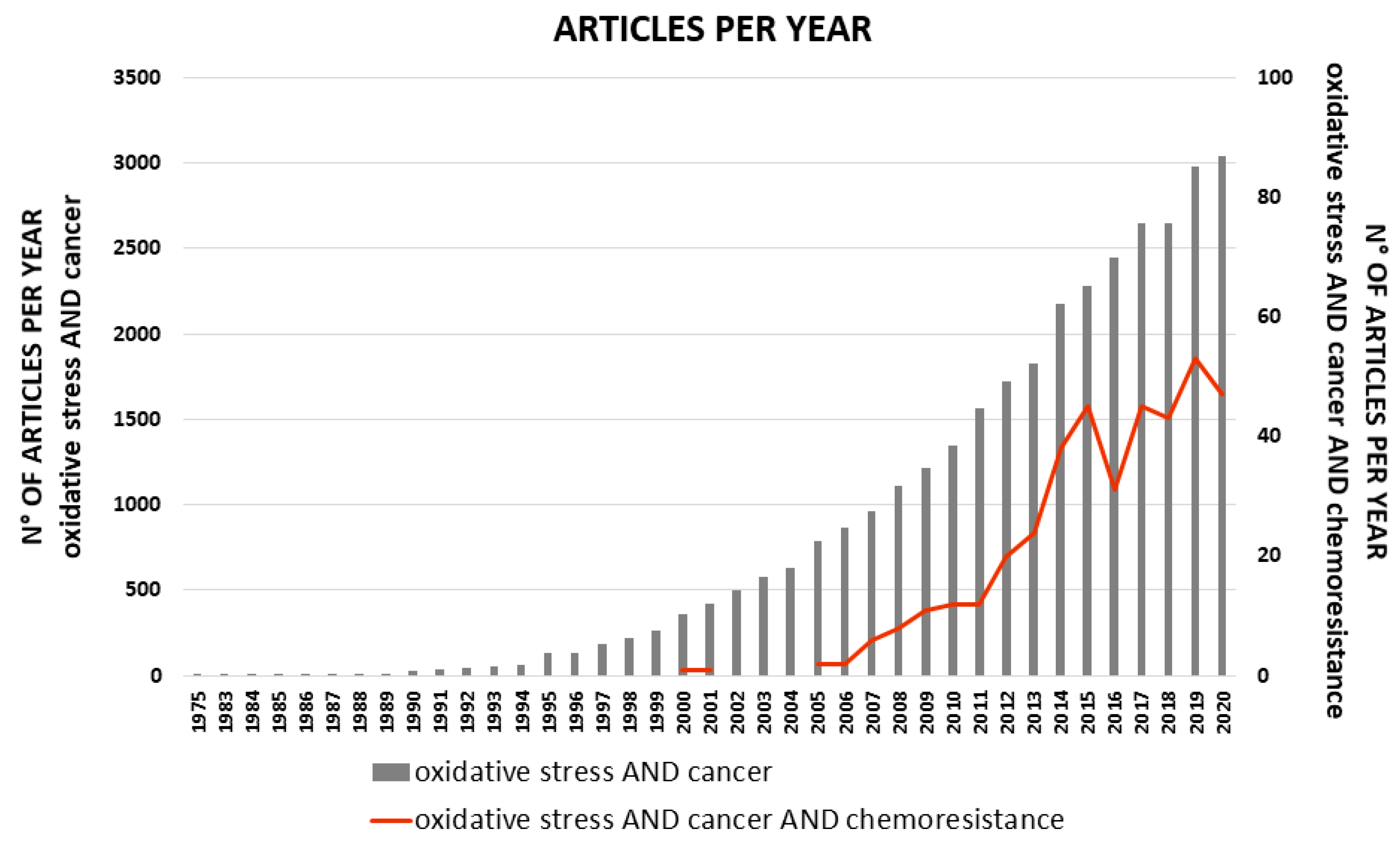
3. Oxidative Stress and Chemoresistance
4. Control of Nrf2 Transcription Factor in Chemoresistance
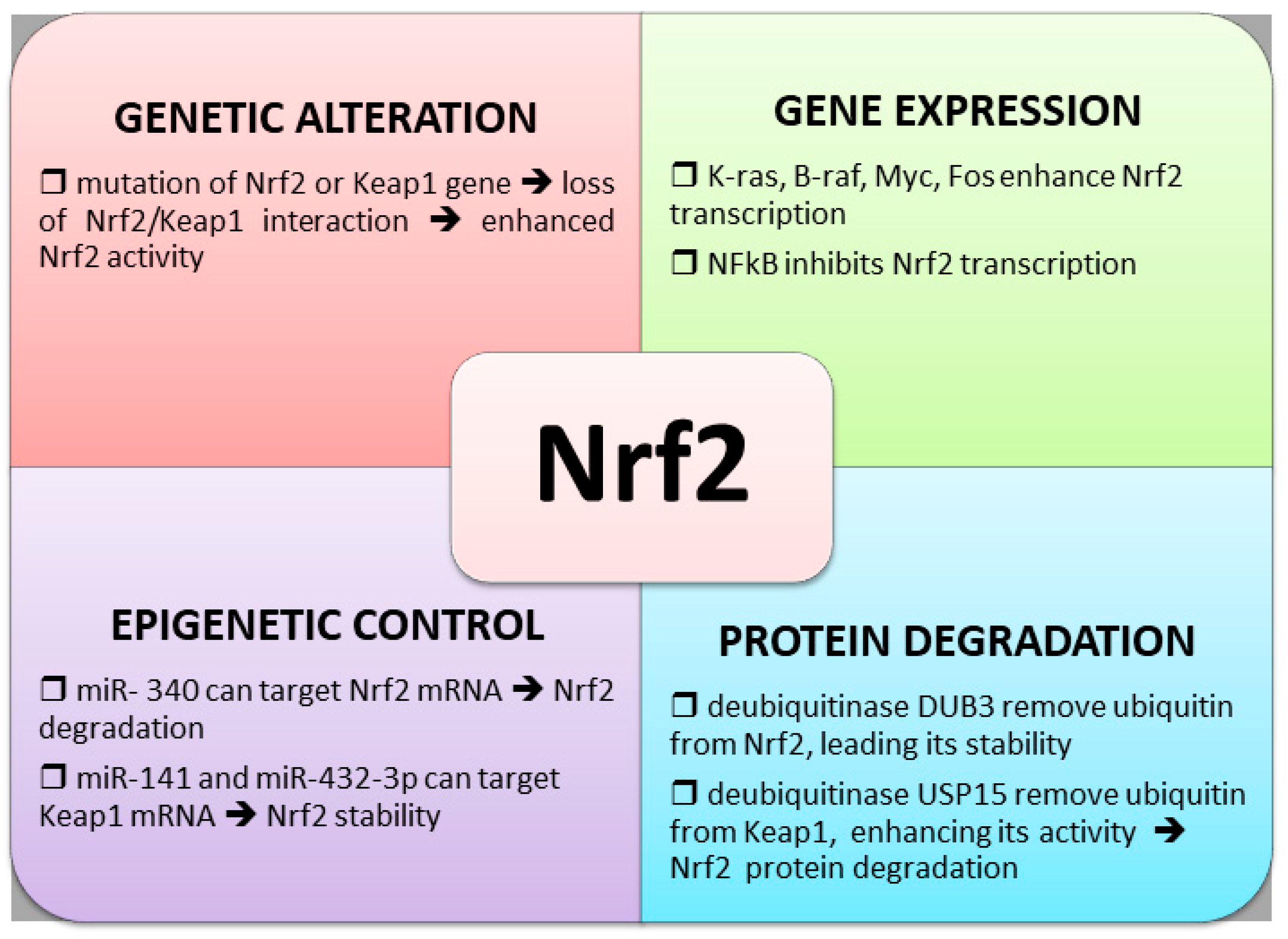
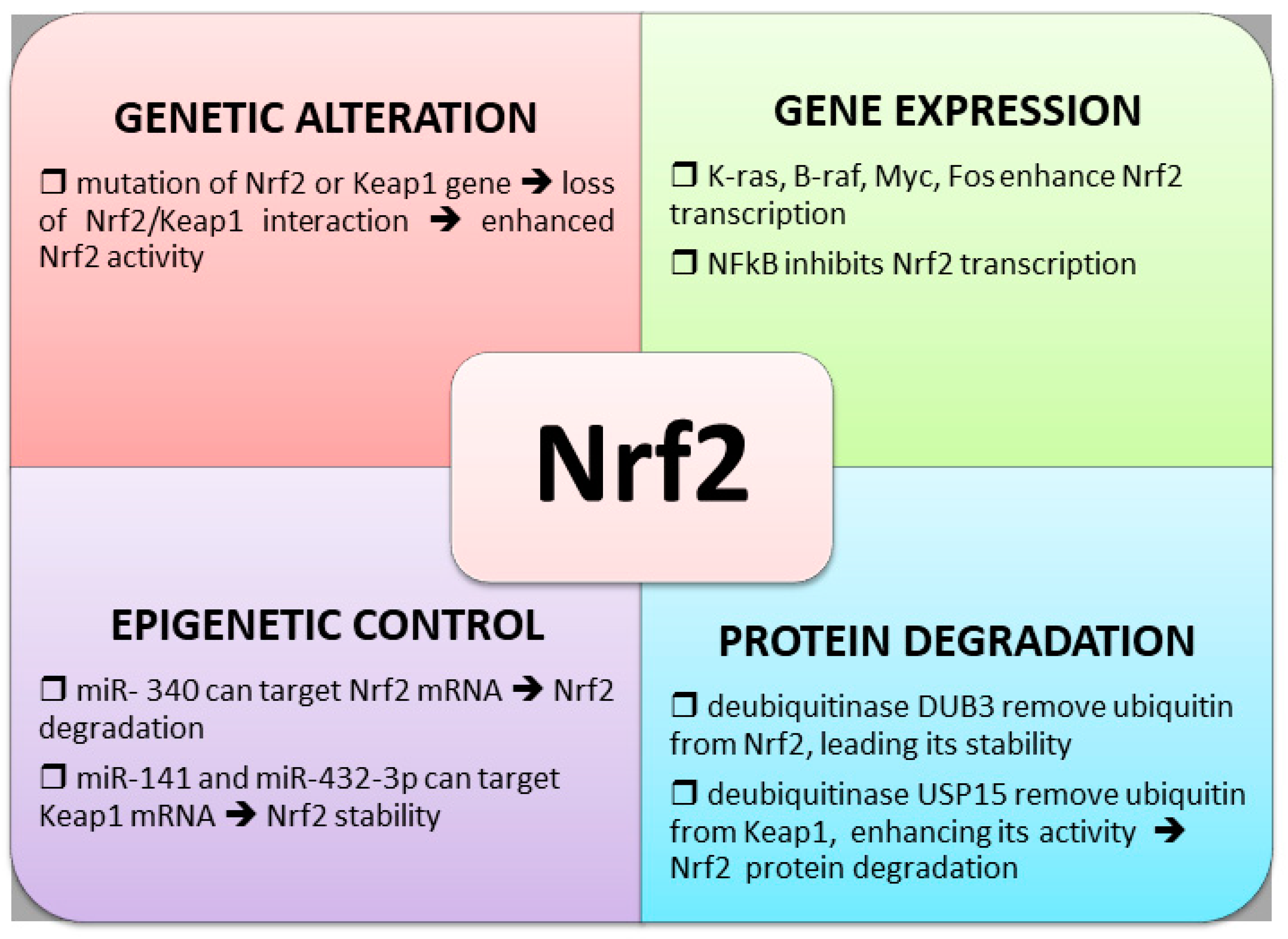
4.1. Nrf2 and Keap1 Genetic Alterations
4.2. Altered Nrf2 Transcription
4.3. Control of Nrf2 Protein Degradation
4.4. Epigenetic Regulation of Nrf2 Expression
4.5. Control of Nrf2 Activity by Bach1
5. Other Transcription Factors Involved in Oxidative Stress Control
5.1. NF-kB Transcription Factor
5.2. FOXO Family of Transcription Factors
5.3. AP-1 Transcription Factors
5.4. P53 Transcription Factor
5.5. PGC-1a and YAP Transcriptional Coactivators
6. Pro-Oxidant Drugs and Antioxidant-Dependent Chemoresistance
7. Nrf2 Inhibitors in the Treatment of Chemoresistant Tumors
8. Conclusions
Funding
Conflicts of Interest
References
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19. [Google Scholar] [CrossRef]
- Asić, K. Dominant mechanisms of primary resistance differ from dominant mechanisms of secondary resistance to targeted therapies. Crit. Rev. Oncol. 2016, 97, 178–196. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Lipinski, B. Hydroxyl Radical and Its Scavengers in Health and Disease. Oxidative Med. Cell. Longev. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxidedismutase: Organellespecificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Liu, R.; Yuan, K.; Yi, T.; Zhao, X.; Huang, C.; Wei, Y. Proteomics analysis of tumor microenvironment: Implications of metabolic and oxidative stresses in tumorigenesis. Mass Spectrom. Rev. 2012, 32, 267–311. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2007, 15, 171–182. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B., 3rd; Wondrak, G.T. Targeting the redox Achilles heel of cancer. Curr. Opin. Investig. Drugs. 2007, 8, 1022–1037. [Google Scholar]
- Wondrak, G.T. Redox-Directed Cancer Therapeutics: Molecular Mechanisms and Opportunities. Antioxid. Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic ap-proach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Mendes, C.; Serpa, J. Metabolic Remodelling: An Accomplice for New Therapeutic Strategies to Fight Lung Cancer. Antioxidants 2019, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Volti, G.L.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme Oxygenase-1 in Central Nervous System Malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Lam, Y.M.; Pym, J.; Campling, B.G. Mechanisms of resistance of human small cell lung cancer lines selected in VP-16 and cisplatin. Cancer 1996, 77, 1797–1808. [Google Scholar] [CrossRef]
- Hospers, G.A.P.; Meijer, C.; De Leij, L.; Uges, D.R.A.; Mulder, N.H.; De Vries, E.G.E.; Meuer, C. A study of human small-cell lung carcinoma (hsclc) cell lines with different sensitivities to detect relevant mechanisms of cisplatin (cddp) resistance. Int. J. Cancer 1990, 46, 138–144. [Google Scholar] [CrossRef]
- Zhan, M.; Liu, X.Y. Schedule-dependent reversion of cisplatin resistance by 5-fluorouracil in a cisplatin-resistant human lung adenocarcinoma cell line A549DDP. Chin. Med. J. 1999, 112, 336–339. [Google Scholar] [PubMed]
- Kinnula, K.; Linnainmaa, K.; Raivio, K.O.; Kinnula, V.L. Endogenous antioxidant enzymes and glutathione S-transferase in protection of mesothelioma cells against hydrogen peroxide and epirubicin toxicity. Br. J. Cancer 1998, 77, 1097–1102. [Google Scholar] [CrossRef] [Green Version]
- Sluiter, W.J.; Mulder, N.H.; Timmer-Bosscha, H.; Jan-Meersma, G.; de Vries, E.G. Relationship of Cellular Glutathione to the Cytotoxicity and Resistance of Seven Platinum Compounds. Cancer Res. 1992, 52, 6885–6889. [Google Scholar]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Yu, K.-D.; Huang, A.-J.; Fan, L.; Li, W.-F.; Shao, Z.-M. Genetic Variants in Oxidative Stress–Related Genes Predict Chemoresistance in Primary Breast Cancer: A Prospective Observational Study and Validation. Cancer Res. 2012, 72, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Lanzardo, S.; Conti, L.; Rooke, R.; Ruiu, R.; Accart, N.; Bolli, E.; Arigoni, M.; Macagno, M.; Barrera, G.; Pizzimenti, S.; et al. Immunotargeting of Antigen xCT Attenuates Stem-like Cell Behavior and Metastatic Progression in Breast Cancer. Cancer Res. 2016, 76, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-Glutamate Transporter SLC7A11 in Cancer Chemosensitivity and Chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, S.; Sato, H.; Kuriyama-Matsumura, K.; Tamba, M.; Wang, H.; Sohda, S.; Hamada, H.; Yoshikawa, H.; Kondo, T.; Bannai, S. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer 2003, 88, 951–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma-On, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, Y.; Yang, M.; Wang, K.; Liu, Y.; Zhang, M.; Yang, Y.; Jin, C.; Wang, R.; Hu, R. Digoxin sensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine via inhibiting Nrf2 signaling pathway. Redox Biol. 2019, 22, 101131. [Google Scholar] [CrossRef]
- Tazehkand, A.P.; Akbarzadeh, M.; Velaie, K.; Sadeghi, M.R.; Samadi, N. The role of Her2-Nrf2 axis in induction of oxaliplatin resistance in colon cancer cells. Biomed. Pharmacother. 2018, 103, 755–766. [Google Scholar] [CrossRef]
- Benassi, B.; Marani, M.; Loda, M.; Blandino, G. USP2a alters chemotherapeutic response by modulating redox. Cell Death Dis. 2013, 4, e812. [Google Scholar] [CrossRef]
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 806–814. [Google Scholar] [CrossRef]
- Daga, M.; Pizzimenti, S.; Dianzani, C.; Cucci, M.A.; Cavalli, R.; Grattarola, M.; Ferrara, B.; Scariot, V.; Trotta, F.; Barrera, G. Ailanthone inhibits cell growth and migration of cisplatin resistant bladder cancer cells through down-regulation of Nrf2, YAP, and c-Myc expression. Phytomedicine 2019, 56, 156–164. [Google Scholar] [CrossRef]
- Cucci, M.A.; Grattarola, M.; Dianzani, C.; Damia, G.; Ricci, F.; Roetto, A.; Trotta, F.; Barrera, G.; Pizzimenti, S. Ailanthone in-creases oxidative stress in CDDP-resistant ovarian and bladder cancer cells by inhibiting of Nrf2 and YAP expression through a post-translational mechanism. Free Radic. Biol. Med. 2020, 150, 125–135. [Google Scholar] [CrossRef]
- Ambrosio, L.; Argenziano, M.; Cucci, M.A.; Grattarola, M.; De Graaf, I.A.; Dianzani, C.; Barrera, G.; Nieves, J.S.; Gomez, R.; Cavalli, R.; et al. Carbosilane Dendrimers Loaded with siRNA Targeting Nrf2 as a Tool to Overcome Cisplatin Chemoresistance in Bladder Cancer Cells. Antioxidants 2020, 9, 993. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Z.; Li, M.; Chai, J.; He, S.; Wu, J.; Xu, J. Icariside II overcomes BRAF inhibitor resistance in melanoma by inducing ROS production and inhibiting MITF. Oncol. Rep. 2020, 44, 360–370. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, X.Q.; Delaidelli, A.; Rafn, B.; Cran, J.; et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef]
- Karathedath, S.; Rajamani, B.M.; Aalam, S.M.M.; Abraham, A.; Varatharajan, S.; Krishnamurthy, P.; Mathews, V.; Velayudhan, S.R.; Balasubramanian, P. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS ONE 2017, 12, e0177227. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase to Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nat. Cell Biol. 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Lignitto, L.; Leboeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e18. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.-Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updates 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 Function Activates Nrf2 and Provides Advantages for Lung Cancer Cell Growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic Alteration of Keap1 Confers Constitutive Nrf2 Activation and Resistance to Chemotherapy in Gallbladder Cancer. Gastroenterology 2008, 135, 1358–1368.e4. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 Mutations and Nrf2 Pathway Activation in Epithelial Ovarian Cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietlein, F.; Weghorn, D.; Taylor-Weiner, A.; Richters, A.; Reardon, B.; Liu, D.; Lander, E.S.; Van Allen, E.M.; Sunyaev, S.R. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 2020, 52, 208–218. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative Haploinsufficiency and Triplosensitivity Drive Aneuploidy Patterns and Shape the Cancer Genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS Confers Chemoresistance by Upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Shi, S.; Liu, M.; Qin, Y.; Meng, Q.; Hua, J.; Ji, S.; Zhang, Y.; Yang, J.; Xu, J.; et al. PIN1 Maintains Redox Balance via the c-Myc/NRF2 Axis to Counteract Kras-Induced Mitochondrial Respiratory Injury in Pancreatic Cancer Cells. Cancer Res. 2019, 79, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Shin, H.-J.; Bak, I.S.; Bak, Y.; Jeong, Y.-L.; Kwon, T.; Park, Y.-H.; Sun, H.-N.; Kim, C.-H.; Yu, D.-Y. Peroxiredoxin I is important for cancer-cell survival in Ras-induced hepatic tumorigenesis. Oncotarget 2016, 7, 68044–68056. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic value of FOXM1 in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 32298–32308. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, G.L.; Chen, Y.; Teng, N.N. Targeting FOXM1 Improves Cytotoxicity of Paclitaxel and Cisplatinum in Platinum-Resistant Ovarian Cancer. Int. J. Gynecol. Cancer 2017, 27, 1602–1609. [Google Scholar] [CrossRef]
- de Moraes, G.N.; Delbue, D.; Silva, K.L.; Robaina, M.C.; Khongkow, P.; Gomes, A.R.; Zona, S.; Crocamo, S.; Mencalha, A.L.; Magalhães, L.M.; et al. FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cell. Signal. 2015, 27, 2496–2505. [Google Scholar] [CrossRef]
- Lin, J.-Z.; Wang, W.-W.; Hu, T.-T.; Zhu, G.-Y.; Li, L.-N.; Zhang, C.-Y.; Xu, Z.; Yu, H.-B.; Wu, H.-F.; Zhu, J.-G. FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy. Cancer Lett. 2020, 469, 481–489. [Google Scholar] [CrossRef]
- Hou, Y.; Zhu, Q.; Li, Z.; Peng, Y.; Yu, X.; Yuan, B.; Liu, Y.; Liu, Y.; Yin, L.; Peng, Y.; et al. The FOXM1–ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017, 8, e2659. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Halasi, M.; Zia, M.F.; Gann, P.; Gaitonde, S.; Mahmud, N.; Gartel, A.L. Nuclear FOXM1 drives chemoresistance in AML. Leukemia 2016, 31, 251–255. [Google Scholar] [CrossRef]
- Varghese, V.; Magnani, L.; Harada-Shoji, N.; Mauri, F.; Szydlo, R.M.; Yao, S.; Lam, E.W.-F.; Kenny, L.M. FOXM1 modulates 5-FU resistance in colorectal cancer through regulating TYMS expression. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- De La Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2–Keap1 Antioxidant Response by the Ubiquitin Proteasome System: An Insight into Cullin-Ring Ubiquitin Ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Zhang, Z.-Y.; Du, H.; Li, S.-Z.; Tu, R.; Jia, Y.-F.; Zheng, Z.; Song, X.-M.; Du, R.-L.; Zhang, X.-D. DUB3 deubiquitinates and stabilizes NRF2 in chemotherapy resistance of colorectal cancer. Cell Death Differ. 2019, 26, 2300–2313. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Tian, W.; Wu, T.; Sun, Z.; Lau, A.; Chapman, E.; Fang, D.; Zhang, D.D. USP15 Negatively Regulates Nrf2 through Deubiquitination of Keap1. Mol. Cell 2013, 51, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Chen, Z.-G.; Wu, L.-L.; Zheng, J.-J.; Yang, J.-R.; Chen, X.-F.; Chen, Z.-Q.; Liu, C.-L.; Chi, S.-Y.; Zheng, J.-Y.; et al. miR-340 Reverses Cisplatin Resistance of Hepatocellular Carcinoma Cell Lines by Targeting Nrf2-dependent Antioxidant Pathway. Asian Pac. J. Cancer Prev. 2015, 15, 10439–10444. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Chen, X.; Yu, F.; Zheng, F.; Lin, X. MiR-141 Activates Nrf2-Dependent Antioxidant Pathway via Down-Regulating the Expression of Keap1 Conferring the Resistance of Hepatocellular Carcinoma Cells to 5-Fluorouracil. Cell. Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef] [Green Version]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme Induces Ubiquitination and Degradation of the Transcription Factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Wu, H.; Lei, R.; Chong, R.A.; Wei, Y.; Lu, X.; Tagkopoulos, I.; Kung, S.-Y.; Yang, Q.; Hu, G.; et al. Transcriptional Network Analysis Identifies BACH1 as a Master Regulator of Breast Cancer Bone Metastasis. J. Biol. Chem. 2012, 287, 33533–33544. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- St Clair, D.K.; Porntadavity, S.; Xu, Y.; Kiningham, K. Transcription regulation of human manganese superoxide dismutase gene. Methods Enzymol. 2002, 349, 306–312. [Google Scholar]
- Lingappan, K. NF-kappaB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Scoggin, S.; Gaynor, R.B.; Williams, N.S. Identification of NF-κB-regulated genes induced by TNFα utilizing expression profiling and RNA interference. Oncogene 2003, 22, 2054–2064. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.A.; Beug, H.; Wirth, T. Epithelial-Mesenchymal Transition: NF-κB Takes Center Stage. Cell Cycle 2004, 3, 1477–1480. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jia, X.-H.; Chen, J.-R.; Wang, J.-Y.; Li, Y.-J. Osthole shows the potential to overcome P-glycoprotein-mediated multidrug resistance in human myelogenous leukemia K562/ADM cells by inhibiting the PI3K/Akt signaling pathway. Oncol. Rep. 2016, 35, 3659–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFα-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egodwin, P.; Ebaird, A.-M.; Eheavey, S.; Ebarr, M.; O’Byrne, K.J.; Gately, K.A. Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Sethi, G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. et Biophys. Acta (BBA) Rev. Cancer 2010, 1805, 167–180. [Google Scholar] [CrossRef]
- Li, Q.; Yang, G.; Feng, M.; Zheng, S.; Cao, Z.; Qiu, J.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; et al. NF-kappaB in pancreatic cancer: Its key role in chemoresistance. Cancer Lett. 2018, 421, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: Functional validation and clinical significance. Theranostics 2020, 10, 3967–3979. [Google Scholar] [CrossRef]
- Zhan, Y.; Qiu, Y.; Wang, H.; Wang, Z.; Xu, J.; Fan, G.; Xu, J.; Li, W.; Cao, Y.; Le, M.-V.; et al. Bufalin reverses multidrug resistance by regulating stemness through the CD133/nuclear factor-κB/MDR1 pathway in colorectal cancer. Cancer Sci. 2020, 111, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [Green Version]
- Kops, G.J.P.L.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.A.; Coffer, P.J.; Huang, T.-T.; Bos, J.L.; Medema, R.H.; Burgering, B.M.T. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nat. Cell Biol. 2002, 419, 316–321. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Hui, R.C.-Y.; Gomes, A.R.; Constantinidou, D.; Costa, J.R.; Karadedou, C.T.; De Mattos, S.F.; Wymann, M.P.; Brosens, J.J.; Schulze, A.; Lam, E.W.-F. The Forkhead Transcription Factor FOXO3a Increases Phosphoinositide-3 Kinase/Akt Activity in Drug-Resistant Leukemic Cells through Induction of PIK3CA Expression. Mol. Cell. Biol. 2008, 28, 5886–5898. [Google Scholar] [CrossRef] [Green Version]
- Hui, R.C.-Y.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W.-F. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Kumazoe, M.; Takai, M.; Hiroi, S.; Takeuchi, C.; Kadomatsu, M.; Nojiri, T.; Onda, H.; Bae, J.; Huang, Y.; Takamatsu, K.; et al. The FOXO3/PGC-1β signaling axis is essential for cancer stem cell properties of pancreatic ductal adenocarcinoma. J. Biol. Chem. 2017, 292, 10813–10823. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Zhang, Z.; Pei, H.; Wang, H.; Li, L.; Xia, Q. FoxO3a induces temozolomide resistance in glioblastoma cells via the regulation of β-catenin nuclear accumulation. Oncol. Rep. 2017, 37, 2391–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, M.; Hagenbuchner, J.; Rass, B.; Fiegl, H.; Kiechl-Kohlendorfer, U.; Obexer, P.; Ausserlechner, M.J. FOXO3-mediated chemo-protection in high-stage neuroblastoma depends on wild-type TP53 and SESN3. Oncogene 2017, 36, 6190–6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcher, S.; Spoden, G.; Hagenbuchner, J.; Führer, S.; Kaserer, T.; Tollinger, M.; Huber-Cantonati, P.; Gruber, T.; Schuster, D.; Gust, R.; et al. A drug library screen identifies Carbenoxolone as novel FOXO inhibitor that overcomes FOXO3-mediated chemoprotection in high-stage neuroblastoma. Oncogene 2020, 39, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A.; et al. IGF1 Receptor Signaling Regulates Adaptive Radioprotection in Glioma Stem Cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, M.; Wei, Y.; Yu, S.; Li, H.; Wang, Y.; Xu, X.; Cui, Y.; Tian, J.; Liang, L.; et al. FoxO3a confers cetuximab resistance in RAS wild-type metastatic colorectal cancer through c-Myc. Oncotarget 2016, 7, 80888–80900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.S.; Brosens, J.J.; Schwenen, H.D.; Lam, E.W.-F. FOXO and FOXM1 in cancer: The FOXO-FOXM1 axis shapes the outcome of cancer chemotherapy. Curr. Drug Targets 2011, 12, 1256–1266. [Google Scholar] [CrossRef]
- Glorieux, C.; Sandoval, J.M.; Fattaccioli, A.; Dejeans, N.; Garbe, J.C.; Dieu, M.; Verrax, J.; Renard, P.; Huang, P.; Calderon, P.B. Chromatin remodeling regulates catalase expression during cancer cells adaptation to chronic oxidative stress. Free Radic. Biol. Med. 2016, 99, 436–450. [Google Scholar] [CrossRef]
- Amstad, P.A.; Krupitza, G.; Cerutti, P.A. Mechanism of c-fos induction by active oxygen. Cancer Res. 1992, 52, 3952–3960. [Google Scholar] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Hsieh, M.-J.; Chen, J.-C.; Yang, W.-E.; Chien, S.-Y.; Chen, M.-K.; Lo, Y.-S.; Hsi, Y.-T.; Chuang, Y.-C.; Lin, C.-C.; Yang, S.-F. Dehydroandrographolide inhibits oral cancer cell migration and invasion through NF-κB-, AP-1-, and SP-1-modulated matrix metalloproteinase-2 inhibition. Biochem. Pharmacol. 2017, 130, 10–20. [Google Scholar] [CrossRef]
- Wang, F.; Lv, P.; Gu, Y.; Li, L.; Ge, X.; Guo, G. Galectin-1 knockdown improves drug sensitivity of breast cancer by reducing P-glycoprotein expression through inhibiting the Raf-1/AP-1 signaling pathway. Oncotarget 2017, 8, 25097–25106. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wan, G.-H.; Wu, Y.-M.; Wang, H.-S.; Wang, H.-F.; Zhang, G.; Lu, L.-L.; Li, Z.-Q.; Chan, K.-Y.; Zhou, Y.; et al. AP-1 confers resistance to anti-cancer therapy by activating XIAP. Oncotarget 2018, 9, 14124–14137. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-N.; Jin, H.-O.; Park, J.-A.; Kim, J.-H.; Kim, J.-Y.; Kim, B.; Kim, W.; Hong, S.-E.; Lee, Y.-H.; Chang, Y.H.; et al. Heme oxygenase-1 determines the differential response of breast cancer and normal cells to piperlongumine. Mol. Cells 2015, 38, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamaschi, D.; Gasco, M.; Hiller, L.; Sullivan, A.; Syed, N.; Trigiante, G.; Yulug, I.; Merlano, M.; Numico, G.; Comino, A.; et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 2003, 3, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Chee, J.L.; Saidin, S.; Lane, D.P.; Leong, S.M.; Noll, J.E.; Neilsen, P.M.; Phua, Y.T.; Gabra, H.; Lim, T.M. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 2013, 12, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.K.; Pietsch, E.C.; Dumont, P.; Tao, J.; Murphy, M.E. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol. Ther. 2011, 11, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Goffart, S.; Wiesner, R.J. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp. Physiol. 2003, 88, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.M.; Wingert, R.A. PGC-1α in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef]
- Mastropasqua, F.; Girolimetti, G.; Shoshan, M. PGC1α: Friend or Foe in Cancer? Genes 2018, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Paku, M.; Haraguchi, N.; Takeda, M.; Fujino, S.; Ogino, T.; Takahashi, H.; Miyoshi, N.; Uemura, M.; Mizushima, T.; Yamamoto, H.; et al. SIRT3-Mediated SOD2 and PGC-1α Contribute to Chemoresistance in Colorectal Cancer Cells. Ann. Surg. Oncol. 2021, 1–13. [Google Scholar] [CrossRef]
- Cruz-Bermúdez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; García-Grande, A.; Coronado, M.J.; Laine-Menéndez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepeñas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1α in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Ciamporcero, E.; Daga, M.; Pizzimenti, S.; Roetto, A.; Dianzani, C.; Compagnone, A.; Palmieri, A.; Ullio, C.; Cangemi, L.; Pili, R.; et al. Crosstalk between Nrf2 and YAP contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic. Biol. Med. 2018, 115, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.-S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, Y.; Zhou, W.; Chen, T.; Wu, Q.; Chutturghoon, V.K.; Lin, B.; Geng, L.; Yang, Z.; Zhou, L.; et al. YAP promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1-ROS-mTOR pathway. Cancer Cell Int. 2019, 19, 1–15. [Google Scholar] [CrossRef]
- Bauzone, M.; Souidi, M.; Dessein, A.-F.; Wisztorski, M.; Vincent, A.; Gimeno, J.-P.; Monté, D.; Van Seuningen, I.; Gespach, C.; Huet, G. Cross-talk between YAP and RAR-RXR Drives Expression of Stemness Genes to Promote 5-FU Resistance and Self-Renewal in Colorectal Cancer Cells. Mol. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Buckarma, E.H.; Werneburg, N.W.; Conboy, C.B.; Kabashima, A.; O’Brien, D.R.; Wang, C.; Rizvi, S.H.; Smoot, R.L. The YAP-Interacting Phosphatase SHP2 Can Regulate Transcriptional Coactivity and Modulate Sensitivity to Chemotherapy in Cholangiocarcinoma. Mol. Cancer Res. 2020, 18, 1574–1588. [Google Scholar] [CrossRef]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 Expression Inhibits YAP and TAZ Oncogenic Functions in Pancreatic Cancer. Mol. Cancer Ther. 2020, 19, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Varela-López, A.; Battino, M.; Navarro-Hortal, M.D.; Giampieri, F.; Forbes-Hernández, T.Y.; Romero-Márquez, J.M.; Collado, R.; Quiles, J.L. An update on the mechanisms related to cell death and toxicity of doxorubicin and the protective role of nutrients. Food Chem. Toxicol. 2019, 134, 110834. [Google Scholar] [CrossRef]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Azami, N.; Hamzehlou, S.; Farahani, M.; Hushmandi, K.; Ashrafizadeh, M.; et al. Nrf2 Signaling Pathway in Chemoprotection and Doxorubicin Resistance: Potential Application in Drug Discovery. Antioxidants 2021, 10, 349. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Timerbaev, A.R.; Hartinger, C.G.; Aleksenko, S.S.; Keppler, B.K. Interactions of Antitumor Metallodrugs with Serum Proteins: Advances in Characterization Using Modern Analytical Methodology. Chem. Rev. 2006, 106, 2224–2248. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Salatino, A.; Aversa, I.; Battaglia, A.M.; Sacco, A.; Di Vito, A.; Santamaria, G.; Chirillo, R.; Veltri, P.; Tradigo, G.; Di Cello, A.; et al. H-Ferritin Affects Cisplatin-Induced Cytotoxicity in Ovarian Cancer Cells through the Modulation of ROS. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zhou, X.; Qiu, J. Emerging role of NRF2 in ROS-mediated tumor chemoresistance. Biomed. Pharmacother. 2020, 131, 110676. [Google Scholar] [CrossRef]
- Verstovsek, S.; Giles, F.; Quintás-Cardama, A.; Perez, N.; Ravandi-Kashani, F.; Beran, M.; Freireich, E.; Kantarjian, H. Arsenic derivatives in hematologic malignancies: A role beyond acute promyelocytic leukemia? Hematol. Oncol. 2006, 24, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Sumi, D.; Shinkai, Y.; Kumagai, Y. Signal transduction pathways and transcription factors triggered by arsenic trioxide in leukemia cells. Toxicol. Appl. Pharmacol. 2010, 244, 385–392. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Smeester, L.; Zou, F.; Kesic, M.; Jaspers, I.; Pi, J.; Fry, R.C. The NRF2-mediated oxidative stress response pathway is associated with tumor cell resistance to arsenic trioxide across the NCI-60 panel. BMC Med Genom. 2010, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Zhan, L.; Zhang, H.; Zhang, Q.; Woods, C.G.; Chen, Y.; Xue, P.; Dong, J.; Tokar, E.J.; Xu, Y.; Hou, Y.; et al. Regulatory role of KEAP1 and NRF2 in PPARgamma expression and chemoresistance in human non-small-cell lung carcinoma cells. Free Radic. Biol. Med. 2012, 53, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, S.; Suzuki, T.; Koike, S.; Yuan, B.; Takagi, N.; Ogasawara, Y. Nrf2 activation ameliorates cytotoxic effects of arsenic trioxide in acute promyelocytic leukemia cells through increased glutathione levels and arsenic efflux from cells. Toxicol. Appl. Pharmacol. 2016, 305, 161–168. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Donehower, R.C.; Jones, R.J.; Tucker, R.W. Microtubule changes and cytotoxicity in leukemic cell lines treated with taxol. Cancer Res. 1988, 48, 4093–4100. [Google Scholar]
- Jiang, H.; Zhang, X.; Liao, Q.; Wu, W.; Liu, Y.; Huang, W. Electrochemical Monitoring of Paclitaxel-Induced ROS Release from Mitochondria inside Single Cells. Small 2019, 15, e1901787. [Google Scholar] [CrossRef]
- Chen, L.; He, J.; Zhou, J.; Xiao, Z.; Ding, N.; Duan, Y.; Li, W.; Sun, L. EIF2A promotes cell survival during paclitaxel treatment in vitro and in vivo. J. Cell. Mol. Med. 2019, 23, 6060–6071. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’Ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Kim, M.P.; Gallick, G.E. Gemcitabine Resistance in Pancreatic Cancer: Picking the Key Players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [Green Version]
- Zarei, M.; Lal, S.; Parker, S.J.; Nevler, A.; Vaziri-Gohar, A.; Dukleska, K.; Mambelli-Lisboa, N.C.; Moffat, C.; Blanco, F.F.; Chand, S.N.; et al. Posttranscriptional Upregulation of IDH1 by HuR Establishes a Powerful Survival Phenotype in Pancreatic Cancer Cells. Cancer Res. 2017, 77, 4460–4471. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Duan, Q.; Zhang, Z.; Li, H.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell. Mol. Med. 2017, 21, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, S.; Li, H.; Duan, Q.; Zhang, Z.; Shen, Q.; Wang, C.; Yin, T. ROS/KRAS/AMPK Signaling Contributes to Gemcitabine-Induced Stem-like Cell Properties in Pancreatic Cancer. Mol. Ther. Oncolytics 2019, 14, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Field-Smith, A.; Morgan, G.J.; E Davies, F. Bortezomib (Velcade?) in the treatment of multiple myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. Cross-talk between two antioxidants, thioredoxin reductase and heme oxygenase-1, and therapeutic implications for multiple myeloma. Redox Biol. 2016, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Volti, G.L. Heme oxygenase-1 nuclear translocation regulates bortezomib-induced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Lee, K.-H.; Hayashi, N.; Okano, M.; Nozaki, H.; Ju-Ichi, M. Antitumor Agents, 65. Brusatol and Cleomiscosin-A, Antileukemic Principles from Brucea javanica. J. Nat. Prod. 1984, 47, 550–551. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, X.; Pan, Z.; Wang, D.; Li, M.; Chen, X.; Zhou, L.; Xu, M.; Li, D.; Zheng, Q. Ailanthone Induces Cell Cycle Arrest and Apoptosis in Melanoma B16 and A375 Cells. Biomolecules 2019, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Chen, C.; Cheng, Y.; Zhu, L.; Wang, Y.; Luo, C.; He, Y.; Yang, Z.; Ji, Z. Ailanthone induces autophagic and apoptotic cell death in human promyelocytic leukemia HL-60 cells. Oncol. Lett. 2018, 16, 3569–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhu, L.; Yang, X.; Wei, C.; Chen, C.; He, Y.; Ji, Z. Ailanthone induces G2/M cell cycle arrest and apoptosis of SGC-7901 human gastric cancer cells. Mol. Med. Rep. 2017, 16, 6821–6827. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; Yao, C.; Zhu, X.; Gong, C.; Xu, Z.; Wang, L.; Li, S.; Zou, C.; Zhu, S. Ailanthone inhibits non-small cell lung cancer cell growth through repressing DNA replication via downregulating RPA1. Br. J. Cancer 2017, 117, 1621–1630. [Google Scholar] [CrossRef]
- Gao, W.; Ge, S.; Sun, J. Ailanthone exerts anticancer effect by up-regulating miR-148a expression in MDA-MB-231 breast cancer cells and inhibiting proliferation, migration and invasion. Biomed. Pharmacother. 2019, 109, 1062–1069. [Google Scholar] [CrossRef]
- Han, F.; Liu, G.; Sun, C.; Wei, J. Ailanthone reverses multidrug resistance by inhibiting the P-glycoprotein-mediated efflux in resistant K562/A02 cells. Cell. Mol. Biol. 2018, 64, 55–61. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Peng, S.; Wang, J.; Chen, H.; Cong, X.; Chen, A.; Hu, M.; Qin, M.; Wu, H.; Gao, S.; et al. Ailanthone targets p23 to overcome MDV3100 resistance in castration-resistant prostate cancer. Nat. Commun. 2016, 7, 13122. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Yi, S.; Gong, L.; Liu, W.; Wang, P.; Liu, N.; Zhao, L.; Wang, P. Inhibition of TGF-β signaling with halofuginone can enhance the antitumor effect of irradiation in Lewis lung cancer. OncoTargets Ther. 2015, 8, 3549–3559. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [Green Version]
- Bollong, M.J.; Yun, H.; Sherwood, L.; Woods, A.K.; Lairson, L.L.; Schultz, P.G. A Small Molecule Inhibits Deregulated NRF2 Transcriptional Activity in Cancer. ACS Chem. Biol. 2015, 10, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 19589–19594. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.-J.; Jung, B.-J.; Lee, S.-H.; Yoo, H.-S.; Shin, E.-A.; Ko, H.-J.; Chang, S.; Kim, S.-Y.; Jeon, S.-M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Gao, A.-M.; Ke, Z.-P.; Wang, J.-N.; Yang, J.-Y.; Chen, S.-Y.; Chen, H. Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway. Carcinog 2013, 34, 1806–1814. [Google Scholar] [CrossRef]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.A.; Parama, D.; Daimari, E.; Girisa, S.; Banik, K.; Harsha, C.; Dutta, U.; Kunnumakkara, A.B. Rationalizing the therapeutic potential of apigenin against cancer. Life Sci. 2021, 267, 118814. [Google Scholar] [CrossRef]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510. https://doi.org/10.3390/antiox10040510
Barrera G, Cucci MA, Grattarola M, Dianzani C, Muzio G, Pizzimenti S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants. 2021; 10(4):510. https://doi.org/10.3390/antiox10040510
Chicago/Turabian StyleBarrera, Giuseppina, Marie Angele Cucci, Margherita Grattarola, Chiara Dianzani, Giuliana Muzio, and Stefania Pizzimenti. 2021. "Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role" Antioxidants 10, no. 4: 510. https://doi.org/10.3390/antiox10040510
APA StyleBarrera, G., Cucci, M. A., Grattarola, M., Dianzani, C., Muzio, G., & Pizzimenti, S. (2021). Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants, 10(4), 510. https://doi.org/10.3390/antiox10040510