
Pleiotropic and Potentially Beneficial Effects of Reactive Oxygen Species on the Intracellular Signaling Pathways in Endothelial Cells

 , ,
, ,  , , , and
, , , and
Abstract
:
1. Introduction
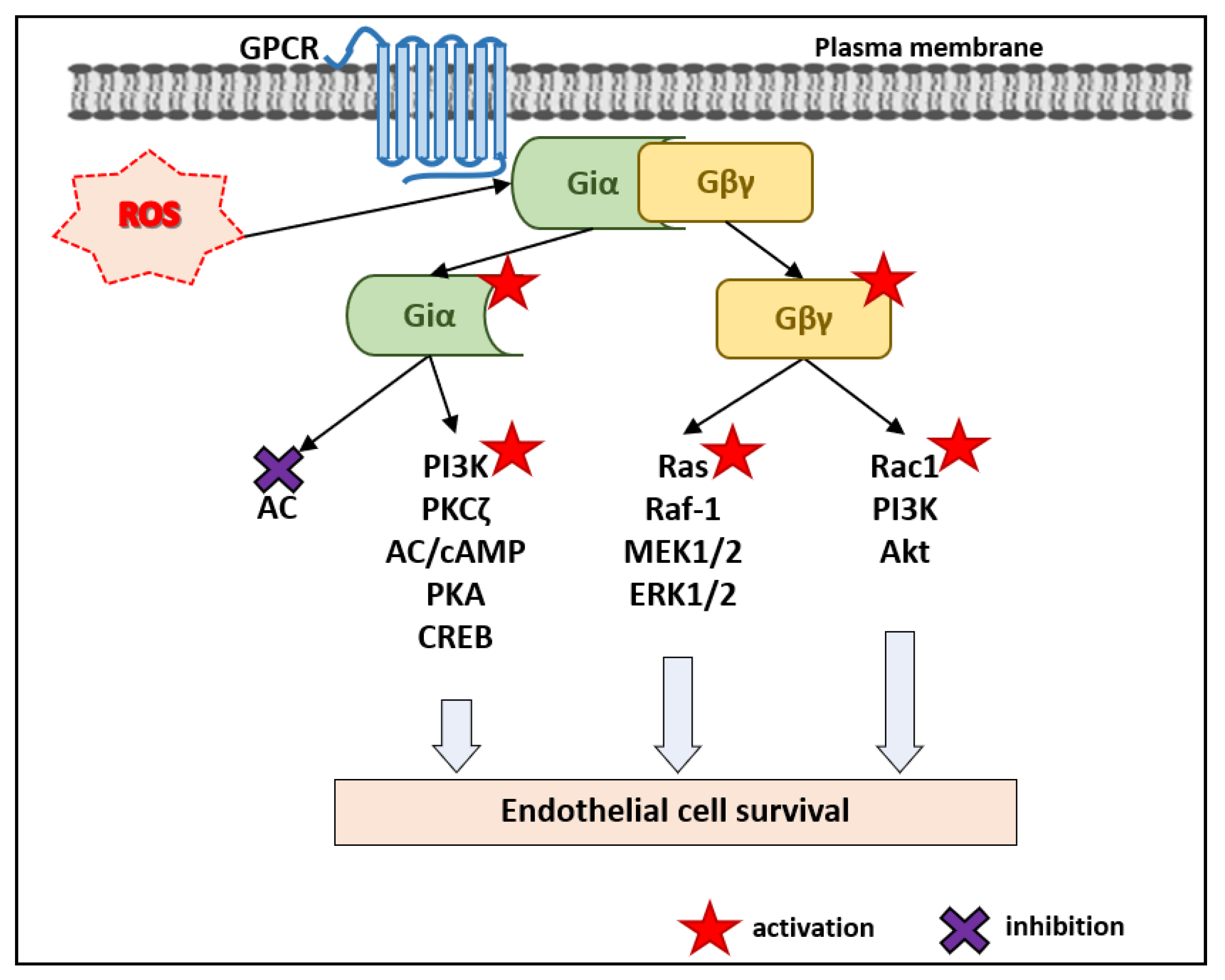
2. ROS, Gαi/o Subunits of the Heterotrimeric G Proteins and EC Survival
2.1. Gi/o Proteins and AC–cAMP–PKA Pathway
2.2. Gi/o Proteins and Ras–Raf–MEK1/2–ERK1/2 Pathway
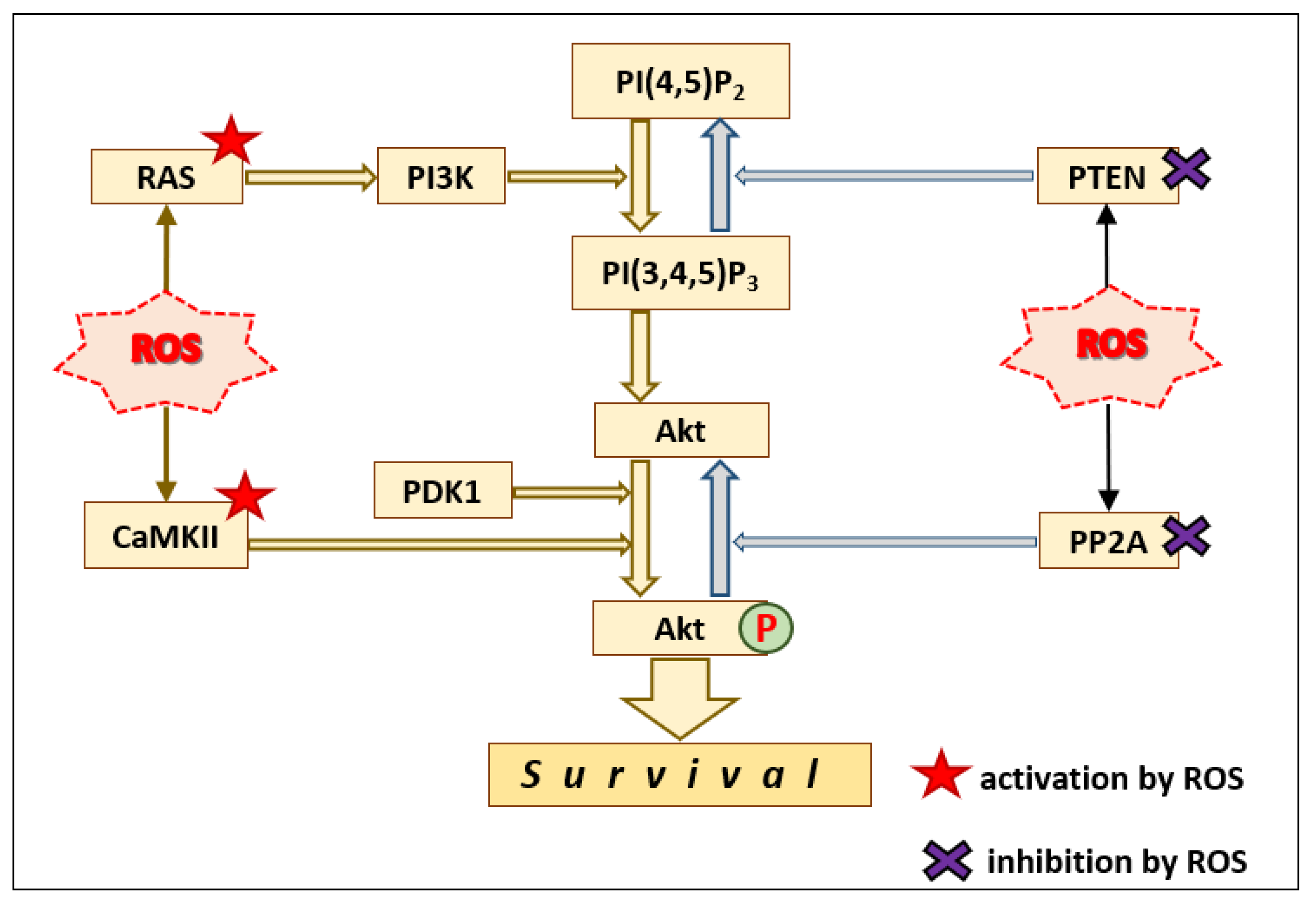
2.3. Gi/o Proteins and the PI3K–Akt Pathway
2.4. Gi/o Proteins and Pro-Apoptotic Pathways in ECs
3. ROS Can Mimic Insulin Signaling
3.1. Reversible Inhibition of Protein Tyrosine Phosphatase 1B (PTP1B) by ROS
3.2. Indirect Activation of the PI3K–Akt Pathway
3.3. Small GTPase Ras and Ras–Raf–MEK–ERK Pathway
4. Endothelial Barrier: Redox Dependence of Some Intracellular Signaling Proteins Involved in Regulation of Endothelial Permeability
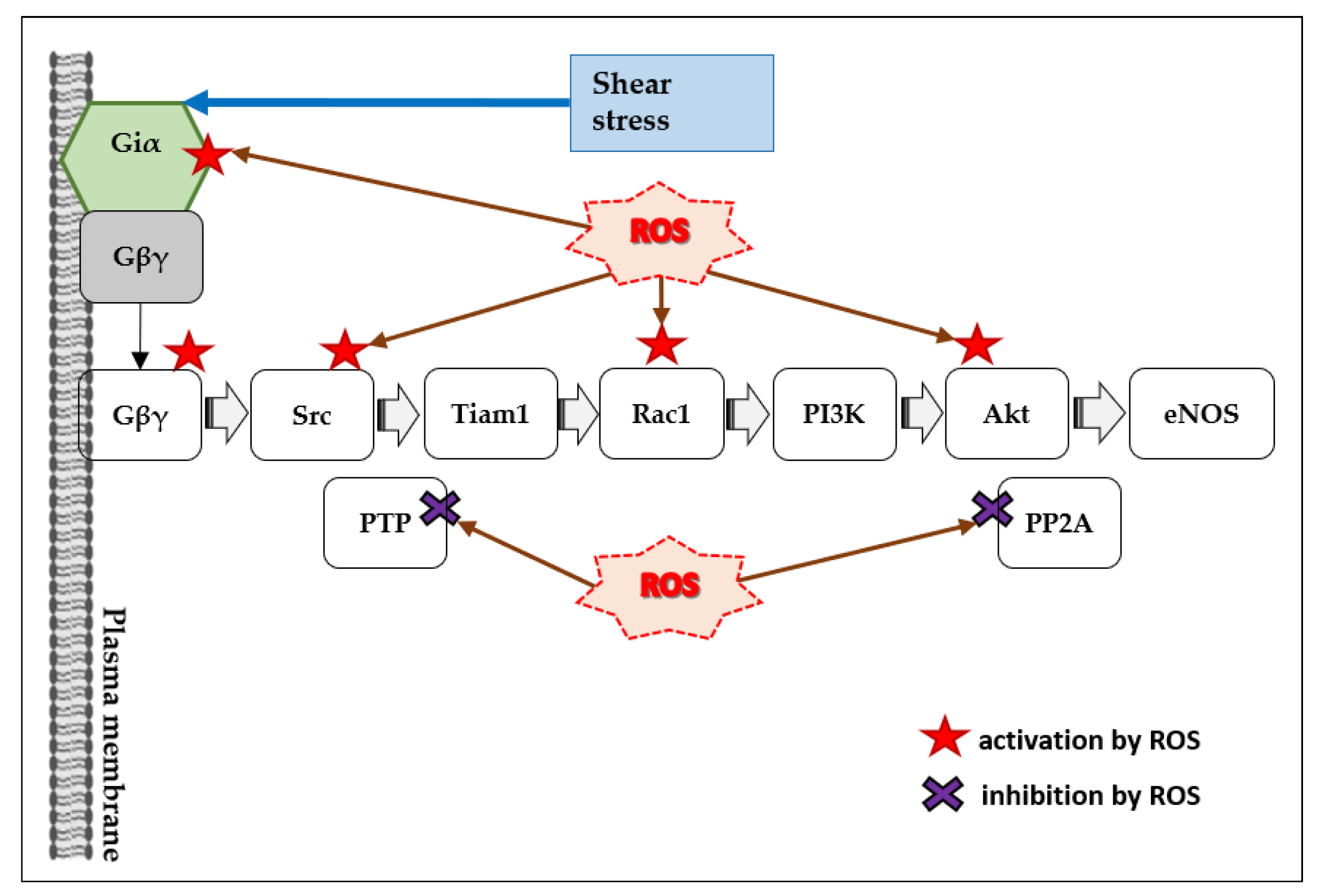
4.1. ROS and Gi/o Proteins
4.2. ROS and Some Branches of Signaling Downstream of Growth Factor Receptors
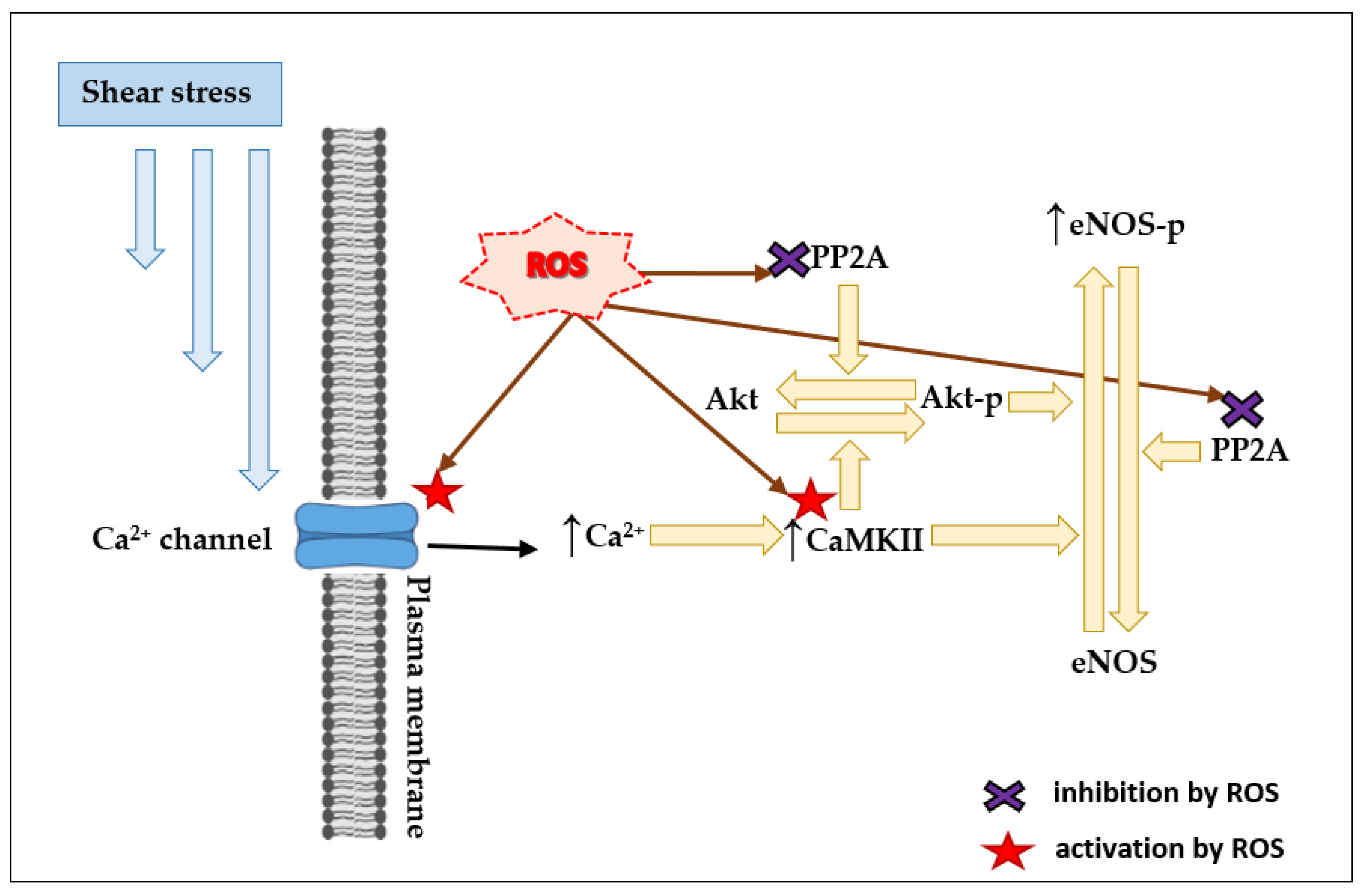
4.3. ROS and Ca2+-Dependent Mechanisms
4.4. ROS and Tyrosine Kinases and Phosphatases
5. Endothelial Dysfunction
5.1. ROS and Flow-Induced Release of Vasodilators by ECs
5.2. ROS, MEK5–ERK5 Module and Transcription Factors (TFs) That Can Alleviate Endothelial Dysfunction
5.2.1. Redox-Sensitive Elements in SS-Induced Signaling Pathway Leading to ERK5 Activation
5.2.2. Krüppel-Like Factors (KLF) Family
5.2.3. MEF2 Family of Transcription Factors
5.2.4. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)
5.2.5. Peroxisome Proliferator-Activated Receptors (PPARs)
6. Role of NADPH Oxidase-Derived ROS in Angiogenesis
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Barvitenko, N.N.; Aslam, M.; Filosa, J.; Matteucci, E.; Nikinmaa, M.; Pantaleo, A.; Saldanha, C.; Baskurt, O.K. Tissue Oxygen Demand in Regulation of the Behavior of the Cells in the Vasculature. Microcirculation 2013, 20, 484–501. [Google Scholar] [CrossRef]
- Wolin, M.S. Interactions of Oxidants with Vascular Signaling Systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2018, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, Physiology, and Pathophysiology of NADPH Oxidases in the Cardiovascular System. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2013, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.-D.V.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Miller, I.P.; Pavlović, I.; Poljšak, B.; Šuput, D.; Milisav, I. Beneficial Role of ROS in Cell Survival: Moderate Increases in H2O2 Production Induced by Hepatocyte Isolation Mediate Stress Adaptation and Enhanced Survival. Antioxidants 2019, 8, 434. [Google Scholar] [CrossRef] [Green Version]
- Tonks, N.K. Redox Redux: Revisiting PTPs and the Control of Cell Signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Hertog, J.; Groen, A.; van der Wijk, T. Redox Regulation of Protein-Tyrosine Phosphatases. Arch. Biochem. Biophys. 2005, 434, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.J.; Parsons, Z.D.; Cummings, A.H.; Zhou, H.; Gates, K.S. Redox Regulation of Protein Tyrosine Phosphatases: Structural and Chemical Aspects. Antioxid. Redox Signal. 2010, 15, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, A.; Cotter, T.G. Redox Regulation of Protein Kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Carroll, K.S. Redox Regulation of Protein Kinases. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 332–356. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Rios, F.J.; Alves-Lopes, R.; Neves, K.B.; Camargo, L.L.; Montezano, A.C. Oxidative Stress: A Unifying Paradigm in Hypertension. Can. J. Cardiol. 2020, 36, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R.; Keaney, J.F., Jr. Role of Oxidative Modifications in Atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive Species-Induced Microvascular Dysfunction in Ischemia/Reperfusion. Free Radic. Biol. Med. 2019, 135, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Pula, G. Reactive Oxygen Species: Physiological Roles in the Regulation of Vascular Cells. Curr. Mol. Med. 2014, 14, 1103–1125. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive Oxygen Species: Key Regulators in Vascular Health and Diseases. Br. J. Pharmacol. 2018, 175, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K. NADPH Oxidase-derived Reactive Oxygen Species: Dosis Facit Venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of Mitochondria with NADPH Oxidase via Reactive Oxygen and Nitrogen Species Signalling and Its Role for Vascular Function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barvitenko, N.; Lawen, A.; Aslam, M.; Pantaleo, A.; Saldanha, C.; Skverchinskaya, E.; Regolini, M.; Tuszynski, J.A. Integration of Intracellular Signaling: Biological Analogues of Wires, Processors and Memories Organized by a Centrosome 3D Reference System. Biosystems 2018, 173, 191–206. [Google Scholar] [CrossRef]
- Nishida, M.; Maruyama, Y.; Tanaka, R.; Kontani, K.; Nagao, T.; Kurose, H. Gαi and Gαo Are Target Proteins of Reactive Oxygen Species. Nature 2000, 408, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Schey, K.L.; Takagahara, S.; Kontani, K.; Katada, T.; Urano, Y.; Nagano, T.; Nagao, T.; Kurose, H. Activation Mechanism of Gi and Go by Reactive Oxygen Species. J. Biol. Chem. 2002, 277, 9036–9042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettschureck, N.; Offermanns, S. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, E.; Granata, R.; Miceli, I.; Baragli, A.; Settanni, F.; Cavallo Perin, P.; Ghigo, E.; Camussi, G.; Zanone, M.M. The Ghrelin Gene Products and Exendin-4 Promote Survival of Human Pancreatic Islet Endothelial Cells in Hyperglycaemic Conditions, through Phosphoinositide 3-Kinase/Akt, Extracellular Signal-Related Kinase (ERK)1/2 and CAMP/Protein Kinase A (PKA) Signalling Pathways. Diabetologia 2012, 55, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Liu, J.; Tian, Z.; Gao, B.; Kunos, G. Dose-Dependent Activation of Antiapoptotic and Proapoptotic Pathways by Ethanol Treatment in Human Vascular Endothelial Cells: Differential involvement of adenosine*. J. Biol. Chem. 2002, 277, 20927–20933. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharm. Rev. 2010, 62, 588. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, K.; Sarker, K.P.; Kawahara, K.; Iino, S.; Yamakuchi, M.; Abeyama, K.; Hashiguchi, T.; Maruyama, I. Anandamide Induces Apoptosis in Human Endothelial Cells: Its Regulation System and Clinical Implications. Thromb. Haemost. 2003, 89, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Haskó, G.; Liaudet, L.; Mackie, K.; Pacher, P. Cannabinoid-1 Receptor Activation Induces Reactive Oxygen Species-Dependent and -Independent Mitogen-Activated Protein Kinase Activation and Cell Death in Human Coronary Artery Endothelial Cells. Br. J. Pharmacol. 2010, 160, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Mary, S.; Damian, M.; Louet, M.; Floquet, N.; Fehrentz, J.-A.; Marie, J.; Martinez, J.; Banères, J.-L. Ligands and Signaling Proteins Govern the Conformational Landscape Explored by a G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 8304. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.; Hla, T.; Lynch, K.R.; Spiegel, S.; Moolenaar, W.H. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid Receptor Nomenclature. Pharm. Rev. 2010, 62, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular Endothelial Cell Adherens Junction Assembly and Morphogenesis Induced by Sphingosine-1-Phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional Regulation by the Phosphorylation-Dependent Factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-T.; Chang, Y.-C.; Tu, Y.-F.; Huang, C.-C. VEGF-A/VEGFR-2 Signaling Leading to CAMP Response Element-Binding Protein Phosphorylation Is a Shared Pathway Underlying the Protective Effect of Preconditioning on Neurons and Endothelial Cells. J. Neurosci. 2009, 29, 4356. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Woo, J.-S.; Shin, Y.-W. Cilostazol Protects Endothelial Cells against Lipopolysaccharide-Induced Apoptosis through ERK1/2-and P38 MAPK-Dependent Pathways. Korean J. Intern. Med. 2009, 24, 113. [Google Scholar] [CrossRef]
- Halls, M.L.; Bathgate, R.A.; Summers, R.J. Relaxin Family Peptide Receptors RXFP1 and RXFP2 Modulate CAMP Signaling by Distinct Mechanisms. Mol. Pharmacol. 2006, 70, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-P38 MAP Kinases on Apoptosis. Science 1995, 270, 1326. [Google Scholar] [CrossRef]
- Verin, A.D.; Liu, F.; Bogatcheva, N.; Borbiev, T.; Hershenson, M.B.; Wang, P.; Garcia, J.G.N. Role of Ras-Dependent ERK Activation in Phorbol Ester-Induced Endothelial Cell Barrier Dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L360–L370. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-J.; Evans, M.; Hla, T. The Inducible G Protein-Coupled Receptor EDG-1 Signals via the G/Mitogen-Activated Protein Kinase Pathway. J. Biol. Chem. 1996, 271, 11272–11279. [Google Scholar] [CrossRef] [Green Version]
- Crespo, P.; Xu, N.; Simonds, W.F.; Gutkind, J.S. Ras-Dependent Activation of MAP Kinase Pathway Mediated by G-Protein Βγ Subunits. Nature 1994, 369, 418–420. [Google Scholar] [CrossRef]
- Koch, W.J.; Hawes, B.E.; Allen, L.F.; Lefkowitz, R.J. Direct Evidence That Gi-Coupled Receptor Stimulation of Mitogen-Activated Protein Kinase Is Mediated by G Beta Gamma Activation of P21ras. Proc. Natl. Acad. Sci. USA 1994, 91, 12706–12710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular Survival: A Play in Three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Assmus, B.; Hermann, C.; Haendeler, J.; Zeiher, A.M. Fluid Shear Stress Stimulates Phosphorylation of Akt in Human Endothelial Cells: Involvement in Suppression of Apoptosis. Circ. Res. 1998, 83, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Hermann, C.; Assmus, B.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Insulin-Mediated Stimulation of Protein Kinase Akt: A Potent Survival Signaling Cascade for Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 402–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, P.A.; Dudek, S.M.; Chiang, E.T.; Garcia, J.G.N. Regulation of Sphingosine 1-Phosphate-Induced Endothelial Cytoskeletal Rearrangement and Barrier Enhancement by S1P1 Receptor, PI3 Kinase, Tiam1/Rac1, and α-Actinin. FASEB J. 2005, 19, 1646–1656. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kou, R.; Michel, T. Rac1 Modulates Sphingosine 1-Phosphate-Mediated Activation of Phosphoinositide 3-Kinase/Akt Signaling Pathways in Vascular Endothelial Cells*. J. Biol. Chem. 2006, 281, 3210–3216. [Google Scholar] [CrossRef] [Green Version]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial Dysfunction—A Major Mediator of Diabetic Vascular Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [Green Version]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive Oxygen Species Have a Causal Role in Multiple Forms of Insulin Resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Bashan, N.; Kovsan, J.; Kachko, I.; Ovadia, H.; Rudich, A. Positive and Negative Regulation of Insulin Signaling by Reactive Oxygen and Nitrogen Species. Physiol. Rev. 2009, 89, 27–71. [Google Scholar] [CrossRef] [Green Version]
- Krieger-Brauer, H.I.; Medda, P.K.; Kather, H. Insulin-Induced Activation of NADPH-Dependent H2O2 Generation in Human Adipocyte Plasma Membranes Is Mediated by Gαi2. J. Biol. Chem. 1997, 272, 10135–10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H Oxidase Homolog Nox4 Modulates Insulin-Stimulated Generation of H2O2 and Plays an Integral Role in Insulin Signal Transduction. Mol. Cell. Biol. 2004, 24, 1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-Stimulated Hydrogen Peroxide Reversibly Inhibits Protein-Tyrosine Phosphatase 1B in Vivo and Enhances the Early Insulin Action Cascade*. J. Biol. Chem. 2001, 276, 21938–21942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.-C. Increased Insulin Sensitivity and Obesity Resistance in Mice Lacking the Protein Tyrosine Phosphatase-1B Gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.-B.; Sharpe, A.H. Increased Energy Expenditure, Decreased Adiposity, and Tissue-Specific Insulin Sensitivity in Protein-Tyrosine Phosphatase 1B-Deficient Mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, H.; Figueroa, A.L.C.; Garcia, A.; Fernandez-Ruiz, R.; Broca, C.; Wojtusciszyn, A.; Malpique, R.; Gasa, R.; Gomis, R. Targeting Pancreatic Islet PTP1B Improves Islet Graft Revascularization and Transplant Outcomes. Sci. Transl. Med. 2019, 11, eaar6294. [Google Scholar] [CrossRef] [Green Version]
- Messina, S.; De Simone, G.; Ascenzi, P. Cysteine-Based Regulation of Redox-Sensitive Ras Small GTPases. Redox Biol. 2019, 26, 101282. [Google Scholar] [CrossRef]
- Clavreul, N.; Adachi, T.; Pimental, D.R.; Ido, Y.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by Peroxynitrite of P21ras at Cysteine-118 Mediates Its Direct Activation and Downstream Signaling in Endothelial Cells. FASEB J. 2006, 20, 518–520. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium Promotes Cell Survival through CaM-K Kinase Activation of the Protein-Kinase-B Pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef]
- Erickson, J.R.; He, B.J.; Grumbach, I.M.; Anderson, M.E. CaMKII in the Cardiovascular System: Sensing Redox States. Physiol. Rev. 2011, 91, 889–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, H.E.; McGarry Houghton, A. Insulin Receptor Substrate Regulation of Phosphoinositide 3-Kinase. Clin. Cancer Res. 2011, 17, 206. [Google Scholar] [CrossRef] [Green Version]
- Stambolic, V.; Suzuki, A.; De La Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative Regulation of PKB/Akt-Dependent Cell Survival by the Tumor Suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Lee, S.-R.; Yang, K.-S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible Oxidation and Inactivation of the Tumor Suppressor PTEN in Cells Stimulated with Peptide Growth Factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox Regulation of PI 3-kinase Signalling via Inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Seo, J.H.; Ahn, Y.; Lee, S.-R.; Yeo, C.Y.; Hur, K.C. The Major Target of the Endogenously Generated Reactive Oxygen Species in Response to Insulin Stimulation Is Phosphatase and Tensin Homolog and Not Phosphoinositide-3 Kinase (PI-3 Kinase) in the PI-3 Kinase/Akt Pathway. Mol. Biol. Cell 2005, 16, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial Reactive Oxygen Species Perturb AKT/Cyclin D1 Cell Cycle Signaling via Oxidative Inactivation of PP2A in Lowdose Irradiated Human Fibroblasts. Oncotarget 2015, 7, 3559–3570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Vogel, R.O.; Puigserver, P. Clk2 and B56β Mediate Insulin-Regulated Assembly of the PP2A Phosphatase Holoenzyme Complex on Akt. Mol. Cell 2011, 41, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical Nodes in Signalling Pathways: Insights into Insulin Action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Haeussler, D.J.; Kumar, V.; Ji, Y.; Pimental, D.R.; Zee, R.S.; Costello, C.E.; Lin, C.; McComb, M.E.; Cohen, R.A.; et al. Oxidation of HRas Cysteine Thiols by Metabolic Stress Prevents Palmitoylation in Vivo and Contributes to Endothelial Cell Apoptosis. FASEB J. 2012, 26, 832–841. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Talukder, M.A.H.; Gao, F. Oxidative Stress and Microvessel Barrier Dysfunction. Front. Physiol. 2020, 11, 472. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Hall, D.A.; Beresford, I.J.M.; Browning, C.; Giles, H. Signalling by CXC-Chemokine Receptors 1 and 2 Expressed in CHO Cells: A Comparison of Calcium Mobilization, Inhibition of Adenylyl Cyclase and Stimulation of GTPγS Binding Induced by IL-8 and GROα. Br. J. Pharmacol. 1999, 126, 810–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reutershan, J.; Morris, M.A.; Burcin, T.L.; Smith, D.F.; Chang, D.; Saprito, M.S.; Ley, K. Critical Role of Endothelial CXCR2 in LPS-Induced Neutrophil Migration into the Lung. J. Clin. Investig. 2006, 116, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.; Hebda, J.K.; Le Guelte, A.; Galan-Moya, E.-M.; Smith, S.S.; Azzi, S.; Bidere, N.; Gavard, J. Glioblastoma Cell-Secreted Interleukin-8 Induces Brain Endothelial Cell Permeability via CXCR2. PLoS ONE 2012, 7, e45562. [Google Scholar] [CrossRef]
- Gündüz, D.; Thom, J.; Hussain, I.; Lopez, D.; Härtel, F.V.; Erdogan, A.; Grebe, M.; Sedding, D.; Piper, H.M.; Tillmanns, H.; et al. Insulin Stabilizes Microvascular Endothelial Barrier Function via Phosphatidylinositol 3-Kinase/Akt-Mediated Rac1 Activation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Siddaramappa Umapathy, N.; Kaczmarek, E.; Fatteh, N.; Burns, N.; Lucas, R.; Stenmark, K.R.; Verin, A.D.; Gerasimovskaya, E.V. Adenosine A1 Receptors Promote Vasa Vasorum Endothelial Cell Barrier Integrity via Gi and Akt-Dependent Actin Cytoskeleton Remodeling. PLoS ONE 2013, 8, e59733. [Google Scholar] [CrossRef] [Green Version]
- Verin, A.D.; Batori, R.; Kovacs-Kasa, A.; Cherian-Shaw, M.; Kumar, S.; Czikora, I.; Karoor, V.; Strassheim, D.; Stenmark, K.R.; Gerasimovskaya, E.V. Extracellular Adenosine Enhances Pulmonary Artery Vasa Vasorum Endothelial Cell Barrier Function via Gi/ELMO1/Rac1/PKA-Dependent Signaling Mechanisms. Am. J. Physiol. Cell Physiol. 2020, 319, C183–C193. [Google Scholar] [CrossRef]
- Aslam, M.; Schluter, K.-D.; Rohrbach, S.; Rafiq, A.; Nazli, S.; Piper, H.M.; Noll, T.; Schulz, R.; Gündüz, D. Hypoxia–Reoxygenation-Induced Endothelial Barrier Failure: Role of RhoA, Rac1 and Myosin Light Chain Kinase. J. Physiol. 2013, 591, 461–473. [Google Scholar] [CrossRef]
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. cAMP Controls the Restoration of Endothelial Barrier Function after Thrombin-induced Hyperpermeability via Rac1 Activation. Physiol. Rep. 2014, 2, e12175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolosova, I.A.; Mirzapoiazova, T.; Adyshev, D.; Usatyuk, P.; Romer, L.H.; Jacobson, J.R.; Natarajan, V.; Pearse, D.B.; Garcia, J.G.; Verin, A.D. Signaling Pathways Involved in Adenosine Triphosphate-Induced Endothelial Cell Barrier Enhancement. Circ. Res. 2005, 97, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bátori, R.; Kumar, S.; Bordán, Z.; Cherian-Shaw, M.; Kovács-Kása, A.; MacDonald, J.A.; Fulton, D.J.; Erdődi, F.; Verin, A.D. Differential Mechanisms of Adenosine-and ATPγS-induced Microvascular Endothelial Barrier Strengthening. J. Cell. Physiol. 2019, 234, 5863–5879. [Google Scholar] [CrossRef]
- Borbiev, T.; Verin, A.D.; Birukova, A.; Liu, F.; Crow, M.T.; Garcia, J.G. Role of CaM Kinase II and ERK Activation in Thrombin-Induced Endothelial Cell Barrier Dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L43–L54. [Google Scholar] [CrossRef] [Green Version]
- Abraham, S.T.; Benscoter, H.A.; Schworer, C.M.; Singer, H.A. A Role for Ca2+/Calmodulin-Dependent Protein Kinase II in the Mitogen-Activated Protein Kinase Signaling Cascade of Cultured Rat Aortic Vascular Smooth Muscle Cells. Circ. Res. 1997, 81, 575–584. [Google Scholar] [CrossRef]
- Heo, J.; Raines, K.W.; Mocanu, V.; Campbell, S.L. Redox Regulation of RhoA. Biochemistry 2006, 45, 14481–14489. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Zhou, B.; Cox, A.D.; Campbell, S.L. Rho GTPases, Oxidation, and Cell Redox Control. Null 2014, 5, e28579. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and Activation of Myosin by Rho-Associated Kinase (Rho-Kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Rokita, A.G.; Anderson, M.E.; Maier, L.S. Redox Regulation of Sodium and Calcium Handling. Antioxid. Redox Signal. 2013, 18, 1063–1077. [Google Scholar] [CrossRef] [Green Version]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-Induced Activation of Type I Protein Kinase A Is Mediated by RI Subunit Interprotein Disulfide Bond Formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishna, R.; Anderson, W.B. Ca2+-and Phospholipid-Independent Activation of Protein Kinase C by Selective Oxidative Modification of the Regulatory Domain. Proc. Natl. Acad. Sci. USA 1989, 86, 6758–6762. [Google Scholar] [CrossRef] [Green Version]
- Klomp, J.E.; Shaaya, M.; Matsche, J.; Rebiai, R.; Aaron, J.S.; Collins, K.B.; Huyot, V.; Gonzalez, A.M.; Muller, W.A.; Chew, T.-L.; et al. Time-Variant SRC Kinase Activation Determines Endothelial Permeability Response. Cell Chem. Biol. 2019, 26, 1081–1094. [Google Scholar] [CrossRef]
- Heppner, D.E.; Dustin, C.M.; Liao, C.; Hristova, M.; Veith, C.; Little, A.C.; Ahlers, B.A.; White, S.L.; Deng, B.; Lam, Y.-W. Direct Cysteine Sulfenylation Drives Activation of the Src Kinase. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Wang, L.; Chiang, E.T.; Simmons, J.T.; Garcia, J.G.N.; Dudek, S.M. FTY720-Induced Human Pulmonary Endothelial Barrier Enhancement Is Mediated by c-Abl. Eur. Respir. J. 2011, 38, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Majumder, P.; Shioya, H.; Wu, F.; Kumar, S.; Weichselbaum, R.; Kharbanda, S.; Kufe, D. Activation of the Cytoplasmic C-Abl Tyrosine Kinase by Reactive Oxygen Species. J. Biol. Chem. 2000, 275, 17237–17240. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Zhang, G.; Welch, E.J.; Liang, Y.; Fu, J.; Vogel, S.M.; Lowell, C.A.; Du, X.; Cheresh, D.A.; Malik, A.B.; et al. A Critical Role for Lyn Kinase in Strengthening Endothelial Integrity and Barrier Function. Blood 2013, 122, 4140–4149. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.K.; Starnes, T.W.; Deng, Q.; Huttenlocher, A. Lyn Is a Redox Sensor That Mediates Leukocyte Wound Attraction in Vivo. Nature 2011, 480, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Patrushev, N.; Inomata, H.; Mehta, D.; Urao, N.; Kim, H.W.; Razvi, M.; Kini, V.; Mahadev, K.; Goldstein, B.J.; et al. Role of Protein Tyrosine Phosphatase 1B in Vascular Endothelial Growth Factor Signaling and Cell–Cell Adhesions in Endothelial Cells. Circ. Res. 2008, 102, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [Green Version]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A Mechanosensory Complex That Mediates the Endothelial Cell Response to Fluid Shear Stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Niu, N.; Xu, S.; Xu, Y.; Little, P.J.; Jin, Z.-G. Targeting Mechanosensitive Transcription Factors in Atherosclerosis. Trends Pharmacol. Sci. 2019, 40, 253–266. [Google Scholar] [CrossRef]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef] [Green Version]
- Feelisch, M.; Akaike, T.; Griffiths, K.; Ida, T.; Prysyazhna, O.; Goodwin, J.J.; Gollop, N.D.; Fernandez, B.O.; Minnion, M.; Cortese-Krott, M.M. Long-Lasting Blood Pressure Lowering Effects of Nitrite Are NO-Independent and Mediated by Hydrogen Peroxide, Persulfides, and Oxidation of Protein Kinase G1α Redox Signalling. Cardiovasc. Res. 2020, 116, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine Redox Sensor in PKGIa Enables Oxidant-Induced Activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. Targeting the Microcirculation by Indole-3-Carbinol and Its Main Derivate 3,3,′-Diindolylmethane: Effects on Angiogenesis, Thrombosis and Inflammation. Mini Rev. Med. Chem. 2018, 18, 962–968. [Google Scholar] [CrossRef]
- Ampofo, E.; Berg, J.J.; Menger, M.D.; Laschke, M.W. Maslinic Acid Alleviates Ischemia/Reperfusion-Induced Inflammation by Downregulation of NFκB-Mediated Adhesion Molecule Expression. Sci. Rep. 2019, 9, 6119. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, K.A.; Schwartz, M.A. Ion Channels in Endothelial Responses to Fluid Shear Stress. Physiology 2016, 31, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Veit, F.; Pak, O.; Brandes, R.P.; Weissmann, N. Hypoxia-Dependent Reactive Oxygen Species Signaling in the Pulmonary Circulation: Focus on Ion Channels. Antioxid. Redox Signal. 2014, 22, 537–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, P.; Folgering, J.; Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Bowman, C.; Bichet, D.; Patel, A.; Sachs, F. Revisiting TRPC1 and TRPC6 Mechanosensitivity. Pflügers Arch. Eur. J. Physiol. 2008, 455, 1097–1103. [Google Scholar] [CrossRef]
- Inoue, R.; Jensen, L.J.; Jian, Z.; Shi, J.; Hai, L.; Lurie, A.I.; Henriksen, F.H.; Salomonsson, M.; Morita, H.; Kawarabayashi, Y.; et al. Synergistic Activation of Vascular TRPC6 Channel by Receptor and Mechanical Stimulation via Phospholipase C/Diacylglycerol and Phospholipase A2/ω-Hydroxylase/20-HETE Pathways. Circ. Res. 2009, 104, 1399–1409. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.; Ding, M.; Ding, Y.; Sours-Brothers, S.; Luchowski, R.; Gryczynski, Z.; Yorio, T.; Ma, H.; Ma, R. Canonical Transient Receptor Potential 6 (TRPC6), a Redox-Regulated Cation Channel. J. Biol. Chem. 2010, 285, 23466–23476. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.-C.; Song, X.; Lu, X.-Y.; Li, D.T.; Eaton, D.C.; Shen, B.-Z.; Li, X.-Q.; Ma, H.-P. High Glucose Induces Podocyte Apoptosis by Stimulating TRPC6 via Elevation of Reactive Oxygen Species. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 1434–1442. [Google Scholar] [CrossRef] [Green Version]
- Hool, L.C. Evidence for the Regulation of L-Type Ca2+ Channels in the Heart by Reactive Oxygen Species–Mechanism for Mediating Pathology. Clin. Exp. Pharmacol. Physiol.. 2008, 35, 229–234. [Google Scholar] [CrossRef]
- Keeley, T.P.; Siow, R.C.M.; Jacob, R.; Mann, G.E. A PP2A-Mediated Feedback Mechanism Controls Ca2+-Dependent NO Synthesis under Physiological Oxygen. FASEB J. 2017, 31, 5172–5183. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of Nitric Oxide Synthase in Endothelial Cells by Akt-Dependent Phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudi, S.R.; Clark, C.B.; Frangos, J.A. Fluid Flow Rapidly Activates G Proteins in Human Endothelial Cells. Circ. Res. 1996, 79, 834–839. [Google Scholar] [CrossRef]
- Gudi, S.; Nolan, J.P.; Frangos, J.A. Modulation of GTPase Activity of G Proteins by Fluid Shear Stress and Phospholipid Composition. Proc. Natl. Acad. Sci. USA 1998, 95, 2515–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyckoff, M.H.; Chambliss, K.L.; Mineo, C.; Yuhanna, I.S.; Mendelsohn, M.E.; Mumby, S.M.; Shaul, P.W. Plasma Membrane Estrogen Receptors Are Coupled to Endothelial Nitric-Oxide Synthase through Gαi. J. Biol. Chem. 2001, 276, 27071–27076. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Dillon, J.S. Dehydroepiandrosterone Activates Endothelial Cell Nitric-Oxide Synthase by a Specific Plasma Membrane Receptor Coupled to Gαi2,3. J. Biol. Chem. 2002, 277, 21379–21388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.R.; Prossnitz, E.R.; Barton, M. The G Protein-Coupled Estrogen Receptor GPER/GPR30 as a Regulator of Cardiovascular Function. Vasc. Pharmacol. 2011, 55, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Chambliss, K.L.; Yuhanna, I.S.; Mineo, C.; Liu, P.; German, Z.; Sherman, T.S.; Mendelsohn, M.E.; Anderson, R.G.W.; Shaul, P.W. Estrogen Receptor α and Endothelial Nitric Oxide Synthase Are Organized Into a Functional Signaling Module in Caveolae. Circ. Res. 2000, 87, e44–e52. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Libby Peter Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [CrossRef] [Green Version]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Kvietys, P.R.; Granger, D.N. Role of Reactive Oxygen and Nitrogen Species in the Vascular Responses to Inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.M.; Larman, H.B.; Dai, G.; Zhang, Y.; Wang, E.T.; Moorthy, S.N.; Kratz, J.R.; Lin, Z.; Jain, M.K.; Gimbrone Jr, M.A. Integration of Flow-Dependent Endothelial Phenotypes by Kruppel-like Factor 2. J. Clin. Investig. 2006, 116, 49–58. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.; Lim, J.H.; Lee, C.; Choi, H.C.; Woo, C.-H. Laminar Flow Activation of ERK5 Protein in Vascular Endothelium Leads to Atheroprotective Effect via NF-E2-Related Factor 2 (Nrf2) Activation*. J. Biol. Chem. 2012, 287, 40722–40731. [Google Scholar] [CrossRef] [Green Version]
- Suzaki, Y.; Yoshizumi, M.; Kagami, S.; Koyama, A.H.; Taketani, Y.; Houchi, H.; Tsuchiya, K.; Takeda, E.; Tamaki, T. Hydrogen Peroxide Stimulates C-Src-Mediated Big Mitogen-Activated Protein Kinase 1 (BMK1) and the MEF2C Signaling Pathway in PC12 Cells: POTENTIAL ROLE IN CELL SURVIVAL FOLLOWING OXIDATIVE INSULTS. J. Biol. Chem. 2002, 277, 9614–9621. [Google Scholar] [CrossRef] [Green Version]
- Abe, J.; Takahashi, M.; Ishida, M.; Lee, J.-D.; Berk, B.C. c-Src Is Required for Oxidative Stress-Mediated Activation of Big Mitogen-Activated Protein Kinase 1 (BMK1). J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Takahashi, M.; Okuda, M.; Lee, J.; Berk, B. Fluid Shear Stress Stimulates Big Mitogen-Activated Protein Kinase 1 (BMK1) Activity in Endothelial Cells. Dependence on Tyrosine Kinases and Intracellular Calcium. J. Biol. Chem. 1999, 274, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaike, M.; Che, W.; Marmarosh, N.-L.; Ohta, S.; Osawa, M.; Ding, B.; Berk, B.C.; Yan, C.; Abe, J. The Hinge-Helix 1 Region of Peroxisome Proliferator-Activated Receptor Γ1 (PPARγ1) Mediates Interaction with Extracellular Signal-Regulated Kinase 5 and PPARγ1 Transcriptional Activation: Involvement in Flow-Induced PPARγ Activation in Endothelial Cells. Mol. Cell. Biol. 2004, 24, 8691–8704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, G.B.; Jain, M.K. Role of Krüppel-Like Transcription Factors in Endothelial Biology. Circ. Res. 2007, 100, 1686–1695. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian Krüppel-like Factors in Health and Diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Dekker, R.J.; van Thienen, J.V.; Rohlena, J.; de Jager, S.C.; Elderkamp, Y.W.; Seppen, J.; de Vries, C.J.M.; Biessen, E.A.L.; van Berkel, T.J.C.; Pannekoek, H.; et al. Endothelial KLF2 Links Local Arterial Shear Stress Levels to the Expression of Vascular Tone-Regulating Genes. Am. J. Pathol. 2005, 167, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Zhao, M.; Morikawa, A.; Sugiyama, T.; Chakravortty, D.; Koide, N.; Yoshida, T.; Tapping, R.I.; Yang, Y.; Yokochi, T. Big Mitogen-Activated Kinase Regulates Multiple Members of the MEF2 Protein Family. J. Biol. Chem. 2000, 275, 18534–18540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Yoshida, T.; Wu, L.; Maiti, D.; Cebotaru, L.; Duh, E.J. Transcription Factor MEF2C Suppresses Endothelial Cell Inflammation via Regulation of NF-ΚB and KLF2. J. Cell. Physiol. 2015, 230, 1310–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen Peroxide Sensing, Signaling and Regulation of Transcription Factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Flohé, L. Basic Principles and Emerging Concepts in the Redox Control of Transcription Factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.B.; Chowdhury, N.S.; Sohrab, M.H.; Rana, M.S.; Hasan, C.M.; Lee, S.-H. Cerevisterol Alleviates Inflammation via Suppression of MAPK/NF-ΚB/AP-1 and Activation of the Nrf2/HO-1 Signaling Cascade. Biomolecules 2020, 10, 199. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase to Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Siow, R.C.M.; Mann, G.E. Impaired Redox Signaling and Antioxidant Gene Expression in Endothelial Cells in Diabetes: A Role for Mitochondria and the Nuclear Factor-E2-Related Factor 2-Kelch-Like ECH-Associated Protein 1 Defense Pathway. Antioxid. Redox Signal. 2011, 14, 469–487. [Google Scholar] [CrossRef]
- Yang, K.; Song, H.; Yin, D. PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis via Activating Nrf2. J. Cardiovasc. Pharmacol. 2021, 77, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, Y.; Xu, K.; Lin, L. Protective Effects of 1,25 Dihydroxyvitamin D3 against High-Glucose-Induced Damage in Human Umbilical Vein Endothelial Cells Involve Activation of Nrf2 Antioxidant Signaling. J. Vasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARγ Signaling and Emerging Opportunities for Improved Therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Verna, L.; Chen, N.-G.; Chen, J.; Li, H.; Forman, B.M.; Stemerman, M.B. Constitutive Activation of Peroxisome Proliferator-Activated Receptor-γ Suppresses Pro-Inflammatory Adhesion Molecules in Human Vascular Endothelial Cells. J. Biol. Chem. 2002, 277, 34176–34181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W.R. Structural Basis for the Activation of PPARγ by Oxidized Fatty Acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD (P) H Oxidase: Role in Cardiovascular Biology and Disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M. Redox Signaling in Angiogenesis: Role of NADPH Oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Youn, S.-W.; Li, Y.; Kim, Y.-M.; Sudhahar, V.; Abdelsaid, K.; Kim, H.W.; Liu, Y.; Fulton, D.J.R.; Ashraf, M.; Tang, Y.; et al. Modification of Cardiac Progenitor Cell-Derived Exosomes by MiR-322 Provides Protection against Myocardial Infarction through Nox2-Dependent Angiogenesis. Antioxidants 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Harijith, A.; Natarajan, V.; Fu, P. The Role of Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Lung Architecture Remodeling. Antioxidants 2017, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Manuneedhi Cholan, P.; Cartland, S.P.; Kavurma, M.M. NADPH Oxidases, Angiogenesis, and Peripheral Artery Disease. Antioxidants 2017, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, L.; Zhou, H.J.; Min, W. The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk. Antioxidants 2017, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hartnett, M.E. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants 2017, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Bermejo, R.; Hernández-Hernández, A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants 2017, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Ushio–Fukai, M. VEGF Signaling through NADPH Oxidase-Derived ROS. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Compartmentalization of Redox Signaling Through NADPH Oxidase–Derived ROS. Antioxid. Redox Signal. 2008, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, M.; Lemtalsi, T.; Toque, H.A.; Xu, Z.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Kaplan, N.; Urao, N.; Furuta, E.; Kim, S.-J.; Razvi, M.; Nakamura, Y.; McKinney, R.D.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Localized Cysteine Sulfenic Acid Formation by Vascular Endothelial Growth Factor: Role in Endothelial Cell Migration and Angiogenesis. Free Radic. Res. 2011, 45, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Colavitti, R.; Pani, G.; Bedogni, B.; Anzevino, R.; Borrello, S.; Waltenberger, J.; Galeotti, T. Reactive Oxygen Species as Downstream Mediators of Angiogenic Signaling by Vascular Endothelial Growth Factor Receptor-2/KDR*. J. Biol. Chem. 2002, 277, 3101–3108. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Jiang, W.G.; Ahmed, A.; Boulton, M. Vascular Endothelial Growth Factor-Induced Endothelial Cell Proliferation Is Regulated by Interaction between VEGFR-2, SH-PTP1 and ENOS. Microvasc. Res. 2006, 71, 20–31. [Google Scholar] [CrossRef]
- Abdelsaid, M.A.; El-Remessy, A.B. S-Glutathionylation of LMW-PTP Regulates VEGF-Mediated FAK Activation and Endothelial Cell Migration. J. Cell Sci. 2012, 125, 4751–4760. [Google Scholar] [CrossRef] [Green Version]
- Oshikawa, J.; Urao, N.; Kim, H.W.; Kaplan, N.; Razvi, M.; McKinney, R.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Extracellular SOD-Derived H 2 O 2 Promotes VEGF Signaling in Caveolae/Lipid Rafts and Post-Ischemic Angiogenesis in Mice. PLoS ONE 2010, 5, e10189. [Google Scholar] [CrossRef] [Green Version]
- Urao, N.; McKinney, R.D.; Fukai, T.; Ushio-Fukai, M. NADPH Oxidase 2 Regulates Bone Marrow Microenvironment Following Hindlimb Ischemia: Role in Reparative Mobilization of Progenitor Cells. Stem Cells 2012, 30, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harel, S.; Mayaki, D.; Sanchez, V.; Hussain, S.N.A. NOX2, NOX4, and Mitochondrial-Derived Reactive Oxygen Species Contribute to Angiopoietin-1 Signaling and Angiogenic Responses in Endothelial Cells. Vasc. Pharmacol. 2017, 92, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-Induced ROS Release Orchestrated by Nox4, Nox2, and Mitochondria in VEGF Signaling and Angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agonist | Receptor(s) | Coupling of Receptor to Gi Proteins | Effect on EC Survival and Apoptosis | ||
|---|---|---|---|---|---|
| Pro-Survival or Pro-Apoptotic, EC Type | Signaling Pathway | Reference(s) | |||
| Adenosine | A1AR | [28] | Pro-survival, HUVEC | PI3K—Akt | [29] |
| Anandamide | CB1, CB2 | [30] | Pro-apoptotic, HUVEC | Activation of JNK and p38 but not ERK | [31] |
| Anandamide | CB1 | [30] | Pro-apoptotic, HCAEC | JNK and p38 | [32] |
| Ghrelin gene products | GHS-R1a | Gq/11, Gi [33] | Pro-survival, human pancreatic islet microvascular ECs | AC–cAMP–PKA | [27] |
| S1P | S1P1 | Gi [34] | Pro-survival, HUVEC | ERK1/2 | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barvitenko, N.; Skverchinskaya, E.; Lawen, A.; Matteucci, E.; Saldanha, C.; Uras, G.; Manca, A.; Aslam, M.; Pantaleo, A. Pleiotropic and Potentially Beneficial Effects of Reactive Oxygen Species on the Intracellular Signaling Pathways in Endothelial Cells. Antioxidants 2021, 10, 904. https://doi.org/10.3390/antiox10060904
Barvitenko N, Skverchinskaya E, Lawen A, Matteucci E, Saldanha C, Uras G, Manca A, Aslam M, Pantaleo A. Pleiotropic and Potentially Beneficial Effects of Reactive Oxygen Species on the Intracellular Signaling Pathways in Endothelial Cells. Antioxidants. 2021; 10(6):904. https://doi.org/10.3390/antiox10060904
Chicago/Turabian StyleBarvitenko, Nadezhda, Elisaveta Skverchinskaya, Alfons Lawen, Elena Matteucci, Carlota Saldanha, Giuseppe Uras, Alessia Manca, Muhammad Aslam, and Antonella Pantaleo. 2021. "Pleiotropic and Potentially Beneficial Effects of Reactive Oxygen Species on the Intracellular Signaling Pathways in Endothelial Cells" Antioxidants 10, no. 6: 904. https://doi.org/10.3390/antiox10060904
APA StyleBarvitenko, N., Skverchinskaya, E., Lawen, A., Matteucci, E., Saldanha, C., Uras, G., Manca, A., Aslam, M., & Pantaleo, A. (2021). Pleiotropic and Potentially Beneficial Effects of Reactive Oxygen Species on the Intracellular Signaling Pathways in Endothelial Cells. Antioxidants, 10(6), 904. https://doi.org/10.3390/antiox10060904