
Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan


Abstract
:1. Introduction
2. Acetyl-CoA and CoA-SH Levels
3. CR Appears to Increase Hepatic Acetyl-CoA without Decreasing Autophagy, While Long Term Fasting Appears to Decrease Hepatic Acetyl-CoA to Stimulate Autophagy
4. ACLY Synthesizes the Majority of Nucleocytoplasmic Acetyl-CoA, While Acetyl-CoA Synthetase 2 (ACSS2) Plays a Smaller but Important Role
5. ACSS2 Associates with Chromatin in Neural Cells and Provides Acetyl-CoA for Histone Acetylation That Induces Autophagy
6. Hypothalamic AMPK Increases with Fasting to Inhibit Fatty Acid Synthesis and Stimulate Fatty Acid Oxidation That Could Increase Nuclear Acetyl-CoA Levels to Stimulate HAT Function
7. Brain Histone Acetylation Changes with Aging
8. CR Increases HAT and Decreases HDAC Activity in Brain Cortex
9. CBP Levels in the Hypothalamus Positively Correlate with Longevity and This Is Likely Due to the Role of CBP in the Mitochondrial Unfolded Protein Response
10. Consumption of the Acetate Precursors GTA or Ethanol Increases Histone Acetylation in the Brain
11. Pyruvate, Ketone Bodies, Citric Acid Cycle Metabolites, or Pantothenate Supplementation May Increase Nucleocytoplasmic Acetyl-CoA Levels and Are Protective in Many Rodent Models of Human Aging-Related Disease
12. Mammalian Changes in Histone Lysine Acetylation Marks with Aging outside the Brain
13. Yeast Changes in Histone Lysine Acetylation Marks with Aging and Anti-Aging CR
14. Fruit Fly Changes in Histone Lysine Acetylation Marks with Aging and Anti-Aging Dietary Restriction
15. Nematode Changes in Histone Lysine Acetylation Marks with Aging and Anti-Aging Dietary Restriction
16. Experiments Using C. elegans Suggest That Increased Acetyl-CoA Synthesis Stimulates Longevity
17. Decreased Acetyl-CoA during C. elegans Development Is Required for the UPRmt to Increase Lifespan
18. Conclusions
Funding
Conflicts of Interest
References
- Balasubramanian, P.; Howell, P.R.; Anderson, R.M. Aging and Caloric Restriction Research: A Biological Perspective with Translational Potential. EBioMedicine 2017, 21, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e548. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546.e535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, D.; Coogan, S.C.; Solon-Biet, S.M.; De Cabo, R.; Haran, J.B.; Raubenheimer, D.; Cogger, V.C.; Mattson, M.P.; Simpson, S.J.; Le Couteur, D.G. Cognitive and behavioral evaluation of nutritional interventions in rodent models of brain aging and dementia. Clin. Interv. Aging 2017, 12, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Peng, L.; Cline, G.W.; Petersen, K.F.; Shulman, G.I. A Non-invasive Method to Assess Hepatic Acetyl-CoA In Vivo. Cell Metab. 2017, 25, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell 2012, 11, 371–377. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 2020, 81, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Lan, Y.; Donahue, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 2019, 8, e47866. [Google Scholar] [CrossRef]
- Liu, X.; Cooper, D.E.; Cluntun, A.A.; Warmoes, M.O.; Zhao, S.; Reid, M.A.; Liu, J.; Lund, P.J.; Lopes, M.; Garcia, B.A.; et al. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell 2018, 175, 502–513.e513. [Google Scholar] [CrossRef] [Green Version]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “great” controlling nucleotide coenzymes. IUBMB Life 2019, 71, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hinc, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The Regulatory Effects of Acetyl-CoA Distribution in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2018, 12, 169. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, K.; Ramsey, J.J.; Weindruch, R. Caloric restriction increases gluconeogenic and transaminase enzyme activities in mouse liver. Exp. Gerontol. 2003, 38, 267–278. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell. Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Shurubor, Y.I.; D’Aurelio, M.; Clark-Matott, J.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Cooper, A.J.L.; Krasnikov, B.F. Determination of Coenzyme A and Acetyl-Coenzyme A in Biological Samples Using HPLC with UV Detection. Molecules 2017, 22, 1388. [Google Scholar] [CrossRef] [Green Version]
- Marriage, B.J.; Clandinin, M.T.; Macdonald, I.M.; Glerum, D.M. Cofactor treatment improves ATP synthetic capacity in patients with oxidative phosphorylation disorders. Mol. Genet. Metab. 2004, 81, 263–272. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Starvation in man. Clin. Endocrinol. Metab. 1976, 5, 397–415. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr.; Marliss, E.B.; Aoki, T.T. Fat and nitrogen metabolism in fasting man. Horm. Metab. Res. Horm. Stoffwechs. Horm. Metab. 1970, 2 (Suppl. 2), 181–185. [Google Scholar]
- Cahill Jr, G.F. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnoni, G.V.; Priore, P.; Geelen, M.J.; Siculella, L. The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 2009, 61, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.L.; Xiong, Y.; Lei, Q.Y. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell. 2013, 51, 506–518. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherrier, M.; Li, H. The impact of keto-adaptation on exercise performance and the role of metabolic-regulating cytokines. Am. J. Clin. Nutr. 2019, 110, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kawakami, K.; Kametani, F.; Nakamoto, H.; Goto, S. Biological significance of protein modifications in aging and calorie restriction. Ann. N. Y. Acad Sci. 2010, 1197, 33–39. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Ji, L.L. Aging alters acetylation status in skeletal and cardiac muscles. GeroScience 2020, 42, 963–976. [Google Scholar] [CrossRef]
- Mezhnina, V.; Pearce, R.; Poe, A.; Velingkaar, N.; Astafev, A.; Ebeigbe, O.P.; Makwana, K.; Sandlers, Y.; Kondratov, R.V. CR reprograms acetyl-CoA metabolism and induces long-chain acyl-CoA dehydrogenase and CrAT expression. Aging Cell 2020, 19, e13266. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.L.; László, L.; Fellinger, E.; Jakab, A.; Orosz, A.; Réz, G.; Kovács, J. Combined effects of fasting and vinblastine treatment on serum insulin level, the size of autophagic-lysosomal compartment, protein content and lysosomal enzyme activities of liver and exocrine pancreatic cells of the mouse. Comp. Biochem. Physiol. B Comp. Biochem. 1989, 94, 505–510. [Google Scholar] [CrossRef]
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, A.; Cavallini, G.; Bergamini, E. Effects of aging, antiaging calorie restriction and in vivo stimulation of autophagy on the urinary excretion of 8OHdG in male Sprague-Dawley rats. Age 2013, 35, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, S.E.; Julian, D.; Akin, D.E.; Fried, J.; Toscano, K.; Leeuwenburgh, C.; Dunn, W.A., Jr. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007, 10, 281–292. [Google Scholar] [CrossRef]
- Houston, R.; Sekine, S.; Calderon, M.J.; Seifuddin, F.; Wang, G.; Kawagishi, H.; Malide, D.A.; Li, Y.; Gucek, M.; Pirooznia, M.; et al. Acetylation-mediated remodeling of the nucleolus regulates cellular acetyl-CoA responses. PLoS Biol. 2020, 18, e3000981. [Google Scholar] [CrossRef] [PubMed]
- Beigneux, A.P.; Kosinski, C.; Gavino, B.; Horton, J.D.; Skarnes, W.C.; Young, S.G. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 2004, 279, 9557–9564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Zhang, M.; Plec, A.A.; Estill, S.J.; Cai, L.; Repa, J.J.; McKnight, S.L.; Tu, B.P. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, E9499–e9506. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Torres, A.; Henry, R.A.; Trefely, S.; Wallace, M.; Lee, J.V.; Carrer, A.; Sengupta, A.; Campbell, S.L.; Kuo, Y.-M.; et al. ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 2016, 17, 1037–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265.e256. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189. [Google Scholar] [CrossRef]
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002, 277, 9520–9528. [Google Scholar] [CrossRef] [Green Version]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.-Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, A.C.; Huff, M.W. ATP-citrate lyase: Genetics, molecular biology and therapeutic target for dyslipidemia. Curr. Opin. Lipidol. 2017, 28, 193–200. [Google Scholar] [CrossRef]
- Khwairakpam, A.D.; Banik, K.; Girisa, S.; Shabnam, B.; Shakibaei, M.; Fan, L.; Arfuso, F.; Monisha, J.; Wang, H.; Mao, X.; et al. The vital role of ATP citrate lyase in chronic diseases. J. Mol. Med. 2020, 98, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Pouikli, A.; Parekh, S.; Maleszewska, M.; Baghdadi, M.; Tripodi, I.; Nikopoulou, C.; Folz-Donahue, K.; Hinze, Y.; Mesaros, A.; Giavalisco, P.; et al. Deregulated mito-nuclear communication alters chromatin plasticity and differentiation potential of mesenchymal stem cells upon ageing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics and Oncogenesis-Part 1: Acetyl-CoA, Acetogenesis and Acyl-CoA Short-Chain Synthetases. Front. Physiol. 2020, 11, 580167. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.E.; Jarrett, I.G.; Filsell, O.H.; Ballard, F.J. Production and utilization of acetate in mammals. Biochem. J. 1974, 142, 401–411. [Google Scholar] [CrossRef]
- Skutches, C.L.; Holroyde, C.P.; Myers, R.N.; Paul, P.; Reichard, G.A. Plasma acetate turnover and oxidation. J. Clin. Investig. 1979, 64, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Akanji, A.O.; Humphreys, S.; Thursfield, V.; Hockaday, T.D. The relationship of plasma acetate with glucose and other blood intermediary metabolites in non-diabetic and diabetic subjects. Clin. Chim. Acta 1989, 185, 25–34. [Google Scholar] [CrossRef]
- Smith, R.F.; Humphreys, S.; Hockaday, T.D. The measurement of plasma acetate by a manual or automated technique in diabetic and non-diabetic subjects. Ann. Clin. Biochem. 1986, 23, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Jackowski, S.; Leonardi, R. Deregulated coenzyme A, loss of metabolic flexibility and diabetes. Biochem. Soc. Trans. 2014, 42, 1118–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Zhang, X.M.; Zhang, D.; Kumashiro, N.; Camporez, J.P.; Cline, G.W.; Rothman, D.L.; Shulman, G.I. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat. Med. 2014, 20, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; Van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Brierley, G.P.; Jurkowitz, M.; Scott, K.M.; Merola, A.J. Ion transport by heart mitochondria. XXII. Spontaneous, energy-linked accumulation of acetate and phosphate salts of monovalent cations. Arch. Biochem. Biophys. 1971, 147, 545–556. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate dependence of tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lin, S.-H.; Ren, F.; Li, J.-T.; Chen, J.-J.; Yao, C.-B.; Yang, H.-B.; Jiang, S.-X.; Yan, G.-Q.; Wang, D.; et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 2016, 7, 11960. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Shao, F.; Shi, S.; Feng, X.; Wang, W.; Wang, Y.; Guo, W.; Wang, J.; Gao, S.; Gao, Y.; et al. Prognostic Impact of Metabolism Reprogramming Markers Acetyl-CoA Synthetase 2 Phosphorylation and Ketohexokinase-A Expression in Non-Small-Cell Lung Carcinoma. Front. Oncol. 2019, 9, 1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Kong, Y.; Cao, M.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; Li, J.; Zhu, X.; et al. Decreased expression of acetyl-CoA synthase 2 promotes metastasis and predicts poor prognosis in hepatocellular carcinoma. Cancer Sci. 2017, 108, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, D.M.; Namboodiri, A.M.; Moffett, J.R. Acetate as a Metabolic and Epigenetic Modifier of Cancer Therapy. J. Cell Biochem. 2016, 117, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Tsen, A.R.; Long, P.M.; Driscoll, H.E.; Davies, M.T.; Teasdale, B.A.; Penar, P.L.; Pendlebury, W.W.; Spees, J.L.; Lawler, S.E.; Viapiano, M.S.; et al. Triacetin-based acetate supplementation as a chemotherapeutic adjuvant therapy in glioma. Int. J. Cancer 2014, 134, 1300–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, P.M.; Tighe, S.W.; Driscoll, H.E.; Fortner, K.A.; Viapiano, M.S.; Jaworski, D.M. Acetate supplementation as a means of inducing glioblastoma stem-like cell growth arrest. J. Cell. Physiol. 2015, 230, 1929–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong, A.; Hannah, V.C.; Brown, M.S.; Goldstein, J.L. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2000, 275, 26458–26466. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Yamamoto, J.; Okamura, M.; Fujino, T.; Takahashi, S.; Takeuchi, K.; Osborne, T.F.; Yamamoto, T.T.; Ito, S.; Sakai, J. Transcriptional regulation of the murine acetyl-CoA synthetase 1 gene through multiple clustered binding sites for sterol regulatory element-binding proteins and a single neighboring site for Sp1. J. Biol. Chem. 2001, 276, 34259–34269. [Google Scholar] [CrossRef] [Green Version]
- Sone, H.; Shimano, H.; Sakakura, Y.; Inoue, N.; Amemiya-Kudo, M.; Yahagi, N.; Osawa, M.; Suzuki, H.; Yokoo, T.; Takahashi, A.; et al. Acetyl-coenzyme A synthetase is a lipogenic enzyme controlled by SREBP-1 and energy status. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E222–E230. [Google Scholar] [CrossRef] [Green Version]
- Carrer, A.; Parris, J.L.D.; Trefely, S.; Henry, R.A.; Montgomery, D.C.; Torres, A.; Viola, J.M.; Kuo, Y.-M.; Blair, I.A.; Meier, J.L.; et al. Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. J. Biol. Chem. 2017, 292, 3312–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.T.; Magistretti, P.J.; Buck, A.; Weber, B. Labeled acetate as a marker of astrocytic metabolism. J. Cereb. Blood Flow Metab. 2011, 31, 1668–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Gulanski, B.I.; De Feyter, H.M.; Weinzimer, S.A.; Pittman, B.; Guidone, E.; Koretski, J.; Harman, S.; Petrakis, I.L.; Krystal, J.H.; et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Investig. 2013, 123, 1605–1614. [Google Scholar] [CrossRef] [Green Version]
- Ariyannur, P.S.; Moffett, J.R.; Madhavarao, C.N.; Arun, P.; Vishnu, N.; Jacobowitz, D.M.; Hallows, W.C.; Denu, J.M.; Namboodiri, A.M. Nuclear-cytoplasmic localization of acetyl coenzyme a synthetase-1 in the rat brain. J. Comp. Neurol. 2010, 518, 2952–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowlands, B.D.; Klugmann, M.; Rae, C.D. Acetate metabolism does not reflect astrocytic activity, contributes directly to GABA synthesis, and is increased by silent information regulator 1 activation. J. Neurochem. 2017, 140, 903–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medrano-Fernández, A.; Delgado-Garcia, J.M.; Del Blanco, B.; Llinares, M.; Sánchez-Campusano, R.; Olivares, R.; Gruart, A.; Barco, A. The Epigenetic Factor CBP Is Required for the Differentiation and Function of Medial Ganglionic Eminence-Derived Interneurons. Mol. Neurobiol. 2019, 56, 4440–4454. [Google Scholar] [CrossRef] [Green Version]
- Tsui, D.; Voronova, A.; Gallagher, D.; Kaplan, D.R.; Miller, F.D.; Wang, J. CBP regulates the differentiation of interneurons from ventral forebrain neural precursors during murine development. Dev. Biol. 2014, 385, 230–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Huang, S.; Zhu, L.; Yang, Q.; Yang, X.M.; Gu, J.R.; Zhang, Z.G.; Nie, H.Z.; Li, J. Alternative transcription start site selection in ACSS2 controls its nuclear localization and promotes ribosome biosynthesis in hepatocellular carcinoma. Biochem. Biophys Res. Commun. 2019, 514, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.H.; Van Dam, S.; Craig, T.; Tacutu, R.; O’Toole, A.; Merry, B.J.; De Magalhães, J.P. Transcriptome analysis in calorie-restricted rats implicates epigenetic and post-translational mechanisms in neuroprotection and aging. Genome. Biol. 2015, 16, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffett, J.R.; Puthillathu, N.; Vengilote, R.; Jaworski, D.M.; Namboodiri, A.M. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics, and Oncogenesis–Part 2: Acetate and ACSS2 in Health and Disease. Front. Physiol. 2020, 11, 1451. [Google Scholar]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Yang, L.; Dacks, P.; Isoda, F.; Poplawski, M.; Mobbs, C.V. Regulation of peripheral metabolism by substrate partitioning in the brain. Endocrinol. Metab. Clin. N. Am. 2013, 42, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, M.V.; Zhu, Y.; López, M.; Yin, L.; Wozniak, D.F.; Coleman, T.; Hu, Z.; Wolfgang, M.; Vidal-Puig, A.; Lane, M.D.; et al. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J. Clin. Investig. 2007, 117, 2539–2552. [Google Scholar] [CrossRef]
- Jia, Y.L.; Xu, M.; Dou, C.W.; Liu, Z.K.; Xue, Y.M.; Yao, B.W.; Ding, L.L.; Tu, K.S.; Zheng, X.; Liu, Q.G. P300/CBP-associated factor (PCAF) inhibits the growth of hepatocellular carcinoma by promoting cell autophagy. Cell Death Dis. 2016, 7, e2400. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Ying, B.; Zhang, J.; Ying, H. PCAF regulates H3 phosphorylation and promotes autophagy in osteosarcoma cells. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 118, 109395. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Xu, C.; Xia, H.; Chen, J.; Liu, H.; Jiang, H. Downregulation of P300/CBP-Associated Factor Attenuates Myocardial Ischemia-Reperfusion Injury Via Inhibiting Autophagy. Int. J. Med. Sci. 2020, 17, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, Y.; Park, S.Y.; Seo, Y.W.; Jung, S.C.; Kim, K.K.; Kim, K.; Kim, H. p300/CBP-associated factor promotes autophagic degradation of δ-catenin through acetylation and decreases prostate cancer tumorigenicity. Sci. Rep. 2019, 9, 3351. [Google Scholar] [CrossRef]
- Chen, R.; Xu, M.; Nagati, J.; Garcia, J.A. Coordinate regulation of stress signaling and epigenetic events by Acss2 and HIF-2 in cancer cells. PLoS ONE 2017, 12, e0190241. [Google Scholar] [CrossRef] [Green Version]
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [Green Version]
- Ramadori, G.; Coppari, R. Does hypothalamic SIRT1 regulate aging? Aging 2011, 3, 325–328. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Ben-Josef, G.; West, T.; Wozniak, D.F.; Holtzman, D.M.; Herzog, E.D.; Imai, S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J. Neurosci. 2010, 30, 10220–10232. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Imai, S. Hypothalamic Sirt1 in aging. Aging 2014, 6, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Mews, P.; Egervari, G.; Nativio, R.; Sidoli, S.; Donahue, G.; Lombroso, S.I.; Alexander, D.C.; Riesche, S.L.; Heller, E.A.; Nestler, E.J.; et al. Alcohol metabolism contributes to brain histone acetylation. Nature 2019, 574, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Long, P.M.; Moffett, J.R.; Namboodiri, A.M.; Viapiano, M.S.; Lawler, S.E.; Jaworski, D.M. N-acetylaspartate (NAA) and N-acetylaspartylglutamate (NAAG) promote growth and inhibit differentiation of glioma stem-like cells. J. Biol. Chem. 2013, 288, 26188–26200. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, Y.; Liu, J.; Zhang, J.; Xu, M.; You, Z.; Peng, C.; Gong, Z.; Liu, W. Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep. 2020, 21, e48335. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, S.; Xia, Y.; Lu, Y.; Xiao, S.; Cao, Y.; Zhuang, S.; Tan, X.; Fu, Q.; Xie, L.; et al. Loss of GCN5 leads to increased neuronal apoptosis by upregulating E2F1- and Egr-1-dependent BH3-only protein Bim. Cell Death Dis. 2017, 8, e2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutlu, B.; Puigserver, P. GCN5 acetyltransferase in cellular energetic and metabolic processes. Biochim. Biophys. Acta. Gene Regul. Mech. 2021, 1864, 194626. [Google Scholar] [CrossRef]
- Kelly, T.J.; Lerin, C.; Haas, W.; Gygi, S.P.; Puigserver, P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 2009, 284, 19945–19952. [Google Scholar] [CrossRef] [Green Version]
- Lerin, C.; Rodgers, J.T.; Kalume, D.E.; Kim, S.H.; Pandey, A.; Puigserver, P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006, 3, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Li, G.; Guo, H.; Liu, X. Forkhead transcription factor FOXO1 is involved in hypoxia/reoxygenation-induced gonadotropin-releasing hormone decline. Neuroreport 2020, 31, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Shi, R.; Guo, H. Tumor necrosis factor α reduces gonadotropin-releasing hormone release through increase of forkhead box protein O1 activity. Neuroreport 2020, 31, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Wang, Y.; Li, D.; Song, Z.; Jiao, H.; Lin, H. A central role for the mammalian target of rapamycin in LPS-induced anorexia in mice. J. Endocrinol. 2015, 224, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 2007, 317, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Enriori, P.J.; Evans, A.E.; Sinnayah, P.; Jobst, E.E.; Tonelli-Lemos, L.; Billes, S.K.; Glavas, M.M.; Grayson, B.E.; Perello, M.; Nillni, E.A.; et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007, 5, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Sadagurski, M.; Cady, G.; Miller, R.A. Anti-aging drugs reduce hypothalamic inflammation in a sex-specific manner. Aging Cell 2017, 16, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Yavari, A.; Stocker, C.J.; Ghaffari, S.; Wargent, E.T.; Steeples, V.; Czibik, G.; Pinter, K.; Bellahcene, M.; Woods, A.; Martínez de Morentin, P.B.; et al. Chronic Activation of γ2 AMPK Induces Obesity and Reduces β Cell Function. Cell Metab. 2016, 23, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Auwerx, J. Calorie restriction: Is AMPK a key sensor and effector? Physiology 2011, 26, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.L.; Alquier, T.; Asakura, K.; Furukawa, N.; Preitner, F.; Kahn, B.B. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J. Biol. Chem. 2006, 281, 18933–18941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCrimmon, R.J.; Fan, X.; Ding, Y.; Zhu, W.; Jacob, R.J.; Sherwin, R.S. Potential role for AMP-activated protein kinase in hypoglycemia sensing in the ventromedial hypothalamus. Diabetes 2004, 53, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Cheng, K.K.-Y. Hypothalamic AMPK as a Mediator of Hormonal Regulation of Energy Balance. Int. J. Mol. Sci. 2018, 19, 3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Dagon, Y.; Campbell, J.N.; Guo, Y.; Yang, Z.; Yi, X.; Aryal, P.; Wellenstein, K.; Kahn, B.B.; Sabatini, B.L.; et al. A Postsynaptic AMPK→p21-Activated Kinase Pathway Drives Fasting-Induced Synaptic Plasticity in AgRP Neurons. Neuron 2016, 91, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, U.; Filipsson, K.; Abbott, C.R.; Woods, A.; Smith, K.; Bloom, S.R.; Carling, D.; Small, C.J. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004, 279, 12005–12008. [Google Scholar] [CrossRef] [Green Version]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Y.; Sleiman, M.B.; Li, H.; Gao, A.W.; Mottis, A.; Bachmann, A.M.; Alam, G.E.; Li, X.; Goeminne, L.J.E.; Schoonjans, K.; et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 2021, 1, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Poplawski, M.; Yen, K.; Cheng, H.; Bloss, E.; Zhu, X.; Patel, H.; Mobbs, C.V. Role of CBP and SATB-1 in aging, dietary restriction, and insulin-like signaling. PLoS Biol. 2009, 7, e1000245. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. Biomed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme, A. Cell Metab. 2019, 29, 192–201.e197. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.-X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Jang, H.; Lee, S.M.; Lee, J.E.; Choi, J.; Kim, T.W.; Cho, E.J.; Youn, H.D. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J. 2015, 282, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, J.A.B.; Wasinski, F.; Donato, J., Jr. Prolonged fasting induces long-lasting metabolic consequences in mice. J. Nutr. Biochem. 2020, 84, 108457. [Google Scholar] [CrossRef]
- Funato, H.; Oda, S.; Yokofujita, J.; Igarashi, H.; Kuroda, M. Fasting and high-fat diet alter histone deacetylase expression in the medial hypothalamus. PLoS ONE 2011, 6, e18950. [Google Scholar] [CrossRef] [PubMed]
- Sternson, S.M.; Shepherd, G.M.; Friedman, J.M. Topographic mapping of VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 2005, 8, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- López, M.; Lage, R.; Saha, A.K.; Pérez-Tilve, D.; Vázquez, M.J.; Varela, L.; Sangiao-Alvarellos, S.; Tovar, S.; Raghay, K.; Rodríguez-Cuenca, S.; et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 2008, 7, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poplawski, M.M.; Mastaitis, J.W.; Yang, X.J.; Mobbs, C.V. Hypothalamic responses to fasting indicate metabolic reprogramming away from glycolysis toward lipid oxidation. Endocrinology 2010, 151, 5206–5217. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Currais, A.; Huang, L.; Petrascheck, M.; Maher, P.; Schubert, D. A chemical biology approach to identifying molecular pathways associated with aging. GeroScience 2020. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Prieto, G.A.; Petrosyan, A.; Loertscher, B.M.; Dieskau, A.P.; Overman, L.E.; Cotman, C.W. H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J. Neurosci. 2016, 36, 3611–3622. [Google Scholar] [CrossRef]
- Molina-Serrano, D.; Kyriakou, D.; Kirmizis, A. Histone Modifications as an Intersection Between Diet and Longevity. Front. Genet. 2019, 10, 192. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [Green Version]
- Creighton, S.D.; Stefanelli, G.; Reda, A.; Zovkic, I.B. Epigenetic Mechanisms of Learning and Memory: Implications for Aging. Int. J. Mol. Sci. 2020, 21, 6918. [Google Scholar] [CrossRef] [PubMed]
- Piña, B.; Martínez, P.; Suau, P. Differential acetylation of core histones in rat cerebral cortex neurons during development and aging. Eur. J. Biochem. 1988, 174, 311–315. [Google Scholar] [CrossRef]
- Lovatel, G.A.; Elsner, V.R.; Bertoldi, K.; Vanzella, C.; Moysés Fdos, S.; Vizuete, A.; Spindler, C.; Cechinel, L.R.; Netto, C.A.; Muotri, A.R.; et al. Treadmill exercise induces age-related changes in aversive memory, neuroinflammatory and epigenetic processes in the rat hippocampus. Neurobiol. Learn. Mem. 2013, 101, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.F.; Fletcher, B.R.; Kelley-Bell, B.; Kim, D.H.; Gallagher, M.; Rapp, P.R. Age-related memory impairment is associated with disrupted multivariate epigenetic coordination in the hippocampus. PLoS ONE 2012, 7, e33249. [Google Scholar] [CrossRef] [Green Version]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleff, S.; Andrulis, E.D.; Anderson, C.W.; Sternglanz, R. Identification of a gene encoding a yeast histone H4 acetyltransferase. J. Biol. Chem. 1995, 270, 24674–24677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, R.E.; Cook, R.G.; Perry, C.A.; Annunziato, A.T.; Allis, C.D. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc. Natl. Acad. Sci. USA 1995, 92, 1237–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, P.; Agudelo Garcia, P.A.; Iyer, C.C.; Popova, L.V.; Arnold, W.D.; Parthun, M.R. Early-onset aging and mitochondrial defects associated with loss of histone acetyltransferase 1 (Hat1). Aging Cell 2019, 18, e12992. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Xuan, H.; Green, C.D.; Han, Y.; Sun, N.; Shen, H.; McDermott, J.; Bennett, D.A.; Lan, F.; Han, J.-D.J. Repression of human and mouse brain inflammaging transcriptome by broad gene-body histone hyperacetylation. Proc. Natl. Acad. Sci. USA 2018, 115, 7611–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzus, E.; Rosenfeld, M.G.; Mayford, M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 2004, 42, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.N.; Pahan, K. HATs and HDACs in neurodegeneration: A tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006, 13, 539–550. [Google Scholar] [CrossRef]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.S.; Wang, W.Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Hu, E.; Du, H.; Shang, S.; Zhang, Y.; Lu, X. Beta-Hydroxybutyrate Enhances BDNF Expression by Increasing H3K4me3 and Decreasing H2AK119ub in Hippocampal Neurons. Front. Neurosci. 2020, 14, 591177. [Google Scholar] [CrossRef] [PubMed]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef]
- Sen, P.; Lan, Y.; Li, C.Y.; Sidoli, S.; Donahue, G.; Dou, Z.; Frederick, B.; Chen, Q.; Luense, L.J.; Garcia, B.A.; et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol. Cell 2019, 73, 684–698.e688. [Google Scholar] [CrossRef] [Green Version]
- Sławińska, N.; Krupa, R. Molecular Aspects of Senescence and Organismal Ageing-DNA Damage Response, Telomeres, Inflammation and Chromatin. Int. J. Mol. Sci. 2021, 22, 590. [Google Scholar] [CrossRef]
- Lipinski, M.; Del Blanco, B.; Barco, A. CBP/p300 in brain development and plasticity: Disentangling the KAT’s cradle. Curr. Opin. Neurobiol. 2019, 59, 1–8. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, H.S.; Park, S.A.; Ryu, S.H.; Meng, W.; Jürgensmeier, J.M.; Kurie, J.M.; Hong, W.K.; Boyer, J.L.; Herbst, R.S.; et al. A Novel Small-Molecule Inhibitor Targeting CREB-CBP Complex Possesses Anti-Cancer Effects along with Cell Cycle Regulation, Autophagy Suppression and Endoplasmic Reticulum Stress. PLoS ONE 2015, 10, e0122628. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Shi, Y.; Liu, J.; Su, H.; Huang, J.; Zhang, Y.; Peng, C.; Zhou, T.; Sun, Q.; Wan, W.; et al. Acetylation of STX17 (syntaxin 17) controls autophagosome maturation. Autophagy 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar]
- Caccamo, A.; Maldonado, M.A.; Bokov, A.F.; Majumder, S.; Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 22687–22692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Günther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.L.; Ehrlich, M.E.; Mobbs, C.V. Protection by dietary restriction in the YAC128 mouse model of Huntington’s disease: Relation to genes regulating histone acetylation and HTT. Neurobiol. Dis. 2016, 85, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouliaras, L.; van den Hove, D.L.; Kenis, G.; Draanen, M.; Hof, P.R.; van Os, J.; Steinbusch, H.W.; Schmitz, C.; Rutten, B.P. Histone deacetylase 2 in the mouse hippocampus: Attenuation of age-related increase by caloric restriction. Curr. Alzheimer Res. 2013, 10, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Zupkovitz, G.; Lagger, S.; Martin, D.; Steiner, M.; Hagelkruys, A.; Seiser, C.; Schöfer, C.; Pusch, O. Histone deacetylase 1 expression is inversely correlated with age in the short-lived fish Nothobranchius furzeri. Histochem. Cell Biol. 2018, 150, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Liu, A.; Li, J.; Wolubah, C.; Casaccia-Bonnefil, P. Epigenetic memory loss in aging oligodendrocytes in the corpus callosum. Neurobiol. Aging 2008, 29, 452–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, T.M.; Zürcher, N.R.; Catanese, M.C.; Tseng, C.J.; Di Biase, M.A.; Lyall, A.E.; Hightower, B.G.; Parmar, A.J.; Bhanot, A.; Wu, C.J.; et al. Neuroepigenetic signatures of age and sex in the living human brain. Nat. Commun. 2019, 10, 2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomás Pereira, I.; Coletta, C.E.; Perez, E.V.; Kim, D.H.; Gallagher, M.; Goldberg, I.G.; Rapp, P.R. CREB-binding protein levels in the rat hippocampus fail to predict chronological or cognitive aging. Neurobiol. Aging 2013, 34, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Ericsson, J.; Edwards, P.A. CBP is required for sterol-regulated and sterol regulatory element-binding protein-regulated transcription. J. Biol. Chem. 1998, 273, 17865–17870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas Fernández, M.A.; Concha Vilca, C.M.; Batista, L.O.; Ramos, V.W.; Cinelli, L.P.; Tibau de Albuquerque, K. Intermittent food restriction in female rats induces SREBP high expression in hypothalamus and immediately postfasting hyperphagia. Nutrition 2018, 48, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kakuma, T.; Fukuchi, S.; Masaki, T.; Sakata, T.; Yoshimatsu, H. Sterol regulatory element binding protein (SREBP)-1 expression in brain is affected by age but not by hormones or metabolic changes. Brain Res. 2006, 1081, 19–27. [Google Scholar] [CrossRef]
- Senyuk, V.; Sinha, K.K.; Nucifora, G. Corepressor CtBP1 interacts with and specifically inhibits CBP activity. Arch. Biochem. Biophys. 2005, 441, 168–173. [Google Scholar] [CrossRef]
- Moreno, C.L.; Mobbs, C.V. Epigenetic mechanisms underlying lifespan and age-related effects of dietary restriction and the ketogenic diet. Mol. Cell Endocrinol. 2017, 455, 33–40. [Google Scholar] [CrossRef]
- Li, Q.; Xiao, H.; Isobe, K. Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B93–B98. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.H.; Kim, E.J.; Shin, C.M.; Joo, K.M.; Kim, M.J.; Woo, H.W.; Cha, C.I. Age-related changes in CREB binding protein immunoreactivity in the cerebral cortex and hippocampus of rats. Brain Res. 2002, 956, 312–318. [Google Scholar] [CrossRef]
- Tanaka, Y.; Naruse, I.; Maekawa, T.; Masuya, H.; Shiroishi, T.; Ishii, S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: A partial similarity with Rubinstein-Taybi syndrome. Proc. Natl. Acad. Sci. USA 1997, 94, 10215–10220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudarzi, A.; Zhang, D.; Huang, H.; Barral, S.; Kwon, O.K.; Qi, S.; Tang, Z.; Buchou, T.; Vitte, A.L.; He, T.; et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol. Cell 2016, 62, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarska, Z.; Ortega, E.; Goudarzi, A.; Huang, H.; Kim, S.; Márquez, J.A.; Zhao, Y.; Khochbin, S.; Panne, D. Structure of p300 in complex with acyl-CoA variants. Nat. Chem. Biol. 2017, 13, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Zhang, D.; Weng, Y.; Delaney, K.; Tang, Z.; Yan, C.; Qi, S.; Peng, C.; Cole, P.A.; Roeder, R.G.; et al. The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv. 2021, 7, eabe2771. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, C.L.; Yang, L.; Dacks, P.A.; Isoda, F.; Deursen, J.M.; Mobbs, C.V. Role of Hypothalamic Creb-Binding Protein in Obesity and Molecular Reprogramming of Metabolic Substrates. PLoS ONE 2016, 11, e0166381. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.M.; Kleopoulos, S.P.; Bergen, H.T.; Roberts, J.L.; Priest, C.A.; Mobbs, C.V. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and in ob/ob and db/db mice, but is stimulated by leptin. Diabetes 1998, 47, 294–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daitoku, H.; Sakamaki, J.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Pak, Y.K.; Jang, P.G.; Namkoong, C.; Choi, Y.S.; Won, J.C.; Kim, K.S.; Kim, S.W.; Kim, H.S.; Park, J.Y.; et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 2006, 9, 901–906. [Google Scholar] [CrossRef]
- Ropelle, E.R.; Pauli, J.R.; Prada, P.; Cintra, D.E.; Rocha, G.Z.; Moraes, J.C.; Frederico, M.J.; da Luz, G.; Pinho, R.A.; Carvalheira, J.B.; et al. Inhibition of hypothalamic Foxo1 expression reduced food intake in diet-induced obesity rats. J. Physiol. 2009, 587, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Connaughton, S.; Chowdhury, F.; Attia, R.R.; Song, S.; Zhang, Y.; Elam, M.B.; Cook, G.A.; Park, E.A. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol. Cell Endocrinol. 2010, 315, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuyama, T.; Kitayama, K.; Yamashita, H.; Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003, 375, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Iskandar, K.; Cao, Y.; Hayashi, Y.; Nakata, M.; Takano, E.; Yada, T.; Zhang, C.; Ogawa, W.; Oki, M.; Chua, S., Jr.; et al. PDK-1/FoxO1 pathway in POMC neurons regulates Pomc expression and food intake. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E787–E798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Feng, Y.; Kitamura, Y.I.; Chua, S.C., Jr.; Xu, A.W.; Barsh, G.S.; Rossetti, L.; Accili, D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat. Med. 2006, 12, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Lee, K.-S.; Kwak, S.-J.; Kim, A.-K.; Bai, H.; Jung, M.-S.; Kwon, O.Y.; Song, W.-J.; Tatar, M.; Yu, K. Minibrain/Dyrk1a regulates food intake through the Sir2-FOXO-sNPF/NPY pathway in Drosophila and mammals. PLoS Genet. 2012, 8, e1002857. [Google Scholar] [CrossRef] [Green Version]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.; Zilberfarb, V.; Gangneux, N.; Christeff, N.; Issad, T. O-GlcNAc modification of FoxO1 increases its transcriptional activity: A role in the glucotoxicity phenomenon? Biochimie 2008, 90, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Dietrich, M.O.; Liu, Z.W.; Zimmer, M.R.; Li, M.D.; Singh, J.P.; Zhang, K.; Yin, R.; Wu, J.; Horvath, T.L.; et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 2014, 159, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.R.; Pissios, P.; Otu, H.; Roberson, R.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.F.; Kahn, B.B.; et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1724–E1739. [Google Scholar] [CrossRef]
- Srivastava, S.; Baxa, U.; Niu, G.; Chen, X.; Veech, R.L. A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life 2013, 65, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, S.; Fornari, A.; Pedrazzi, P.; Pellegrini, M.; Zoli, M. Developmental overfeeding alters hypothalamic neuropeptide mRNA levels and response to a high-fat diet in adult mice. Peptides 2011, 32, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Cha, S.H.; Chohnan, S.; Lane, M.D. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc. Natl. Acad. Sci. USA 2003, 100, 12624–12629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Dai, Y.; Prentki, M.; Chohnan, S.; Lane, M.D. A role for hypothalamic malonyl-CoA in the control of food intake. J. Biol. Chem. 2005, 280, 39681–39683. [Google Scholar] [CrossRef] [Green Version]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Kinzig, K.P.; Aja, S.; Scott, K.A.; Keung, W.; Kelly, S.; Strynadka, K.; Chohnan, S.; Smith, W.W.; Tamashiro, K.L.; et al. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc. Natl. Acad. Sci. USA 2007, 104, 17358–17363. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.T.; Wang, H.; Henderson, R.D.; Bowers, C.; Steyn, F.J. Ghrelin as a treatment for amyotrophic lateral sclerosis. J. Neuroendocrinol. 2021, e12938. [Google Scholar] [CrossRef]
- Reich, N.; Hölscher, C. Acylated Ghrelin as a Multi-Targeted Therapy for Alzheimer’s and Parkinson’s Disease. Front. Neurosci. 2020, 14, 614828. [Google Scholar] [CrossRef] [PubMed]
- Minor, R.K.; López, M.; Younts, C.M.; Jones, B.; Pearson, K.J.; Anson, R.M.; Diéguez, C.; de Cabo, R. The arcuate nucleus and neuropeptide Y contribute to the antitumorigenic effect of calorie restriction. Aging Cell 2011, 10, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Marques, M.; Aveleira, C.A.; Carmo-Silva, S.; Botelho, M.; Pereira de Almeida, L.; Cavadas, C. Caloric restriction stimulates autophagy in rat cortical neurons through neuropeptide Y and ghrelin receptors activation. Aging 2016, 8, 1470–1484. [Google Scholar] [CrossRef] [Green Version]
- Minor, R.K.; Chang, J.W.; de Cabo, R. Hungry for life: How the arcuate nucleus and neuropeptide Y may play a critical role in mediating the benefits of calorie restriction. Mol. Cell Endocrinol. 2009, 299, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, N.H.; Walsh, H.; Alvarez-Garcia, O.; Park, S.; Gaylinn, B.; Thorner, M.O.; Smith, R.G. Metabolic Benefit of Chronic Caloric Restriction and Activation of Hypothalamic AGRP/NPY Neurons in Male Mice Is Independent of Ghrelin. Endocrinology 2016, 157, 1430–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, N.; Sagarkar, S.; Jadhav, M.; Shahi, N.; Sirmaur, R.; Sakharkar, A.J. Role for histone deacetylation in traumatic brain injury-induced deficits in neuropeptide Y in arcuate nucleus: Possible implications in feeding behaviour. Neuroendocrinology 2020. [Google Scholar] [CrossRef]
- Kang, G.M.; Min, S.H.; Lee, C.H.; Kim, J.Y.; Lim, H.S.; Choi, M.J.; Jung, S.B.; Park, J.W.; Kim, S.; Park, C.B.; et al. Mitohormesis in Hypothalamic POMC Neurons Mediates Regular Exercise-Induced High-Turnover Metabolism. Cell Metab. 2021, 33, 334–349.e336. [Google Scholar] [CrossRef]
- Timper, K.; Paeger, L.; Sánchez-Lasheras, C.; Varela, L.; Jais, A.; Nolte, H.; Vogt, M.C.; Hausen, A.C.; Heilinger, C.; Evers, N.; et al. Mild Impairment of Mitochondrial OXPHOS Promotes Fatty Acid Utilization in POMC Neurons and Improves Glucose Homeostasis in Obesity. Cell Rep. 2018, 25, 383–397.e310. [Google Scholar] [CrossRef] [Green Version]
- Nakai, N.; Obayashi, M.; Nagasaki, M.; Sato, Y.; Fujitsuka, N.; Yoshimura, A.; Miyazaki, Y.; Sugiyama, S.; Shimomura, Y. The abundance of mRNAs for pyruvate dehydrogenase kinase isoenzymes in brain regions of young and aged rats. Life Sci. 2000, 68, 497–503. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bhusal, A.; Kim, J.H.; Jha, M.K.; Song, G.J.; Go, Y.; Jang, I.S.; Lee, I.K.; Suk, K. Astrocytic pyruvate dehydrogenase kinase-2 is involved in hypothalamic inflammation in mouse models of diabetes. Nat. Commun. 2020, 11, 5906. [Google Scholar] [CrossRef]
- Jha, M.K.; Jeon, S.; Suk, K. Pyruvate Dehydrogenase Kinases in the Nervous System: Their Principal Functions in Neuronal-glial Metabolic Interaction and Neuro-metabolic Disorders. Curr. Neuropharmacol. 2012, 10, 393–403. [Google Scholar] [PubMed]
- Kim, Y.J.; Tu, T.H.; Yang, S.; Kim, J.K.; Kim, J.G. Characterization of Fatty Acid Composition Underlying Hypothalamic Inflammation in Aged Mice. Molecules 2020, 25, 3170. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Xie, N.; Cui, H.; Moellering, D.R.; Abraham, E.; Thannickal, V.J.; Liu, G. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J. Immunol. 2015, 194, 6082–6089. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Jung, D.; Song, J.; Park, J.W.; Hong, J.J.; Seok, S.H. Pyruvate dehydrogenase kinase is a negative regulator of interleukin-10 production in macrophages. J. Mol. Cell Biol. 2020, 12, 543–555. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; McQuibban, G.A. The Mitochondrial Rhomboid Protease PARL Is Regulated by PDK2 to Integrate Mitochondrial Quality Control and Metabolism. Cell Rep. 2017, 18, 1458–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Cai, D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J. Biol. Chem. 2011, 286, 32324–32332. [Google Scholar] [CrossRef] [Green Version]
- Naresh, N.U.; Haynes, C.M. Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Hwang, A.B.; Lee, S.-J. Regulation of life span by mitochondrial respiration: The HIF-1 and ROS connection. Aging 2011, 3, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Senchuk, M.M.; Dues, D.J.; Johnson, B.K.; Cooper, J.F.; Lew, L.; Machiela, E.; Schaar, C.E.; DeJonge, H.; Blackwell, T.K.; et al. Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018, 16, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.O.Y.; Soro-Arnaiz, I.; Aragonés, J. Age-dependent obesity and mitochondrial dysfunction. Adipocyte 2017, 6, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. CMLS 2019, 76, 1759–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1α protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Xenaki, G.; Ontikatze, T.; Rajendran, R.; Stratford, I.J.; Dive, C.; Krstic-Demonacos, M.; Demonacos, C. PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene 2008, 27, 5785–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruas, J.L.; Poellinger, L.; Pereira, T. Role of CBP in regulating HIF-1-mediated activation of transcription. J. Cell Sci. 2005, 118, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, G.; Gonzalez, F.J.; Park, S.M.; Cai, D. Hypoxia-inducible factor directs POMC gene to mediate hypothalamic glucose sensing and energy balance regulation. PLoS Biol. 2011, 9, e1001112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Xu, M.; Nagati, J.S.; Hogg, R.T.; Das, A.; Gerard, R.D.; Garcia, J.A. The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment. PLoS ONE 2015, 10, e0116515. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Khor, S.; Cai, D. Age-dependent decline of hypothalamic HIF2α in response to insulin and its contribution to advanced age-associated metabolic disorders in mice. J. Biol. Chem. 2019, 294, 4946–4955. [Google Scholar] [CrossRef]
- Ashok, A.H.; Myers, J.; Frost, G.; Turton, S.; Gunn, R.N.; Passchier, J.; Colasanti, A.; Marques, T.R.; Nutt, D.; Lingford-Hughes, A.; et al. Acute acetate administration increases endogenous opioid levels in the human brain: A [(11)C]carfentanil molecular imaging study. J. Psychopharmacol. 2021. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [Green Version]
- Reisenauer, C.J.; Bhatt, D.P.; Mitteness, D.J.; Slanczka, E.R.; Gienger, H.M.; Watt, J.A.; Rosenberger, T.A. Acetate supplementation attenuates lipopolysaccharide-induced neuroinflammation. J. Neurochem. 2011, 117, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Soliman, M.L.; Rosenberger, T.A. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol. Cell Biochem. 2011, 352, 173–180. [Google Scholar] [CrossRef]
- Soliman, M.L.; Smith, M.D.; Houdek, H.M.; Rosenberger, T.A. Acetate supplementation modulates brain histone acetylation and decreases interleukin-1β expression in a rat model of neuroinflammation. J. Neuroinflamm. 2012, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Gronier, B.; Savignac, H.M.; Di Miceli, M.; Idriss, S.M.; Tzortzis, G.; Anthony, D.; Burnet, P.W.J. Increased cortical neuronal responses to NMDA and improved attentional set-shifting performance in rats following prebiotic (B-GOS(®)) ingestion. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2018, 28, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Choudhury, A.; Gusain, P.; Parvez, S.; Palit, G.; Shukla, S.; Ganguly, S. Oral acetate supplementation attenuates N-methyl D-aspartate receptor hypofunction-induced behavioral phenotypes accompanied by restoration of acetyl-histone homeostasis. Psychopharmacology 2016, 233, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Martín-Segura, A.; Baliyan, S.; Ahmed, T.; Balschun, D.; Venero, C.; Martin, M.G.; Dotti, C.G. Aging Triggers a Repressive Chromatin State at Bdnf Promoters in Hippocampal Neurons. Cell Rep. 2016, 16, 2889–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Chaves, E.P.; Narayanaswami, V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008, 3, 505–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, P.; Gurinovich, A.; Nygaard, M.; Sasaki, T.; Sweigart, B.; Bae, H.; Andersen, S.L.; Villa, F.; Atzmon, G.; Christensen, K.; et al. APOE Alleles and Extreme Human Longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar]
- Chevalier, A.C.; Rosenberger, T.A. Increasing acetyl-CoA metabolism attenuates injury and alters spinal cord lipid content in mice subjected to experimental autoimmune encephalomyelitis. J. Neurochem. 2017, 141, 721–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arun, P.; Ariyannur, P.S.; Moffett, J.R.; Xing, G.; Hamilton, K.; Grunberg, N.E.; Ives, J.A.; Namboodiri, A.M. Metabolic acetate therapy for the treatment of traumatic brain injury. J. Neurotrauma 2010, 27, 293–298. [Google Scholar] [CrossRef]
- Bhatt, D.P.; Houdek, H.M.; Watt, J.A.; Rosenberger, T.A. Acetate supplementation increases brain phosphocreatine and reduces AMP levels with no effect on mitochondrial biogenesis. Neurochem. Int. 2013, 62, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Hipkiss, A.R. Proteotoxicity and the contrasting effects of oxaloacetate and glycerol on Caenorhabditis elegans life span: A role for methylglyoxal? Rejuvenation Res. 2010, 13, 547–551. [Google Scholar] [CrossRef]
- Mathew, R.; Arun, P.; Madhavarao, C.N.; Moffett, J.R.; Namboodiri, M.A. Progress toward acetate supplementation therapy for Canavan disease: Glyceryl triacetate administration increases acetate, but not N-acetylaspartate, levels in brain. J. Pharmacol. Exp. Ther. 2005, 315, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Cyranka, M.; Clarke, K.; de Wet, H. A Ketone Ester Drink Lowers Human Ghrelin and Appetite. Obesity 2018, 26, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Redmann, S.M., Jr.; Argyropoulos, G. AgRP-deficiency could lead to increased lifespan. Biochem. Biophys. Res. Commun. 2006, 351, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Kovac, S.; Abramov, A.Y.; Walker, M.C. Energy depletion in seizures: Anaplerosis as a strategy for future therapies. Neuropharmacology 2013, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, Y.H.; Koh, J.Y. Protection by pyruvate against transient forebrain ischemia in rats. J. Neurosci. 2001, 21, Rc171. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.; Marin, P.; Israel, M.; Glowinski, J.; Premont, J. Pyruvate and lactate protect striatal neurons against N-methyl-D-aspartate-induced neurotoxicity. Eur. J. Neurosci. 1999, 11, 3215–3224. [Google Scholar] [CrossRef] [PubMed]
- Kristo, G.; Yoshimura, Y.; Niu, J.; Keith, B.J.; Mentzer, R.M., Jr.; Bunger, R.; Lasley, R.D. The intermediary metabolite pyruvate attenuates stunning and reduces infarct size in in vivo porcine myocardium. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H517–H524. [Google Scholar] [CrossRef]
- Slovin, P.N.; Huang, C.J.; Cade, J.R.; Wood, C.E.; Nasiroglu, O.; Privette, M.; Orbach, P.; Skimming, J.W. Sodium pyruvate is better than sodium chloride as a resuscitation solution in a rodent model of profound hemorrhagic shock. Resuscitation 2001, 50, 109–115. [Google Scholar] [CrossRef]
- Schillinger, W.; Hunlich, M.; Sossalla, S.; Hermann, H.P.; Hasenfuss, G. Intracoronary pyruvate in cardiogenic shock as an adjunctive therapy to catecholamines and intra-aortic balloon pump shows beneficial effects on hemodynamics. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2011, 100, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Hu, X.; Liu, C.; Wang, S.; Zhang, W.; Gao, H.; Stetler, R.A.; Gao, Y.; Chen, J. Ethyl pyruvate protects against hypoxic-ischemic brain injury via anti-cell death and anti-inflammatory mechanisms. Neurobiol. Dis. 2010, 37, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.A.; Mohanan, P.V. Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Toxicol. Lett. 2003, 143, 115–122. [Google Scholar] [CrossRef]
- Yue, W.; Liu, Y.X.; Zang, D.L.; Zhou, M.; Zhang, F.; Wang, L. Inhibitory effects of succinic acid on chemical kindling and amygdala electrical kindling in rats. Acta Pharmacol. Sin. 2002, 23, 847–850. [Google Scholar] [PubMed]
- Carvalho, A.S.; Torres, L.B.; Persike, D.S.; Fernandes, M.J.; Amado, D.; Naffah-Mazzacoratti Mda, G.; Cavalheiro, E.A.; da Silva, A.V. Neuroprotective effect of pyruvate and oxaloacetate during pilocarpine induced status epilepticus in rats. Neurochem. Int. 2011, 58, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.Y.; Zheng, G.T.; Zhang, S.S. [Effects of L-malate, an inhibitor of glutamate decarboxylase, on learning and memory in mice]. Yao Xue Xue Bao 1996, 31, 897–900. [Google Scholar]
- Wu, J.L.; Wu, Q.P.; Peng, Y.P.; Zhang, J.M. Effects of L-malate on mitochondrial oxidoreductases in liver of aged rats. Physiol. Res. 2011, 60, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hempenstall, S.; Page, M.M.; Wallen, K.R.; Selman, C. Dietary restriction increases skeletal muscle mitochondrial respiration but not mitochondrial content in C57BL/6 mice. Mech. Ageing Dev. 2012, 133, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Satpute, R.; Lomash, V.; Kaushal, M.; Bhattacharya, R. Neuroprotective effects of alpha-ketoglutarate and ethyl pyruvate against motor dysfunction and oxidative changes caused by repeated 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine exposure in mice. Hum. Exp. Toxicol. 2013, 32, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, M.J.; Yoon, W.; Kim, E.Y.; Kim, H.; Lee, Y.; Min, B.; Kang, K.S.; Son, J.H.; Park, H.T.; et al. Isocitrate protects DJ-1 null dopaminergic cells from oxidative stress through NADP+-dependent isocitrate dehydrogenase (IDH). PLoS Genet. 2017, 13, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.C.; Alhasawi, A.; Appanna, V.P.; Auger, C.; Appanna, V.D. Brain metabolism and Alzheimer’s disease: The prospect of a metabolite-based therapy. J. Nutr. Health Aging 2015, 19, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.; Morris, J.C.; Raichle, M.E. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360.e353. [Google Scholar] [CrossRef] [PubMed]
- Pelton, R.B.; Williams, R.J. Effect of Pantothenic Acid on the Longevity of Mice. Proc. Soc. Exp. Biol. Med. 1958, 99, 632–633. [Google Scholar] [CrossRef]
- Jung, S.; Kim, M.K.; Choi, B.Y. The long-term relationship between dietary pantothenic acid (vitamin B(5)) intake and C-reactive protein concentration in adults aged 40 years and older. Nutr. Metab. Cardiovasc. Dis. NMCD 2017, 27, 806–816. [Google Scholar] [CrossRef]
- Moĭseenok, A.G. Pantothenic acid in human nutrition and its importance in stimulating intestinal bifidoflora. Vopr. Pitan. 1982, 1, 9–17. [Google Scholar]
- Chaleckis, R.; Murakami, I.; Takada, J.; Kondoh, H.; Yanagida, M. Individual variability in human blood metabolites identifies age-related differences. Proc. Natl. Acad. Sci. USA 2016, 113, 4252–4259. [Google Scholar] [CrossRef] [Green Version]
- Son, N.; Hur, H.J.; Sung, M.J.; Kim, M.S.; Hwang, J.T.; Park, J.H.; Yang, H.J.; Kwon, D.Y.; Yoon, S.H.; Chung, H.Y.; et al. Liquid chromatography-mass spectrometry-based metabolomic analysis of livers from aged rats. J. Proteome. Res. 2012, 11, 2551–2558. [Google Scholar] [CrossRef]
- Ishiguro, K. Aging effect of blood pantothenic acid content in female. Tohoku J. Exp. Med. 1972, 107, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Czumaj, A.; Szrok-Jurga, S.; Hebanowska, A.; Turyn, J.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The Pathophysiological Role of CoA. Int. J. Mol. Sci. 2020, 21, 9057. [Google Scholar] [CrossRef]
- Feser, J.; Tyler, J. Chromatin structure as a mediator of aging. FEBS Lett. 2011, 585, 2041–2048. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.C.; Yan, L.Y.; Lei, Z.L.; Miao, Y.L.; Shi, L.H.; Yang, J.W.; Wang, Q.; Ouyang, Y.C.; Sun, Q.Y.; Chen, D.Y. Changes in histone acetylation during postovulatory aging of mouse oocyte. Biol. Reprod. 2007, 77, 666–670. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K.; Nakamura, A.; Ishigami, A.; Goto, S.; Takahashi, R. Age-related difference of site-specific histone modifications in rat liver. Biogerontology 2009, 10, 415–421. [Google Scholar] [CrossRef]
- Heit, C.; Dong, H.; Chen, Y.; Thompson, D.C.; Deitrich, R.A.; Vasiliou, V.K. The role of CYP2E1 in alcohol metabolism and sensitivity in the central nervous system. Subcell Biochem. 2013, 67, 235–247. [Google Scholar] [PubMed] [Green Version]
- Kronfol, M.M.; Jahr, F.M.; Dozmorov, M.G.; Phansalkar, P.S.; Xie, L.Y.; Aberg, K.A.; McRae, M.; Price, E.T.; Slattum, P.W.; Gerk, P.M.; et al. DNA methylation and histone acetylation changes to cytochrome P450 2E1 regulation in normal aging and impact on rates of drug metabolism in the liver. GeroScience 2020, 42, 819–832. [Google Scholar] [CrossRef]
- Kozurková, M.; Misúrová, E.; Kropácová, K. Effect of aging and gamma radiation on acetylation of rat liver histones. Mech. Ageing Dev. 1995, 78, 1–14. [Google Scholar] [CrossRef]
- Jiang, N.; Yan, X.; Zhou, W.; Zhang, Q.; Chen, H.; Zhang, Y.; Zhang, X. NMR-based metabonomic investigations into the metabolic profile of the senescence-accelerated mouse. J. Proteome Res. 2008, 7, 3678–3686. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Saavedra, D.; Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.-X. Epigenetic Regulation of Metabolism and Inflammation by Calorie Restriction. Adv. Nutr. 2019, 10, 520–536. [Google Scholar] [CrossRef]
- Tasselli, L.; Xi, Y.; Zheng, W.; Tennen, R.I.; Odrowaz, Z.; Simeoni, F.; Li, W.; Chua, K.F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef]
- Krishnan, V.; Chow, M.Z.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Füllgrabe, J.; Lynch-Day, M.A.; Heldring, N.; Li, W.; Struijk, R.B.; Ma, Q.; Hermanson, O.; Rosenfeld, M.G.; Klionsky, D.J.; Joseph, B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013, 500, 468–471. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Kim, H.; Rubinsztein, D.C.; Lee, J.E. Autophagy, Cellular Aging and Age-related Human Diseases. Exp. Neurobiol. 2019, 28, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Füllgrabe, J.; Heldring, N.; Hermanson, O.; Joseph, B. Cracking the survival code: Autophagy-related histone modifications. Autophagy 2014, 10, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Uno, M.; Honjoh, S.; Nishida, E. The MYST family histone acetyltransferase complex regulates stress resistance and longevity through transcriptional control of DAF-16/FOXO transcription factors. EMBO Rep. 2017, 18, 1716–1726. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzillotta, A.; Sarnico, I.; Ingrassia, R.; Boroni, F.; Branca, C.; Benarese, M.; Faraco, G.; Blasi, F.; Chiarugi, A.; Spano, P.; et al. The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death Dis. 2010, 1, e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Pronk, J.T.; Yde Steensma, H.; Van Dijken, J.P. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 1996, 12, 1607–1633. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y.; Cole, P.A.; Marmorstein, R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: Implications for histone acetyltransferase evolution and function. Curr. Opin. Struct. Biol. 2008, 18, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Villeponteau, B.; Jazwinski, S.M. Effect of replicative age on transcriptional silencing near telomeres in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1996, 219, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.H.; Su, T.; Xue, Y. Histone H3 N-terminal acetylation sites especially K14 are important for rDNA silencing and aging. Sci. Rep. 2016, 6, 21900. [Google Scholar] [CrossRef] [Green Version]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, S.; Gong, X.; Yu, Q.; Zhang, Y.; Luo, M.; Zhang, X.; Workman, J.L.; Yu, X.; Li, S. Glycolysis regulates gene expression by promoting the crosstalk between H3K4 trimethylation and H3K14 acetylation in Saccharomyces cerevisiae. J. Genet. Genom. Yi Chuan Xue Bao 2019, 46, 561–574. [Google Scholar] [CrossRef]
- Friis, R.M.; Wu, B.P.; Reinke, S.N.; Hockman, D.J.; Sykes, B.D.; Schultz, M.C. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic. Acids Res. 2009, 37, 3969–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Rasmussen, L.J. TIP60 in aging and neurodegeneration. Ageing Res. Rev. 2020, 64, 101195. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Laschober, G.T.; Ruli, D.; Hofer, E.; Muck, C.; Carmona-Gutierrez, D.; Ring, J.; Hutter, E.; Ruckenstuhl, C.; Micutkova, L.; Brunauer, R.; et al. Identification of evolutionarily conserved genetic regulators of cellular aging. Aging Cell 2010, 9, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matecic, M.; Smith, D.L.; Pan, X.; Maqani, N.; Bekiranov, S.; Boeke, J.D.; Smith, J.S. A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 2010, 6, e1000921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orozco, H.; Matallana, E.; Aranda, A. Wine yeast sirtuins and Gcn5p control aging and metabolism in a natural growth medium. Mech. Ageing Dev. 2012, 133, 348–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picazo, C.; Orozco, H.; Matallana, E.; Aranda, A. Interplay among Gcn5, Sch9 and mitochondria during chronological aging of wine yeast is dependent on growth conditions. PLoS ONE 2015, 10, e0117267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Zhong, D.; Zhu, J.; An, Y.; Gao, M.; Zhu, S.; Dang, W.; Wang, X.; Yang, B.; Xie, Z. Inhibition of histone acetyltransferase GCN5 extends lifespan in both yeast and human cell lines. Aging Cell 2020, 19, e13129. [Google Scholar] [CrossRef] [Green Version]
- Fillingham, J.; Recht, J.; Silva, A.C.; Suter, B.; Emili, A.; Stagljar, I.; Krogan, N.J.; Allis, C.D.; Keogh, M.C.; Greenblatt, J.F. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol. Cell Biol. 2008, 28, 4342–4353. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.C.; Stumpferl, S.W.; Tiwari, A.; Qin, Q.; Rodriguez-Quiñones, J.F.; Jazwinski, S.M. Identification of the Target of the Retrograde Response that Mediates Replicative Lifespan Extension in Saccharomyces cerevisiae. Genetics 2016, 204, 659–673. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Ohkuni, K.; Couplan, E.; Jazwinski, S.M. The histone acetyltransferase GCN5 modulates the retrograde response and genome stability determining yeast longevity. Biogerontology 2004, 5, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M.; Jiang, J.C.; Kim, S. Adaptation to metabolic dysfunction during aging: Making the best of a bad situation. Exp. Gerontol. 2018, 107, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.N.; Uppuluri, P.; Broggi, A.; Besold, A.; Ryman, K.; Kambara, H.; Solis, N.; Lorenz, V.; Qi, W.; Acosta-Zaldívar, M.; et al. Intersection of phosphate transport, oxidative stress and TOR signalling in Candida albicans virulence. PLoS Pathog. 2018, 14, e1007076. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Cottignie, I.; Van Zeebroeck, G.; Thevelein, J. Nutrient transceptors physically interact with the yeast S6/Protein kinase B homolog, Sch9, a TOR kinase target. Biochem. J. 2021. [Google Scholar] [CrossRef]
- Popova, Y.; Thayumanavan, P.; Lonati, E.; Agrochão, M.; Thevelein, J.M. Transport and signaling through the phosphate-binding site of the yeast Pho84 phosphate transceptor. Proc. Natl. Acad. Sci. USA 2010, 107, 2890–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevelein, J.M.; Geladé, R.; Holsbeeks, I.; Lagatie, O.; Popova, Y.; Rolland, F.; Stolz, F.; Van de Velde, S.; Van Dijck, P.; Vandormael, P.; et al. Nutrient sensing systems for rapid activation of the protein kinase A pathway in yeast. Biochem. Soc. Trans. 2005, 33, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.J.; Liu, X.G.; McCormick, M.; Wasko, B.M.; Zhao, W.; He, X.; Yuan, Y.; Fang, B.X.; Sun, X.R.; Kennedy, B.K.; et al. PMT1 deficiency enhances basal UPR activity and extends replicative lifespan of Saccharomyces cerevisiae. Age 2015, 37, 9788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofiteru, A.M.; Ruta, L.L.; Rotaru, C.; Dumitru, I.; Ene, C.D.; Neagoe, A.; Farcasanu, I.C. Overexpression of the PHO84 gene causes heavy metal accumulation and induces Ire1p-dependent unfolded protein response in Saccharomyces cerevisiae cells. Appl. Microbiol. Biotechnol. 2012, 94, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.W.; Bradshaw, P.C.; Pfeiffer, D.R. Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J. Biol. Chem. 1997, 272, 21104–21112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.J.; Kaeberlein, M.; Andalis, A.A.; Sturtz, L.A.; Defossez, P.A.; Culotta, V.C.; Fink, G.R.; Guarente, L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 2002, 418, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J.D. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wierman, M.B.; Maqani, N.; Strickler, E.; Li, M.; Smith, J.S. Caloric Restriction Extends Yeast Chronological Life Span by Optimizing the Snf1 (AMPK) Signaling Pathway. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Workman, J.J.; Chen, H.; Laribee, R.N. Saccharomyces cerevisiae TORC1 Controls Histone Acetylation by Signaling Through the Sit4/PP6 Phosphatase to Regulate Sirtuin Deacetylase Nuclear Accumulation. Genetics 2016, 203, 1733–1746. [Google Scholar] [CrossRef]
- Robyr, D.; Suka, Y.; Xenarios, I.; Kurdistani, S.K.; Wang, A.; Suka, N.; Grunstein, M. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 2002, 109, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Deprez, M.A.; Eskes, E.; Winderickx, J.; Wilms, T. The TORC1-Sch9 pathway as a crucial mediator of chronological lifespan in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy048. [Google Scholar] [CrossRef] [PubMed]
- Molina-Serrano, D.; Schiza, V.; Demosthenous, C.; Stavrou, E.; Oppelt, J.; Kyriakou, D.; Liu, W.; Zisser, G.; Bergler, H.; Dang, W.; et al. Loss of Nat4 and its associated histone H4 N-terminal acetylation mediates calorie restriction-induced longevity. EMBO Rep. 2016, 17, 1829–1843. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.C.; Wawryn, J.; Shantha Kumara, H.M.; Jazwinski, S.M. Distinct roles of processes modulated by histone deacetylases Rpd3p, Hda1p, and Sir2p in life extension by caloric restriction in yeast. Exp. Gerontol. 2002, 37, 1023–1030. [Google Scholar] [CrossRef]
- Kim, S.; Benguria, A.; Lai, C.Y.; Jazwinski, S.M. Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol. Biol. Cell 1999, 10, 3125–3136. [Google Scholar] [CrossRef] [Green Version]
- Thurtle-Schmidt, D.M.; Dodson, A.E.; Rine, J. Histone Deacetylases with Antagonistic Roles in Saccharomyces cerevisiae Heterochromatin Formation. Genetics 2016, 204, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Suka, N.; Suka, Y.; Carmen, A.A.; Wu, J.; Grunstein, M. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol. Cell 2001, 8, 473–479. [Google Scholar] [CrossRef]
- Kurdistani, S.K.; Robyr, D.; Tavazoie, S.; Grunstein, M. Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat. Genet. 2002, 31, 248–254. [Google Scholar] [CrossRef]
- Vaquero, A.; Sternglanz, R.; Reinberg, D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007, 26, 5505–5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadosh, D.; Struhl, K. Targeted recruitment of the Sin3-Rpd3 histone deacetylase complex generates a highly localized domain of repressed chromatin in vivo. Mol. Cell Biol. 1998, 18, 5121–5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rundlett, S.E.; Carmen, A.A.; Suka, N.; Turner, B.M.; Grunstein, M. Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4 by RPD3. Nature 1998, 392, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Prasad, H.; Rao, R. Histone deacetylase-mediated regulation of endolysosomal pH. J. Biol. Chem. 2018, 293, 6721–6735. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Tong, J.K.; Schreiber, S.L. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 13708–13713. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L.; Gottschling, D.E. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 2012, 492, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.E.; Coody, T.K.; Jeong, M.Y.; Berg, J.A.; Winge, D.R.; Hughes, A.L. Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 2020, 180, 296–310.e218. [Google Scholar] [CrossRef]
- Stephan, J.; Franke, J.; Ehrenhofer-Murray, A.E. Chemical genetic screen in fission yeast reveals roles for vacuolar acidification, mitochondrial fission, and cellular GMP levels in lifespan extension. Aging Cell 2013, 12, 574–583. [Google Scholar] [CrossRef]
- Peng, W.; Togawa, C.; Zhang, K.; Kurdistani, S.K. Regulators of cellular levels of histone acetylation in Saccharomyces cerevisiae. Genetics 2008, 179, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Tanner, K.G.; Trievel, R.C.; Kuo, M.H.; Howard, R.M.; Berger, S.L.; Allis, C.D.; Marmorstein, R.; Denu, J.M. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 1999, 274, 18157–18160. [Google Scholar] [CrossRef] [Green Version]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone acetylation regulates intracellular pH. Mol. Cell 2013, 49, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, C.; Wang, Y.; Renaud, J.; Tian, G.; Kambhampati, S.; Saatian, B.; Nguyen, V.; Hannoufa, A.; Marsolais, F.; et al. Cytosolic acetyl-CoA promotes histone acetylation predominantly at H3K27 in Arabidopsis. Nat. Plants 2017, 3, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Han, S.K.; Song, J.D.; Noh, Y.S.; Noh, B. Role of plant CBP/p300-like genes in the regulation of flowering time. Plant J. Cell Mol. Biol. 2007, 49, 103–114. [Google Scholar] [CrossRef]
- Thompson, P.R.; Kurooka, H.; Nakatani, Y.; Cole, P.A. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J. Biol. Chem. 2001, 276, 33721–33729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatland, B.L.; Ke, J.; Anderson, M.D.; Mentzen, W.I.; Cui, L.W.; Allred, C.C.; Johnston, J.L.; Nikolau, B.J.; Wurtele, E.S. Molecular characterization of a heteromeric ATP-citrate lyase that generates cytosolic acetyl-coenzyme A in Arabidopsis. Plant Physiol. 2002, 130, 740–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, T.S. The use of Drosophila melanogaster as a screening agent for longevity factors; the effects of biotin, pyridoxine, sodium yeast nucleate, and pantothenic acid on the life span of the fruit fly. J. Gerontol. 1948, 3, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Rogina, B.; Helfand, S.L.; Frankel, S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science 2002, 298, 1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Sun, H.; Lu, J.; Li, X.; Chen, X.; Tao, D.; Huang, W.; Huang, B. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J. Exp. Biol. 2005, 208, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munks, R.J.; Moore, J.; O’Neill, L.P.; Turner, B.M. Histone H4 acetylation in Drosophila. Frequency of acetylation at different sites defined by immunolabelling with site-specific antibodies. FEBS Lett. 1991, 284, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Awe, S.; Renkawitz-Pohl, R. Histone H4 acetylation is essential to proceed from a histone- to a protamine-based chromatin structure in spermatid nuclei of Drosophila melanogaster. Syst. Biol. Reprod. Med. 2010, 56, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Vidali, G.; Boffa, L.C.; Bradbury, E.M.; Allfrey, V.G. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc. Natl. Acad. Sci. USA 1978, 75, 2239–2243. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [CrossRef]
- Yang, J.; Kaur, K.; Edwards, J.G.; Eisenberg, C.A.; Eisenberg, L.M. Inhibition of Histone Methyltransferase, Histone Deacetylase, and <i>β<i>-Catenin Synergistically Enhance the Cardiac Potential of Bone Marrow Cells. Stem. Cells Int. 2017, 2017, 3464953. [Google Scholar] [CrossRef] [Green Version]
- Fischer, N.; Sechet, E.; Friedman, R.; Amiot, A.; Sobhani, I.; Nigro, G.; Sansonetti, P.J.; Sperandio, B. Histone deacetylase inhibition enhances antimicrobial peptide but not inflammatory cytokine expression upon bacterial challenge. Proc. Natl. Acad. Sci. USA 2016, 113, E2993–E3001. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Umehara, T.; Sasaki, K.; Nakamura, Y.; Nishino, N.; Terada, T.; Shirouzu, M.; Padmanabhan, B.; Yokoyama, S.; Ito, A.; et al. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem. Biol. 2011, 18, 495–507. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.R.; Kennedy, C.J.; Delmar, V.A.; Forbes, D.J.; Silver, P.A. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev. 2008, 22, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Liu, X.; Zhu, S.; Hu, X.; Niu, H.; Zhang, X.; Zhu, D.; Nesa, E.U.; Tian, K.; Yuan, H. Hyper-acetylation contributes to the sensitivity of chemo-resistant prostate cancer cells to histone deacetylase inhibitor Trichostatin, A. J. Cell Mol. Med. 2018, 22, 1909–1922. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, O.A.; Krejčí, J.; Suchánková, J.; Bártová, E. Deacetylation of Histone H4 Accompanying Cardiomyogenesis is Weakened in HDAC1-Depleted ES Cells. Int. J. Mol. Sci. 2018, 19, 2425. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, Y.; Araki, A.; Maruta, H.; Takahashi, Y.; Yamashita, H. Molecular cloning of rat acss3 and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J. Biochem. 2017, 161, 279–289. [Google Scholar]
- Peleg, S.; Feller, C.; Forne, I.; Schiller, E.; Sevin, D.C.; Schauer, T.; Regnard, C.; Straub, T.; Prestel, M.; Klima, C.; et al. Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep. 2016, 17, 455–469. [Google Scholar] [CrossRef]
- Becker, L.; Nogueira, M.S.; Klima, C.; de Angelis, M.H.; Peleg, S. Rapid and transient oxygen consumption increase following acute HDAC/KDAC inhibition in Drosophila tissue. Sci. Rep. 2018, 8, 4199. [Google Scholar] [CrossRef]
- Cutler, T.; Sarkar, A.; Moran, M.; Steffensmeier, A.; Puli, O.R.; Mancini, G.; Tare, M.; Gogia, N.; Singh, A. Drosophila Eye Model to Study Neuroprotective Role of CREB Binding Protein (CBP) in Alzheimer’s Disease. PLoS ONE 2015, 10, e0137691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.G.; Kwon, M.J.; Jeon, K.H.; Hyeon, D.Y.; Han, M.H.; Park, J.H.; Cha, I.J.; Cho, J.H.; Kim, K.; Rho, S.; et al. Golgi Outpost Synthesis Impaired by Toxic Polyglutamine Proteins Contributes to Dendritic Pathology in Neurons. Cell Rep. 2017, 20, 356–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Taye, A.A.; Campbell, C.; Kazemi-Esfarjani, P.; Fischbeck, K.H.; Min, K.T. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003, 17, 1463–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, Y.; Masuda, T.; Naganos, S.; Matsuno, M.; Ueno, K.; Miyashita, T.; Horiuchi, J.; Saitoe, M. Fasting launches CRTC to facilitate long-term memory formation in Drosophila. Science 2013, 339, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.M.; Lian, G.; Liu, Q.; Ruan, K.; et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012, 336, 477–481. [Google Scholar] [CrossRef]
- Cai, H.; Dhondt, I.; Vandemeulebroucke, L.; Vlaeminck, C.; Rasulova, M.; Braeckman, B.P. CBP-1 Acts in GABAergic Neurons to Double Life Span in Axenically Cultured Caenorhabditis elegans. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef] [Green Version]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Wu, X.; Zhou, J.; Li, X.; Huang, X.; Li, J.; Wu, J.; Bian, Q.; Wang, Y.; Tian, Y. NuRD mediates mitochondrial stress-induced longevity via chromatin remodeling in response to acetyl-CoA level. Sci. Adv. 2020, 6, eabb2529. [Google Scholar] [CrossRef] [PubMed]
- Houthoofd, K.; Braeckman, B.P.; Lenaerts, I.; Brys, K.; De Vreese, A.; Van Eygen, S.; Vanfleteren, J.R. No reduction of metabolic rate in food restricted Caenorhabditis elegans. Exp. Gerontol. 2002, 37, 1359–1369. [Google Scholar] [CrossRef]
- Henry, R.A.; Kuo, Y.-M.; Andrews, A.J. Differences in Specificity and Selectivity Between CBP and p300 Acetylation of Histone H3 and H3/H4. Biochemistry 2013, 52, 5746–5759. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Lucia, M.S.; Hansen, K.C.; Tyler, J.K. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009, 459, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Schroeder, S.; Büttner, S.; Carmona-Gutierrez, D.; Pendl, T.; Andryushkova, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. A histone point mutation that switches on autophagy. Autophagy 2014, 10, 1143–1145. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Ookuma, S.; Nishida, E. Lifespan extension by suppression of autophagy genes in Caenorhabditis elegans. Genes Cells 2009, 14, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Byrne, J.; Medina, R.; Kolundzic, E.; Geisinger, J.; Hajduskova, M.; Tursun, B.; Richly, H. Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev. 2017, 31, 1561–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, W.-C.; Tishkoff, D.X.; Yang, B.; Wilson-Grady, J.; Yu, X.; Mazer, T.; Eckersdorff, M.; Gygi, S.P.; Lombard, D.B.; Hsu, A.-L.C. elegans SIRT6/7 Homolog SIR-2.4 Promotes DAF-16 Relocalization and Function during Stress. PLoS Genet. 2012, 8, e1002948. [Google Scholar] [CrossRef] [PubMed]
- Tezil, T.; Chamoli, M.; Ng, C.-P.; Simon, R.P.; Butler, V.J.; Jung, M.; Andersen, J.; Kao, A.W.; Verdin, E. Lifespan-increasing drug nordihydroguaiaretic acid inhibits p300 and activates autophagy. NPJ Aging Mech. Dis. 2019, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Allison, D.B.; Ames, B.N.; Astle, C.M.; Atamna, H.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nadon, N.L.; et al. Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 2014, 13, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Strong, R.; Miller, R.A.; Antebi, A.; Astle, C.M.; Bogue, M.; Denzel, M.S.; Fernandez, E.; Flurkey, K.; Hamilton, K.L.; Lamming, D.W.; et al. Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 2016, 15, 872–884. [Google Scholar] [CrossRef] [Green Version]
- Spindler, S.R.; Mote, P.L.; Lublin, A.L.; Flegal, J.M.; Dhahbi, J.M.; Li, R. Nordihydroguaiaretic Acid Extends the Lifespan of Drosophila and Mice, Increases Mortality-Related Tumors and Hemorrhagic Diathesis, and Alters Energy Homeostasis in Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1479–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.S.; Pillus, L. Collaboration between the essential Esa1 acetyltransferase and the Rpd3 deacetylase is mediated by H4K12 histone acetylation in Saccharomyces cerevisiae. Genetics 2009, 183, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.B.; Leaver, D.J.; Hermans, S.J.; Kelly, G.L.; Brennan, M.S.; Downer, N.L.; Nguyen, N.; Wichmann, J.; McRae, H.M.; Yang, Y.; et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 2018, 560, 253–257. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, Y.; Sun, S.; Li, W.; Song, M.; Ji, Q.; Wu, Z.; Liu, Z.; Fan, Y.; Liu, F.; et al. A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Sci. Transl. Med. 2021, 13, eabd2655. [Google Scholar] [CrossRef]
- Hachinohe, M.; Yamane, M.; Akazawa, D.; Ohsawa, K.; Ohno, M.; Terashita, Y.; Masumoto, H. A reduction in age-enhanced gluconeogenesis extends lifespan. PLoS ONE 2013, 8, e54011. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, R.L.; Daniels, E.G.; Molenaars, M.; Houtkooper, R.H.; Janssens, G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 2019, 11, e9854. [Google Scholar] [CrossRef]
- Pasyukova, E.G.; Vaiserman, A.M. HDAC inhibitors: A new promising drug class in anti-aging research. Mech. Ageing Dev. 2017, 166, 6–15. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Dang, N.; Kerr, E.O.; Hu, D.; Steffen, K.K.; Oakes, J.A.; Kennedy, B.K.; Kaeberlein, M. Sirtuin-independent effects of nicotinamide on lifespan extension from calorie restriction in yeast. Aging Cell 2006, 5, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L., Jr.; McClure, J.M.; Matecic, M.; Smith, J.S. Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins. Aging Cell 2007, 6, 649–662. [Google Scholar] [CrossRef]
- Viswanathan, M.; Guarente, L. Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature 2011, 477, E1–E2. [Google Scholar] [CrossRef]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker, R.; Faulkner, S.; Miyokawa, R.; Burhenn, L.; Henriksen, M.; Wood, J.G.; Helfand, S.L. Increased expression of Drosophila Sir2 extends life span in a dose-dependent manner. Aging 2013, 5, 682–691. [Google Scholar] [CrossRef] [Green Version]
- Kusama, S.; Ueda, R.; Suda, T.; Nishihara, S.; Matsuura, E.T. Involvement of Drosophila Sir2-like genes in the regulation of life span. Genes Genet. Syst. 2006, 81, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.M.; McReynolds, M.R.; Hanna-Rose, W. Mitochondrial sirtuins sir-2.2 and sir-2.3 regulate lifespan in C. elegans. bioRxiv 2017, 181727. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Nirala, N.K.; Singh, A.; Zhu, L.J.; Taguchi, K.; Bamba, T.; Fukusaki, E.; Shaw, L.M.; Lambright, D.G.; Acharya, J.K.; et al. Drosophila Sirt2/mammalian SIRT3 deacetylates ATP synthase β and regulates complex V activity. J. Cell Biol. 2014, 206, 289–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.G.; Schwer, B.; Wickremesinghe, P.C.; Hartnett, D.A.; Burhenn, L.; Garcia, M.; Li, M.; Verdin, E.; Helfand, S.L. Sirt4 is a mitochondrial regulator of metabolism and lifespan in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2018, 115, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, L.; Molin, L.; Kasturi, P.; Triba, M.N.; Dumas, M.E.; Wilson, M.C.; Halestrap, A.P.; Roussel, D.; Masse, I.; Dallière, N.; et al. Pyruvate imbalance mediates metabolic reprogramming and mimics lifespan extension by dietary restriction in Caenorhabditis elegans. Aging Cell 2011, 10, 39–54. [Google Scholar] [CrossRef]
- Schaffer, S.; Gruber, J.; Ng, L.F.; Fong, S.; Wong, Y.T.; Tang, S.Y.; Halliwell, B. The effect of dichloroacetate on health- and lifespan in C. elegans. Biogerontology 2011, 12, 195–209. [Google Scholar] [CrossRef]
- Narasimhan, S.D.; Yen, K.; Bansal, A.; Kwon, E.S.; Padmanabhan, S.; Tissenbaum, H.A. PDP-1 links the TGF-β and IIS pathways to regulate longevity, development, and metabolism. PLoS Genet. 2011, 7, e1001377. [Google Scholar] [CrossRef] [Green Version]
- Chuang, M.-H.; Chiou, S.-H.; Huang, C.-H.; Yang, W.-B.; Wong, C.-H. The lifespan-promoting effect of acetic acid and Reishi polysaccharide. Bioorg. Med. Chem. 2009, 17, 7831–7840. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, W.; Ma, J.; Fu, X.; Zhao, Z.J. Beneficial and harmful effects of alcohol exposure on Caenorhabditis elegans worms. Biochem. Biophys. Res. Commun. 2011, 412, 757–762. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Zhang, L.; Wang, W.; Wei, S.; Wang, J.; Che, H.; Zhang, Y. Effects of excess sugars and lipids on the growth and development of Caenorhabditis elegans. Genes Nutr. 2020, 15, 1. [Google Scholar] [CrossRef]
- Alcántar-Fernández, J.; González-Maciel, A.; Reynoso-Robles, R.; Pérez Andrade, M.E.; Hernández-Vázquez, A.J.; Velázquez-Arellano, A.; Miranda-Ríos, J. High-glucose diets induce mitochondrial dysfunction in Caenorhabditis elegans. PLoS ONE 2019, 14, e0226652. [Google Scholar] [CrossRef]
- Tauffenberger, A.; Vaccaro, A.; Parker, J.A. Fragile lifespan expansion by dietary mitohormesis in C. elegans. Aging 2016, 8, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, W.; Beaudoin-Chabot, C.; Thibault, G. Glucose increases the lifespan of post-reproductive C. elegans independently of FOXO. bioRxiv 2018, 347435. [Google Scholar] [CrossRef]
- Edwards, C.; Canfield, J.; Copes, N.; Brito, A.; Rehan, M.; Lipps, D.; Brunquell, J.; Westerheide, S.D.; Bradshaw, P.C. Mechanisms of amino acid-mediated lifespan extension in Caenorhabditis elegans. BMC Genet. 2015, 16, 8. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.B.; Copes, N.; Brito, A.G.; Canfield, J.; Bradshaw, P.C. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS ONE 2013, 8, e58345. [Google Scholar] [CrossRef] [Green Version]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Kennedy, S.; Tolonen, A.C.; Ruvkun, G. DAF-16 target genes that control C. elegans life-span and metabolism. Science 2003, 300, 644–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelson, A.V.; Carr, C.E.; Ruvkun, G. Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes Dev. 2007, 21, 2976–2994. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cook, L.F.; Grasso, L.M.; Cao, M.; Dong, Y. Royal Jelly-Mediated Prolongevity and Stress Resistance in Caenorhabditis elegans Is Possibly Modulated by the Interplays of DAF-16, SIR-2.1, HCF-1, and 14-3-3 Proteins. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Vander Wende, H.; Simko, M.; Klum, S.; Barfield, S.; Choi, H.; Pineda, V.V.; Kaeberlein, M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3483. [Google Scholar] [CrossRef]
- Wang, J.; Robida-Stubbs, S.; Tullet, J.M.; Rual, J.F.; Vidal, M.; Blackwell, T.K. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 2010, 6, e1001048. [Google Scholar] [CrossRef]
- Oh, S.W.; Mukhopadhyay, A.; Dixit, B.L.; Raha, T.; Green, M.R.; Tissenbaum, H.A. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat. Genet. 2006, 38, 251–257. [Google Scholar]
- Segref, A.; Kevei, É.; Pokrzywa, W.; Schmeisser, K.; Mansfeld, J.; Livnat-Levanon, N.; Ensenauer, R.; Glickman, M.H.; Ristow, M.; Hoppe, T. Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab. 2014, 19, 642–652. [Google Scholar] [CrossRef] [Green Version]
- McGhee, J.D.; Sleumer, M.C.; Bilenky, M.; Wong, K.; McKay, S.J.; Goszczynski, B.; Tian, H.; Krich, N.D.; Khattra, J.; Holt, R.A.; et al. The ELT-2 GATA-factor and the global regulation of transcription in the C. elegans intestine. Dev. Biol. 2007, 302, 627–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depuydt, G.; Xie, F.; Petyuk, V.A.; Smolders, A.; Brewer, H.M.; Camp, D.G., 2nd; Smith, R.D.; Braeckman, B.P. LC-MS Proteomics Analysis of the Insulin/IGF-1-Deficient Caenorhabditis elegans daf-2(e1370) Mutant Reveals Extensive Restructuring of Intermediary Metabolism. J. Proteome. Res. 2014, 13, 1938–1956. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.W.; Smith, R.L.; van Weeghel, M.; Kamble, R.; Janssens, G.E.; Houtkooper, R.H. Identification of key pathways and metabolic fingerprints of longevity in C. elegans. Exp. Gerontol. 2018, 113, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Ruzanov, P.; Riddle, D.L.; Marra, M.A.; McKay, S.J.; Jones, S.M. Genes that may modulate longevity in C. elegans in both dauer larvae and long-lived daf-2 adults. Exp. Gerontol. 2007, 42, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkut, C.; Gade, V.R.; Laxman, S.; Kurzchalia, T.V. The glyoxylate shunt is essential for desiccation tolerance in C. elegans and budding yeast. eLife 2016, 5, e13614. [Google Scholar] [CrossRef]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Kreuzer, J.; Kumsta, C.; Wu, L.; Kamer, K.J.; Cedillo, L.; Zhang, Y.; Li, S.; Kacergis, M.C.; Webster, C.M.; et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell 2019, 177, 299–314.e216. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.M.; Von Stetina, S.E.; Barlow, S.J.; Shaffer, C.; Olszewski, K.L.; Moore, J.H.; Dupuy, D.; Vidal, M.; Miller, D.M., III. A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genom. 2005, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Burkewitz, K.; Morantte, I.; Weir, H.J.M.; Yeo, R.; Zhang, Y.; Huynh, F.K.; Ilkayeva, O.R.; Hirschey, M.D.; Grant, A.R.; Mair, W.B. Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell 2015, 160, 842–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lanjuin, A.; Chowdhury, S.R.; Mistry, M.; Silva-Garcia, C.G.; Weir, H.J.; Lee, C.L.; Escoubas, C.C.; Tabakovic, E.; Mair, W.B. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. eLife 2019, 8, e49158. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, B.; Deng, J.; Pang, S.; Tang, H. Histone acetylation promotes long-lasting defense responses and longevity following early life heat stress. PLoS Genet. 2019, 15, e1008122. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Rodriguez, A.; Arevalo, M.-A. The Contribution of Astrocyte Autophagy to Systemic Metabolism. Int. J. Mol. Sci. 2020, 21, 2479. [Google Scholar] [CrossRef] [Green Version]
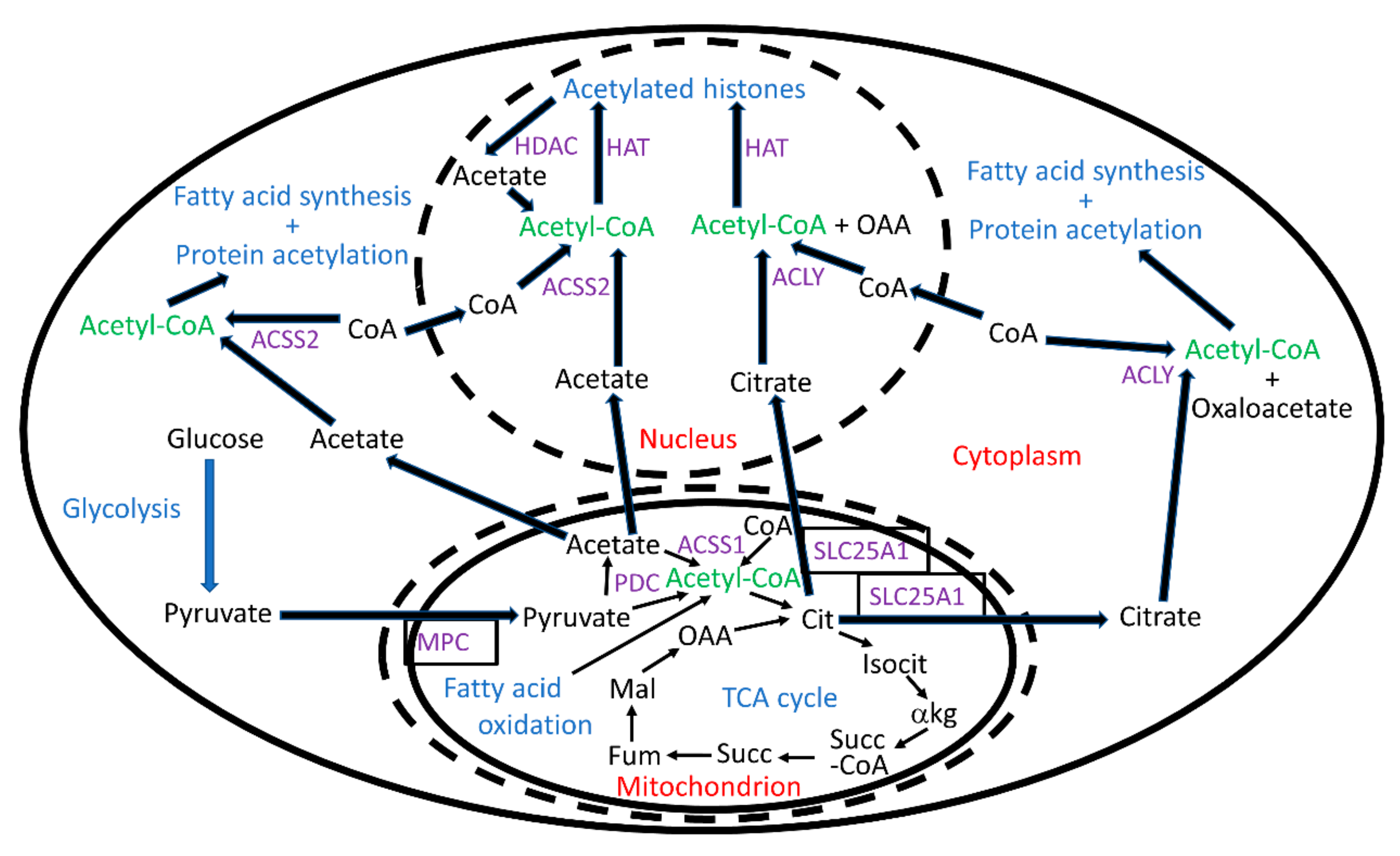

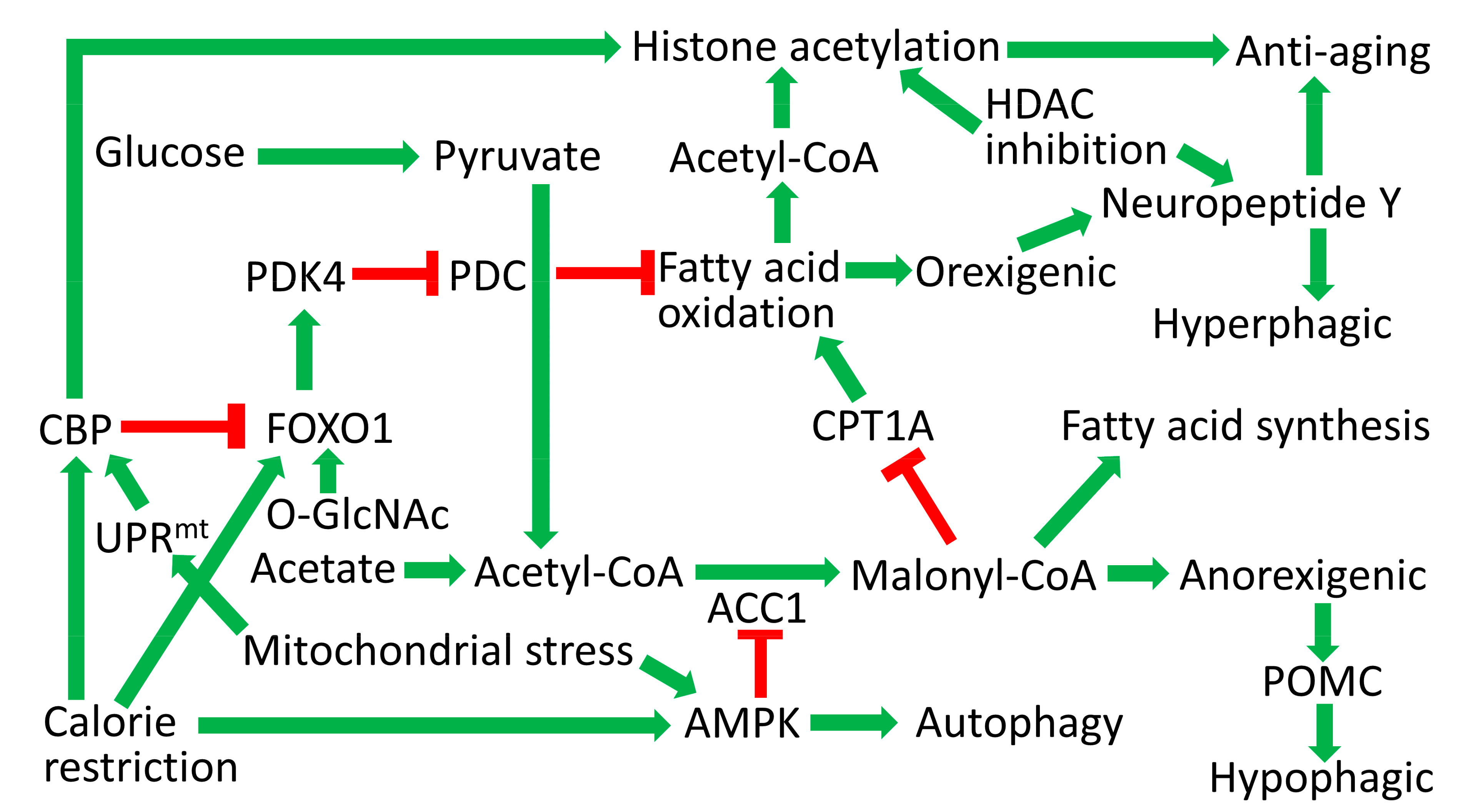

{kind=link}
{kind=link}
{kind=link}
| HAT Protein | Organism | Involvement in Aging |
|---|---|---|
| p300/CBP superfamily | ||
| - | S. cerevisiae | - |
| CBP-1 | C. elegans | DR requires CBP-1 for lifespan extension and increases acetylation of histone H4K5 [127]. NDGA inhibits CBP-1 to increase lifespan and decrease histone H3 acetylation [394]. |
| dCBP/nejire | D. melanogaster | |
| p300, CBP | mammals | Hypothalamic CBP expression correlates with lifespan in studies with five mouse strains [127]. |
| GNAT superfamily | ||
| Gcn5, Hat1, Nut1, Elp3, Hpa2, Hpa3, Nat4 | S. cerevisiae | Deletion of Gcn5 decreases CLS [315,316,317]. Gcn5 hemizygosity increases RLS [320]. Nat4 deletion increases RLS [340]. |
| PCAF-1, HAT-1, ELPC-3 | C. elegans | |
| dGcn5, Elp3, Atac2 | D. melanogaster | |
| GCN5, PCAF, HAT1, ELP3, ATF2, ATAC2 | mammals | HAT1 hemizygosity causes premature aging [151]. |
| MYST superfamily | ||
| Esa1, Sas2, Sas3 | S. cerevisiae | Deletion of Sas2 increases RLS by decreasing histone H4K16 acetylation [311]. Esa1 acetylates histone H4K5 and H4K12 to oppose pro-aging HDAC Rpd3 [398]. |
| MYS-1, MYS-2, LSY-12, MYS-4 | C. elegans | MYS-1 is required for the expression of daf-16 [301]. |
| Mof, Myst5/CG1894, Tip60, Enok, Chameau | D. melanogaster | Chameau hemizygosity increases lifespan and decreases histone H4K12 acetylation [375]. |
| KAT5, KAT6A, KAT6B, KAT7, KAT8 | mammals | Inhibitors of KAT6A/B induce senescence [399], while knockout of KAT7 prevents senescence [400]. |
| Other | ||
| Rtt109 | S. cerevisiae | Deletion of Rtt109 decreases RLS (but not CLS [401]) and histone H3K56 acetylation [311]. |
| HDAC Protein | Organism | Involvement in Aging |
|---|---|---|
| Class I | ||
| Rpd3, Hos1, Hos2, Hos3 | S. cerevisiae | Rpd3 deletion increases RLS [342]. |
| HDA-1, HDA-2, HDA-3 | C. elegans | Knockdown of hda-2 or hda-3 extends lifespan, while knockout decreases lifespan [384]. |
| Rpd3 | D. melanogaster | Rpd3 hemizygosity increases lifespan [362]. |
| HDAC1, HDAC2, HDAC3, HDAC8 | mammals | HDAC inhibitors are protective in many models of aging-related disease [402,403]. |
| Class III (nucleocyto) | ||
| Sir2, Hst1, Hst2, Hst3, Hst4 | S. cerevisiae | Sir2 deletion decreases RLS [311]. Hst3 deletion decreases RLS [404] or CLS [405]. Hst1 deletion decreases CLS [405]. |
| SIR-2.1, SIR-2.4 | C. elegans | SIR-2.1 promotes lifespan extension [406]. SIR-2.4 induces DAF-16 nuclear localization [393]. |
| dSir2, dSirt2, dSirt6, dSirt7 | D. melanogaster | dSir2 was shown to promote longevity [407,408]. Neuronal dSirt6 knockdown shortens lifespan [409]. |
| SIRT1, SIRT2, SIRT6, SIRT7 | mammals | Overexpression of SIRT1 in brain extends lifespan [98]. Overexpression of SIRT6 extends lifespan [290], while knockout decreases lifespan [291]. |
| Class III (mito) | ||
| Hst4 | S. cerevisiae | Hst4 deletion does not affect RLS [404] or CLS [405]. |
| SIR-2.2, SIR-2.3 | C. elegans | Mutants of sir-2.2 and sir-2.3 are long-lived under certain dietary conditions [410]. |
| dSirt2 [411], dSirt4 | D. melanogaster | Knockout of dSirt4 decreases lifespan, while overexpression increases lifespan [412]. Knock-down of dSirt2 in neurons decreases lifespan [409]. |
| SIRT3, SIRT4, SIRT5 | mammals | SIRT3 is required for some of the cytoprotection provided by CR [413]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bradshaw, P.C. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants 2021, 10, 572. https://doi.org/10.3390/antiox10040572
Bradshaw PC. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants. 2021; 10(4):572. https://doi.org/10.3390/antiox10040572
Chicago/Turabian StyleBradshaw, Patrick C. 2021. "Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan" Antioxidants 10, no. 4: 572. https://doi.org/10.3390/antiox10040572
APA StyleBradshaw, P. C. (2021). Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants, 10(4), 572. https://doi.org/10.3390/antiox10040572