
Hidrox® Roles in Neuroprotection: Biochemical Links between Traumatic Brain Injury and Alzheimer’s Disease

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocol
2.3. Experimental Groups
- (1)
- Vehicle group: Rats were subjected to TBI as described above, and vehicle (saline) was administered by gavage for 4 weeks.
- (2)
- Hidrox® group: Rats were subjected to experimental TBI as described above, and Hidrox® (10 mg/Kg) was administered 1 h after TBI and daily by gavage for 4 weeks.
- (3)
- Sham group: Rats were subjected to the surgical procedures (anesthesia, craniectomy, and suturing), and vehicle (saline) was administered by gavage for 4 weeks.
2.4. Morris Water Maze (MWM)
2.5. Elevated Plus Maze Test
2.6. Determination of Reduced Glutathione Levels
2.7. Measurement of Superoxide Dismutase (SOD) Activity
2.8. Measurement of Catalase Activity
2.9. Measurement of Lipid Peroxidation
2.10. Measurement of Nitrite Levels
2.11. Enzyme-Linked Immunosorbent Assay
2.12. Histological Examination
2.13. Western Blot Analysis
2.14. Statistical Evaluation
3. Results
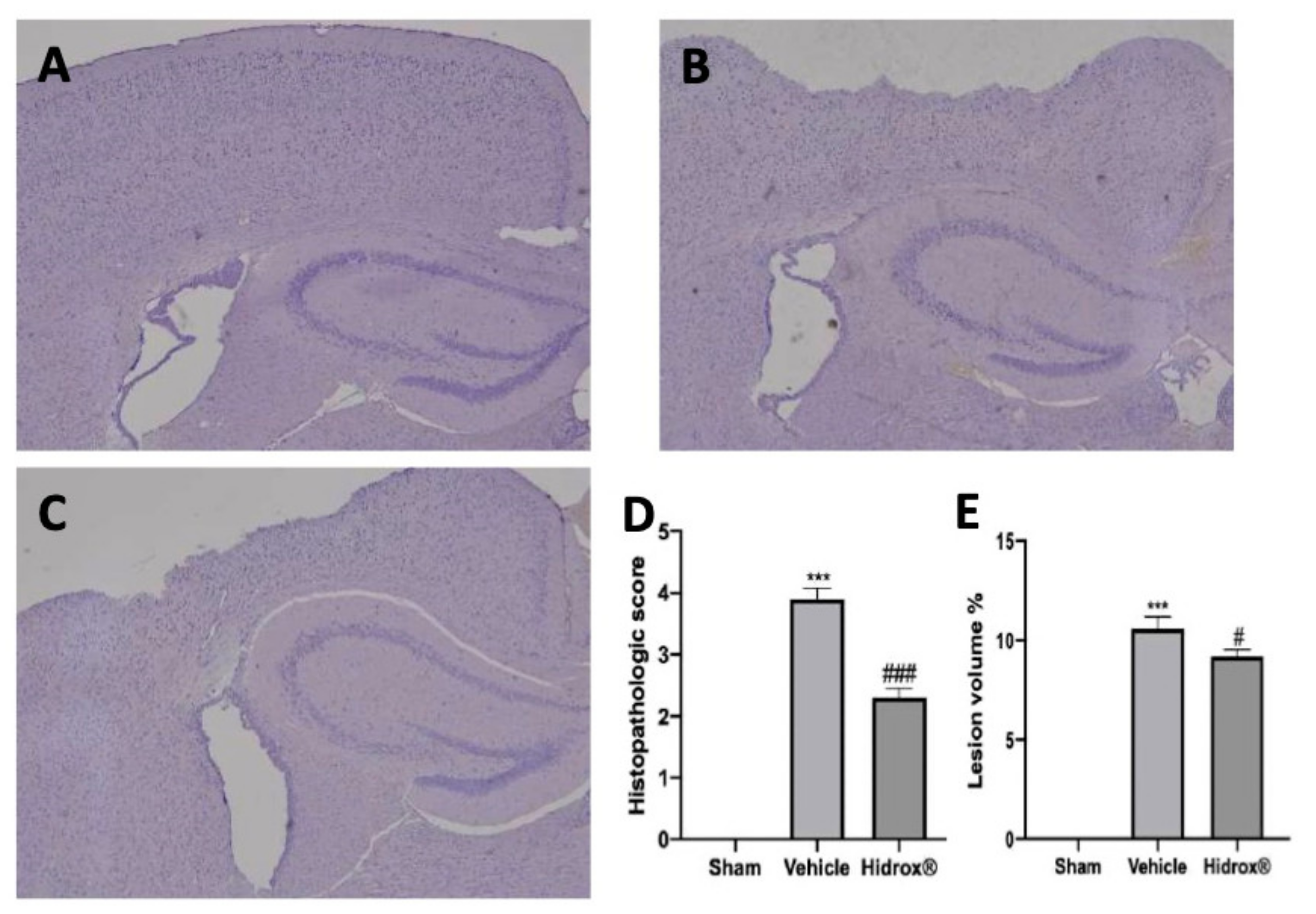
3.1. Effect of Hidrox®—Histological Analysis after TBI
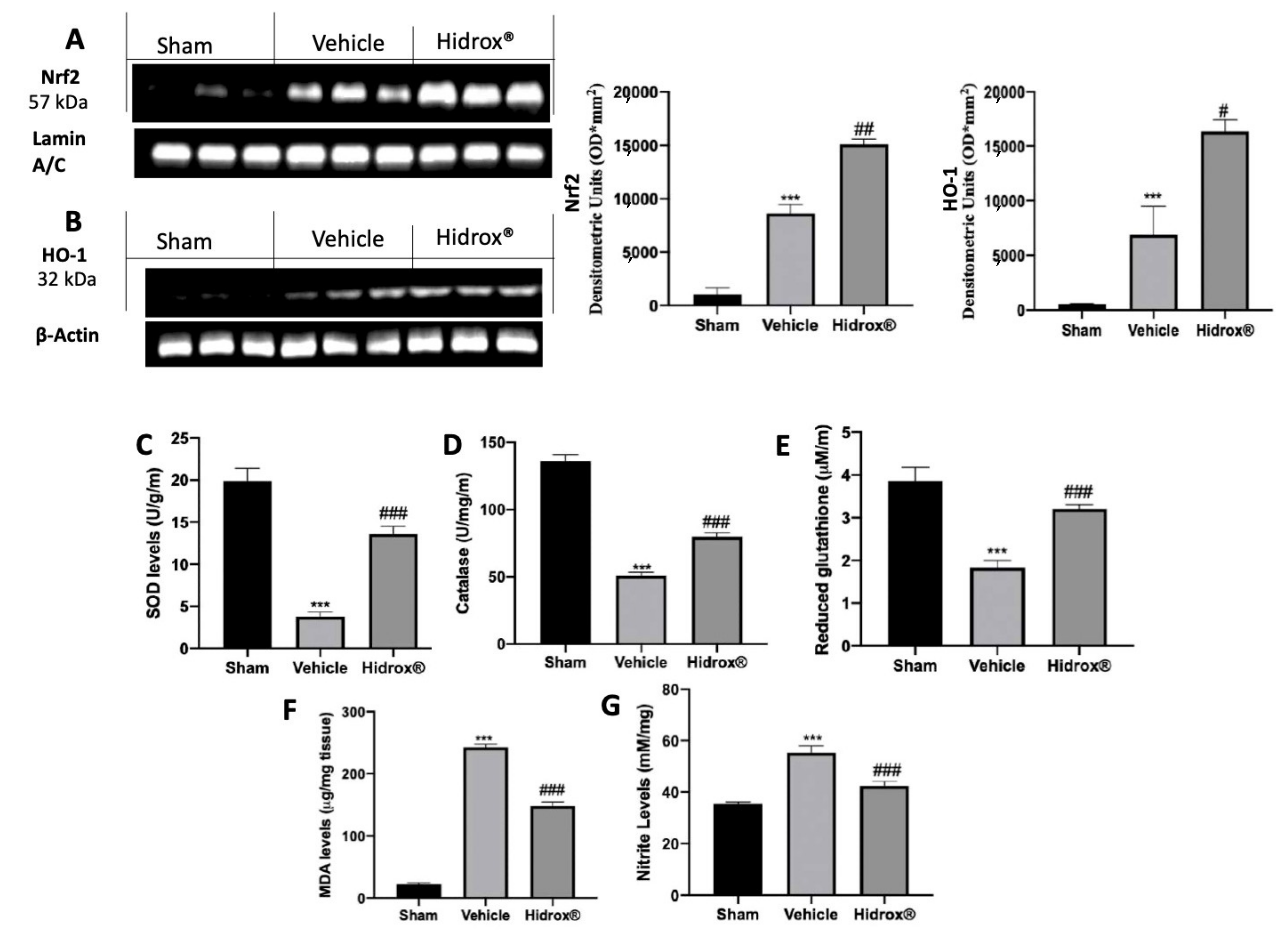
3.2. Effect of Hidrox® Treatment on Oxidative Hippocampal Alterations
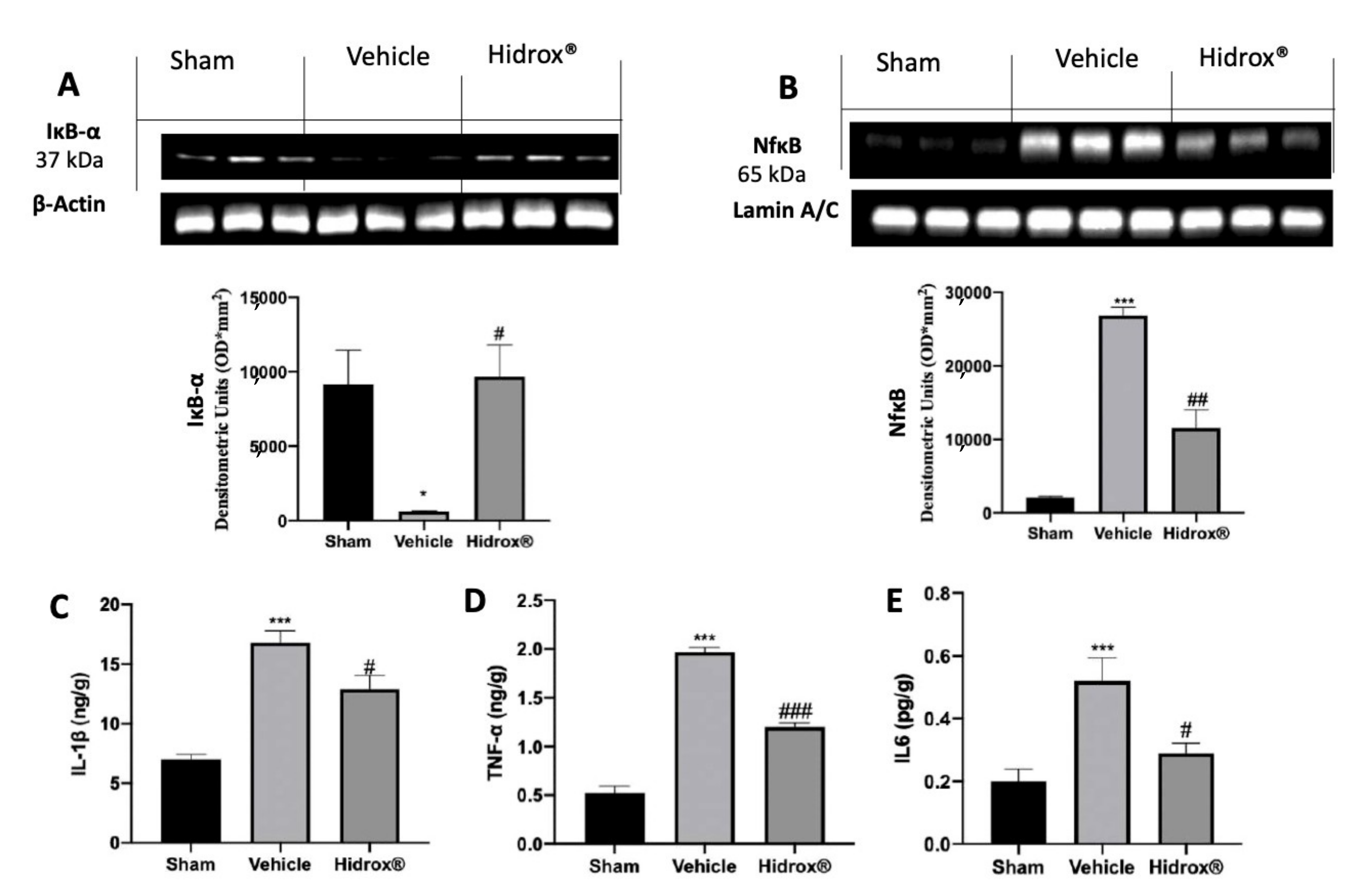
3.3. Effect of Hidrox® Treatment on Cytokine Expression and NFkB Pathway
3.4. Effect of Hidrox® Treatment on Behavioral Alterations
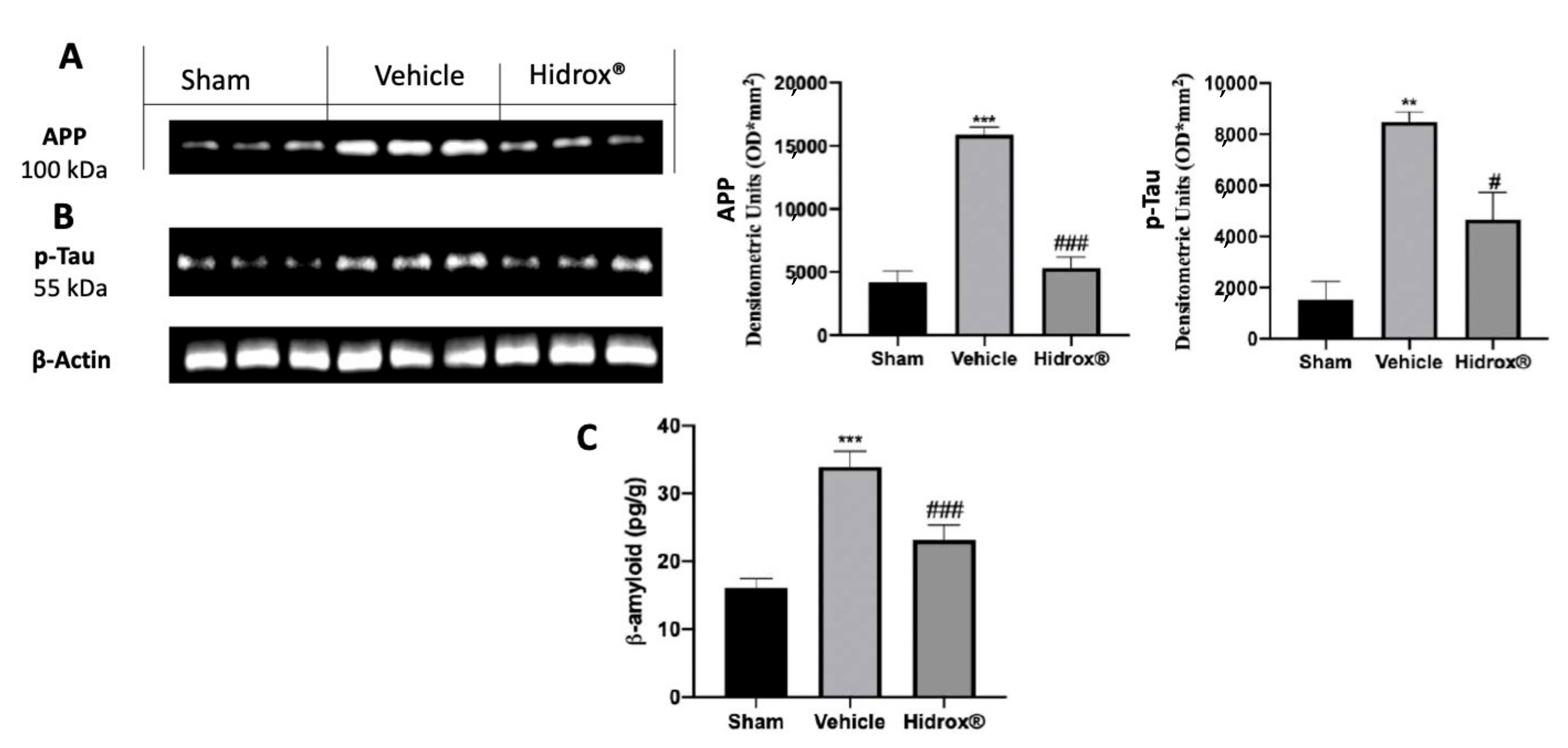
3.5. Effect of the Hidrox® Treatment on AD-Like Neuropathology
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fabrizio, K.S.; Keltner, N.L. Traumatic brain injury in operation enduring freedom/operation iraqi freedom: A primer. Nurs. Clin. N. Am. 2010, 45, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Xu, L.; Wald, M.M.; Coronado, V.; Dellinger, A.M. Traumatic Brain Injury in the United States: National Estimates of Prevalence and Incidence, 2002–2006. Injury Prev. 2010, 16, A268. [Google Scholar] [CrossRef]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, R.; Garakani, A.; Gross, L.S.; Goin, M.K.; Pine, J.; Slaby, A.E.; Sumner, C.R.; Baron, D.A. Neuropsychiatric aspects of concussion. Lancet Psychiatry 2016, 3, 1166–1175. [Google Scholar] [CrossRef]
- Thurman, D.J.; Alverson, C.; Dunn, K.A.; Guerrero, J.; Sniezek, J.E. Traumatic brain injury in the United States: A public health perspective. J. Head Trauma Rehab. 1999, 14, 602–615. [Google Scholar] [CrossRef]
- Wang, G.H.; Jiang, Z.L.; Li, Y.C.; Li, X.; Shi, H.; Gao, Y.Q.; Vosler, P.S.; Chen, J. Free-Radical Scavenger Edaravone Treatment Confers Neuroprotection Against Traumatic Brain Injury in Rats. J. Neurotrauma 2011, 28, 2123–2134. [Google Scholar] [CrossRef] [Green Version]
- Povlishock, J.T.; Christman, C.W. The Pathobiology of Traumatically Induced Axonal Injury in Animals and Humans—A Review of Current Thoughts. J. Neurotrauma 1995, 12, 555–564. [Google Scholar] [CrossRef]
- Ghajar, J. Traumatic brain injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef]
- Borgens, R.B.; Liu-Snyder, P. Understanding secondary injury. Q. Rev. Biol. 2012, 87, 89–127. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.M.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 2013, 8, e53376. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Lou, D.; Du, Y.; Huang, D.; Cai, F.; Zhang, Y.; Li, T.; Zhou, W.; Gao, H.; Song, W. Traumatic Brain Injury Alters the Metabolism and Facilitates Alzheimer’s Disease in a Murine Model. Mol. Neurobiol. 2018, 55, 4928–4939. [Google Scholar] [CrossRef]
- Mannix, R.C.; Whalen, M.J. Traumatic brain injury, microglia, and Beta amyloid. Int. J. Alzheimers Dis. 2012, 2012, 608732. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, W.; McPhie, D.L.; Hassinger, L.; Neve, R.L. APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. J. Cell. Biol. 2003, 163, 27–33. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Sanberg, P.R.; Kaneko, Y.; Borlongan, C.V. Increased Amyloid Precursor Protein and Tau Expression Manifests as Key Secondary Cell Death in Chronic Traumatic Brain Injury. J. Cell. Physiol. 2017, 232, 665–677. [Google Scholar] [CrossRef]
- Wang, L.; Shi, J.X.; Yin, H.X.; Ma, C.Y.; Zhang, Q.R. The influence of subarachnoid hemorrhage on neurons: An animal model. Ann. Clin. Lab. Sci. 2005, 35, 79–85. [Google Scholar]
- Signoretti, S.; Marmarou, A.; Tavazzi, B.; Dunbar, J.; Amorini, A.M.; Lazzarino, G.; Vagnozzi, R. The protective effect of cyclosporin A upon N-acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J. Neurotrauma 2004, 21, 1154–1167. [Google Scholar] [CrossRef]
- Kontos, H.A.; Povlishock, J.T. Oxygen radicals in brain injury. Cent. Nerv. Syst. Trauma 1986, 3, 257–263. [Google Scholar] [CrossRef]
- Cheng, Z.G.; Zhang, G.D.; Shi, P.Q.; Du, B.S. Expression and antioxidation of Nrf2/ARE pathway in traumatic brain injury. Asian Pac. J. Trop. Med. 2013, 6, 305–310. [Google Scholar] [CrossRef]
- Ku, B.M.; Joo, Y.; Mun, J.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Heme oxygenase protects hippocampal neurons from ethanol-induced neurotoxicity. Neurosci. Lett. 2006, 405, 168–171. [Google Scholar] [CrossRef]
- Radjendirane, V.; Joseph, P.; Lee, Y.H.; Kimura, S.; Klein-Szanto, A.J.; Gonzalez, F.J.; Jaiswal, A.K. Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity. J. Biol. Chem. 1998, 273, 7382–7389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Fusco, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. Int. J. Mol. Sci. 2019, 20, 4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’amico, R.; Fusco, R.; Gugliandolo, E.; Cordaro, M.; Siracusa, R.; Impellizzeri, D.; Peritore, A.F.; Crupi, R.; Cuzzocrea, S.; Di Paola, R. Effects of a new compound containing Palmitoylethanolamide and Baicalein in myocardial ischaemia/reperfusion injury in vivo. Phytomedicine 2019, 54, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Effect of PEA-OXA on neuropathic pain and functional recovery after sciatic nerve crush. J. Neuroinflamm. 2018, 15, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirley, R.; Ord, E.N.; Work, L.M. Oxidative Stress and the Use of Antioxidants in Stroke. Antioxidants 2014, 3, 472. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F.; Pettersson, S.; Meyer zum Buschenfelde, K.H.; Strober, W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat. Med. 1996, 2, 998–1004. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Fusco, R.; D’Amico, R.; Militi, A.; Oteri, G.; Wallace, J.L.; Di Paola, R.; Cuzzocrea, S. Anti-inflammatory effect of ATB-352, a H2S -releasing ketoprofen derivative, on lipopolysaccharide-induced periodontitis in rats. Pharmacol. Res. 2018, 132, 220–231. [Google Scholar] [CrossRef]
- Cordaro, M.; Impellizzeri, D.; Siracusa, R.; Gugliandolo, E.; Fusco, R.; Inferrera, A.; Esposito, E.; Di Paola, R.; Cuzzocrea, S. Effects of a co-micronized composite containing palmitoylethanolamide and polydatin in an experimental model of benign prostatic hyperplasia. Toxicol. Appl. Pharmacol. 2017, 329, 231–240. [Google Scholar] [CrossRef]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef]
- Vauzour, D.; Ravaioli, G.; Vafeiadou, K.; Rodriguez-Mateos, A.; Angeloni, C.; Spencer, J.P. Peroxynitrite induced formation of the neurotoxins 5-S-cysteinyl-dopamine and DHBT-1: Implications for Parkinson’s disease and protection by polyphenols. Arch. Biochem. Biophys. 2008, 476, 145–151. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Bahorun, T.; Jen, L.S. Neuroprotection by bioactive components in medicinal and food plant extracts. Mutat. Res. 2003, 544, 203–215. [Google Scholar] [CrossRef]
- Robles-Almazan, M.; Pulido-Moran, M.; Moreno-Fernandez, J.; Ramirez-Tortosa, C.; Rodriguez-Garcia, C.; Quiles, J.L.; Ramirez-Tortosa, M. Hydroxytyrosol: Bioavailability, toxicity, and clinical applications. Food Res. Int. 2018, 105, 654–667. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Barbalace, M.C.; Hrelia, S. Bioactivity of Olive Oil Phenols in Neuroprotection. Int. J. Mol. Sci. 2017, 18, 2230. [Google Scholar] [CrossRef] [Green Version]
- Feart, C.; Samieri, C.; Barberger-Gateau, P. Mediterranean diet and cognitive function in older adults. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Tasset, I.; Pontes, A.J.; Hinojosa, A.J.; de la Torre, R.; Tunez, I. Olive oil reduces oxidative damage in a 3-nitropropionic acid-induced Huntington’s disease-like rat model. Nutr. Neurosci. 2011, 14, 106–111. [Google Scholar] [CrossRef]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The polyphenol oleuropein aglycone protects TgCRND8 mice against Ass plaque pathology. PLoS ONE 2013, 8, e71702. [Google Scholar] [CrossRef]
- Casamenti, F.; Grossi, C.; Rigacci, S.; Pantano, D.; Luccarini, I.; Stefani, M. Oleuropein Aglycone: A Possible Drug against Degenerative Conditions. In Vivo Evidence of its Effectiveness against Alzheimer’s Disease. J. Alzheimers Dis. 2015, 45, 679–688. [Google Scholar] [CrossRef]
- Soni, M.G.; Burdock, G.A.; Christian, M.S.; Bitler, C.M.; Crea, R. Safety assessment of aqueous olive pulp extract as an antioxidant or antimicrobial agent in foods. Food Chem. Toxicol. 2006, 44, 903–915. [Google Scholar] [CrossRef]
- Miraglia, N.; Bianchi, D.; Trentin, A.; Volpi, N.; Soni, M.G. Safety assessment of non-animal chondroitin sulfate sodium: Subchronic study in rats, genotoxicity tests and human bioavailability. Food Chem. Toxicol. 2016, 93, 89–101. [Google Scholar] [CrossRef]
- Siracusa, R.; Scuto, M.; Fusco, R.; Trovato, A.; Ontario, M.L.; Crea, R.; Di Paola, R.; Cuzzocrea, S.; Calabrese, V. Anti-inflammatory and Anti-oxidant Activity of Hidrox((R)) in Rotenone-Induced Parkinson’s Disease in Mice. Antioxidants 2020, 9, 824. [Google Scholar] [CrossRef]
- Mahmoodzadeh, T.; Kashani, M.H.K.; Ramshini, H.; Moslem, A.; Mohammad-Zadeh, M. Effect of Camellia sinensis on Spatial Memory in a Rat Model of Alzheimer’s Disease. J. Biomed. 2016, 1, e5340. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, O.; Akar, F.; Celikyurt, I.K.; Tanyeri, P.; Ulak, G.; Erden, F. 7-NI and ODQ Disturbs Memory in the Elevated Plus Maze, Morris Water Maze, and Radial Arm Maze Tests in Mice. Drug Target Insights 2015, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chavali, V.D.; Agarwal, M.; Vyas, V.K.; Saxena, B. Neuroprotective Effects of Ethyl Pyruvate against Aluminum Chloride-Induced Alzheimer’s Disease in Rats via Inhibiting Toll-Like Receptor 4. J. Mol. Neurosci. 2020, 70, 836–850. [Google Scholar] [CrossRef]
- Chen, G.; Shi, J.; Hu, Z.; Hang, C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: A potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008, 2008, 716458. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.A.; Ahmed, H.I.; Abu-Elfotuh, K. Modeling stages mimic Alzheimer’s disease induced by different doses of aluminum in rats: Focus on progression of the disease in response to time. J. Alzheimers Parkinsonism Dement. 2016, 11, 2. [Google Scholar]
- Fusco, R.; Gugliandolo, E.; Biundo, F.; Campolo, M.; Di Paola, R.; Cuzzocrea, S. Inhibition of inflammasome activation improves lung acute injury induced by carrageenan in a mouse model of pleurisy. FASEB J. 2017, 31, 3497–3511. [Google Scholar] [CrossRef] [Green Version]
- Gugliandolo, E.; Fusco, R.; D’Amico, R.; Peditto, M.; Oteri, G.; Di Paola, R.; Cuzzocrea, S.; Navarra, M. Treatment with a Flavonoid-Rich Fraction of Bergamot Juice Improved Lipopolysaccharide-Induced Periodontitis in Rats. Front. Pharmacol. 2018, 9, 1563. [Google Scholar] [CrossRef] [Green Version]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Front. Neurol. 2018, 9, 590. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, R.; Impellizzeri, D.; Fusco, R.; Cordaro, M.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. Ultramicronized palmitoylethanolamide (PEA-um((R))) in the treatment of idiopathic pulmonary fibrosis. Pharmacol. Res. 2016, 111, 405–412. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Fusco, R.; Ginestra, G.; D’Amico, R.; Bisignano, C.; Mandalari, G.; Cuzzocrea, S.; Di Paola, R. Involvement of TLR4 and PPAR-alpha Receptors in Host Response and NLRP3 Inflammasome Activation, Against Pulmonary Infection with Pseudomonas Aeruginosa. Shock 2019, 51, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Fusco, R.; D’Amico, R.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Absence of formyl peptide receptor 1 causes endometriotic lesion regression in a mouse model of surgically-induced endometriosis. Oncotarget 2018, 9, 31355–31366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siracusa, R.; Fusco, R.; Peritore, A.F.; Cordaro, M.; D’Amico, R.; Genovese, T.; Gugliandolo, E.; Crupi, R.; Smeriglio, A.; Mandalari, G.; et al. The Antioxidant and Anti-Inflammatory Properties of Anacardium occidentale L. Cashew Nuts in a Mouse Model of Colitis. Nutrients 2020, 12, 834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, R.; Fusco, R.; Gugliandolo, E.; D’Amico, R.; Campolo, M.; Latteri, S.; Carughi, A.; Mandalari, G.; Cuzzocrea, S. The Antioxidant Activity of Pistachios Reduces Cardiac Tissue Injury of Acute Ischemia/Reperfusion (I/R) in Diabetic Streptozotocin (STZ)-Induced Hyperglycaemic Rats. Front. Pharmacol. 2018, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Peritore, A.F.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Crupi, R.; D’Amico, R.; Fusco, R.; Evangelista, M.; Cuzzocrea, S.; et al. The neuroprotective effects of micronized PEA (PEA-m) formulation on diabetic peripheral neuropathy in mice. FASEB J. 2019, 33, 11364–11380. [Google Scholar] [CrossRef]
- Di Paola, R.; Cordaro, M.; Crupi, R.; Siracusa, R.; Campolo, M.; Bruschetta, G.; Fusco, R.; Pugliatti, P.; Esposito, E.; Cuzzocrea, S. Protective effects of ultramicronized palmitoylethanolamide (PEA-um) in myocardial ischaemia and reperfusion injury in vivo. Shock 2016, 46, 202–213. [Google Scholar] [CrossRef]
- Buchele, F.; Morawska, M.M.; Schreglmann, S.R.; Penner, M.; Muser, M.; Baumann, C.R.; Noain, D. Novel Rat Model of Weight Drop-Induced Closed Diffuse Traumatic Brain Injury Compatible with Electrophysiological Recordings of Vigilance States. J. Neurotrauma 2016, 33, 1171–1180. [Google Scholar] [CrossRef]
- Ahmad, A.; Crupi, R.; Impellizzeri, D.; Campolo, M.; Marino, A.; Esposito, E.; Cuzzocrea, S. Administration of palmitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain Behav. Immun. 2012, 26, 1310–1321. [Google Scholar] [CrossRef]
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Attenuates Neuroinflammation and Neurobehavioral Deficits Induced by Experimental Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.D.; Hu, Z.G.; Yan, W.; Chen, G.; Yin, H.X. Transcription factor Nrf2 plays a pivotal role in protection against traumatic brain injury-induced acute intestinal mucosal injury in mice. J. Surg. Res. 2009, 157, 251–260. [Google Scholar] [CrossRef]
- Cordaro, M.; D’Amico, R.; Morabito, R.; Fusco, R.; Siracusa, R.; Peritore, A.F.; Impellizzeri, D.; Genovese, T.; Crupi, R.; Gugliandolo, E.; et al. Physiological and Biochemical Changes in NRF2 Pathway in Aged Animals Subjected to Brain Injury. Cell Physiol. Biochem. 2021, 55, 160–179. [Google Scholar]
- Fusco, R.; Cordaro, M.; Siracusa, R.; Peritore, A.F.; D’Amico, R.; Licata, P.; Crupi, R.; Gugliandolo, E. Effects of Hydroxytyrosol against Lipopolysaccharide-Induced Inflammation and Oxidative Stress in Bovine Mammary Epithelial Cells: A Natural Therapeutic Tool for Bovine Mastitis. Antioxidants 2020, 9, 693. [Google Scholar] [CrossRef]
- Fusco, R.; Cordaro, M.; Siracusa, R.; D’Amico, R.; Genovese, T.; Gugliandolo, E.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. Biochemical Evaluation of the Antioxidant Effects of Hydroxytyrosol on Pancreatitis-Associated Gut Injury. Antioxidants 2020, 9, 781. [Google Scholar] [CrossRef]
- Fusco, R.; Cordaro, M.; Siracusa, R.; Peritore, A.F.; Gugliandolo, E.; Genovese, T.; D’Amico, R.; Crupi, R.; Smeriglio, A.; Mandalari, G.; et al. Consumption of Anacardium Occidentale L. (Cashew Nuts) Inhibits Oxidative Stress through Modulation of the Nrf2/HO-1 and NF-kB Pathways. Molecules 2020, 25, 4426. [Google Scholar] [CrossRef]
- Cordaro, M.; Fusco, R.; D’Amico, R.; Siracusa, R.; Peritore, A.F.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Mandalari, G.; Cuzzocrea, S.; et al. Cashew (Anacardium occidentale L.) Nuts Modulate the Nrf2 and NLRP3 Pathways in Pancreas and Lung after Induction of Acute Pancreatitis by Cerulein. Antioxidants 2020, 9, 992. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Fusco, R.; Licata, P.; Peritore, A.F.; D’Amico, R.; Cordaro, M.; Siracusa, R.; Cuzzocrea, S.; Crupi, R. Protective Effect of Hydroxytyrosol on LPS-Induced Inflammation and Oxidative Stress in Bovine Endometrial Epithelial Cell Line. Vet. Sci. 2020, 7, 161. [Google Scholar] [CrossRef]
- Peritore, A.F.; D’Amico, R.; Cordaro, M.; Siracusa, R.; Fusco, R.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Di Paola, R.; Cuzzocrea, S.; et al. PEA/Polydatin: Anti-Inflammatory and Antioxidant Approach to Counteract DNBS-Induced Colitis. Antioxidants 2021, 10, 464. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Ting, S.M.; Gonzales, N.; Aronowski, J. Dimethyl Fumarate Protects Brain from Damage Produced by Intracerebral Hemorrhage by Mechanism Involving Nrf2. Stroke 2015, 46, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, S.; Esposito, E.; Di Paola, R.; Ciampa, A.; Mazzon, E.; Carcereri de Prati, A.; Darra, E.; Vincenzi, S.; Cucinotta, G.; Caminiti, R.; et al. Protective effect of Arbutus unedo aqueous extract in carrageenan-induced lung inflammation in mice. Pharmacol. Res. 2008, 57, 110–124. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Nocentini, G.; Di Paola, R.; Agostini, M.; Mazzon, E.; Ronchetti, S.; Crisafulli, C.; Esposito, E.; Caputi, A.P.; Riccardi, C. Proinflammatory role of glucocorticoid-induced TNF receptor-related gene in acute lung inflammation. J. Immunol. 2006, 177, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; Mazzon, E.; Esposito, E.; Muià, C.; Abdelrahman, M.; Di Paola, R.; Crisafulli, C.; Bramanti, P.; Thiemermann, C. Glycogen synthase kinase-3beta inhibition attenuates the development of ischaemia/reperfusion injury of the gut. Intensive Care Med. 2007, 33, 880–893. [Google Scholar] [CrossRef]
- Cordaro, M.; Impellizzeri, D.; Gugliandolo, E.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. Adelmidrol, a Palmitoylethanolamide Analogue, as a New Pharmacological Treatment for the Management of Inflammatory Bowel Disease. Mol. Pharmacol. 2016, 90, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; Di Paola, R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef]
- Travelli., C.; Aprile, S.; Rahimian, R.; Grolla, A.A.; Rogati, F.; Bertolotti, M.; Malagnino, F.; Di Paola, R.; Impellizzeri, D.; Fusco, R.; et al. Identification of Novel Triazole-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors Endowed with Antiproliferative and Antiinflammatory Activity. J. Med. Chem. 2017, 60, 1768–1792. [Google Scholar] [CrossRef]
- Dixon, C.E.; Kochanek, P.M.; Yan, H.Q.; Schiding, J.K.; Griffith, R.G.; Baum, E.; Marion, D.W.; DeKosky, S.T. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J. Neurotrauma 1999, 16, 109–122. [Google Scholar] [CrossRef]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Vasterling, J.J.; Brailey, K.; Proctor, S.P.; Kane, R.; Heeren, T.; Franz, M. Neuropsychological outcomes of mild traumatic brain injury, post-traumatic stress disorder and depression in Iraq-deployed US Army soldiers. Br. J. Psychiatry 2012, 201, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aungst, S.L.; Kabadi, S.V.; Thompson, S.M.; Stoica, B.A.; Faden, A.I. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 2014, 34, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, D.J.; Scott, G.; Leech, R. Network dysfunction after traumatic brain injury. Nat. Rev. Neurol. 2014, 10, 156–166. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20, S499–S512. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Abeta production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Scoville, W.B. The limbic lobe in man. J. Neurosurg. 1954, 11, 64–66. [Google Scholar] [CrossRef]
- Amaral, D.G.; Witter, M.P. The three-dimensional organization of the hippocampal formation: A review of anatomical data. Neuroscience 1989, 31, 571–591. [Google Scholar] [CrossRef]
- Nakashiba, T.; Young, J.Z.; McHugh, T.J.; Buhl, D.L.; Tonegawa, S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science 2008, 319, 1260–1264. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1995, 92, 4725–4727. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, S. Head injury doubles the risk of Alzheimer’s disease. BMJ 2000, 321, 1100. [Google Scholar]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Soldatovic-Stajic, B.; Misic-Pavkov, G.; Bozic, K.; Novovic, Z.; Gajic, Z. Neuropsychological and neurophysiological evaluation of cognitive deficits related to the severity of traumatic brain injury. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1632–1637. [Google Scholar]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatry Dis. Treat. 2015, 11, 97–106. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordaro, M.; Trovato Salinaro, A.; Siracusa, R.; D’Amico, R.; Impellizzeri, D.; Scuto, M.; Ontario, M.L.; Crea, R.; Cuzzocrea, S.; Di Paola, R.; et al. Hidrox® Roles in Neuroprotection: Biochemical Links between Traumatic Brain Injury and Alzheimer’s Disease. Antioxidants 2021, 10, 818. https://doi.org/10.3390/antiox10050818
Cordaro M, Trovato Salinaro A, Siracusa R, D’Amico R, Impellizzeri D, Scuto M, Ontario ML, Crea R, Cuzzocrea S, Di Paola R, et al. Hidrox® Roles in Neuroprotection: Biochemical Links between Traumatic Brain Injury and Alzheimer’s Disease. Antioxidants. 2021; 10(5):818. https://doi.org/10.3390/antiox10050818
Chicago/Turabian StyleCordaro, Marika, Angela Trovato Salinaro, Rosalba Siracusa, Ramona D’Amico, Daniela Impellizzeri, Maria Scuto, Maria Laura Ontario, Roberto Crea, Salvatore Cuzzocrea, Rosanna Di Paola, and et al. 2021. "Hidrox® Roles in Neuroprotection: Biochemical Links between Traumatic Brain Injury and Alzheimer’s Disease" Antioxidants 10, no. 5: 818. https://doi.org/10.3390/antiox10050818
APA StyleCordaro, M., Trovato Salinaro, A., Siracusa, R., D’Amico, R., Impellizzeri, D., Scuto, M., Ontario, M. L., Crea, R., Cuzzocrea, S., Di Paola, R., Fusco, R., & Calabrese, V. (2021). Hidrox® Roles in Neuroprotection: Biochemical Links between Traumatic Brain Injury and Alzheimer’s Disease. Antioxidants, 10(5), 818. https://doi.org/10.3390/antiox10050818