
Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity

 ,
, 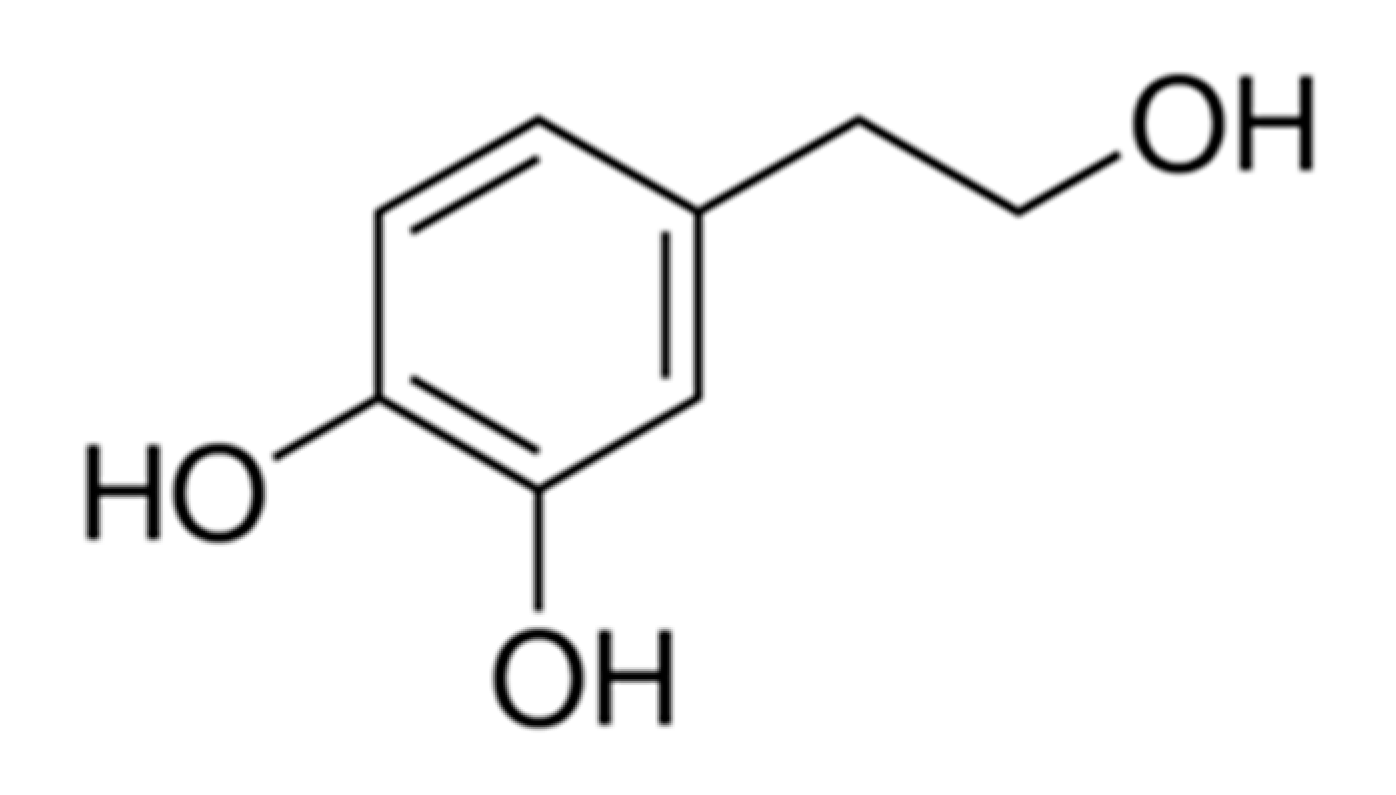{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Insulin Preparation and Glycation
2.3. Fluorescence Measurements
2.4. Cellular Cultures and Treatments
2.5. MTT Assay
2.6. Detection of Intracellular ROS
2.7. Immunoblotting
2.8. Statistical Analysis
3. Results
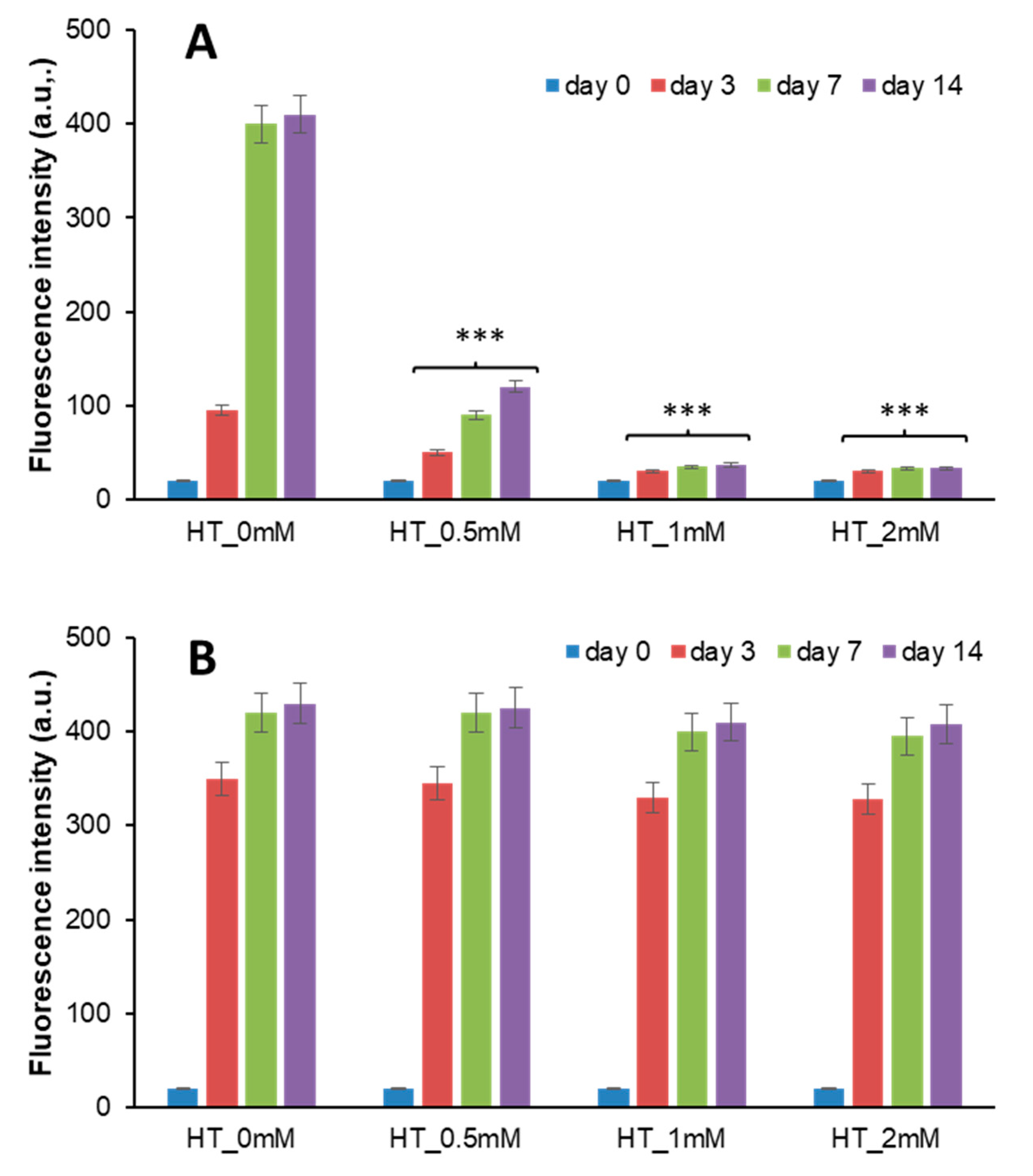
3.1. HT Effect on Insulin-AGE Formation and Cytotoxicity
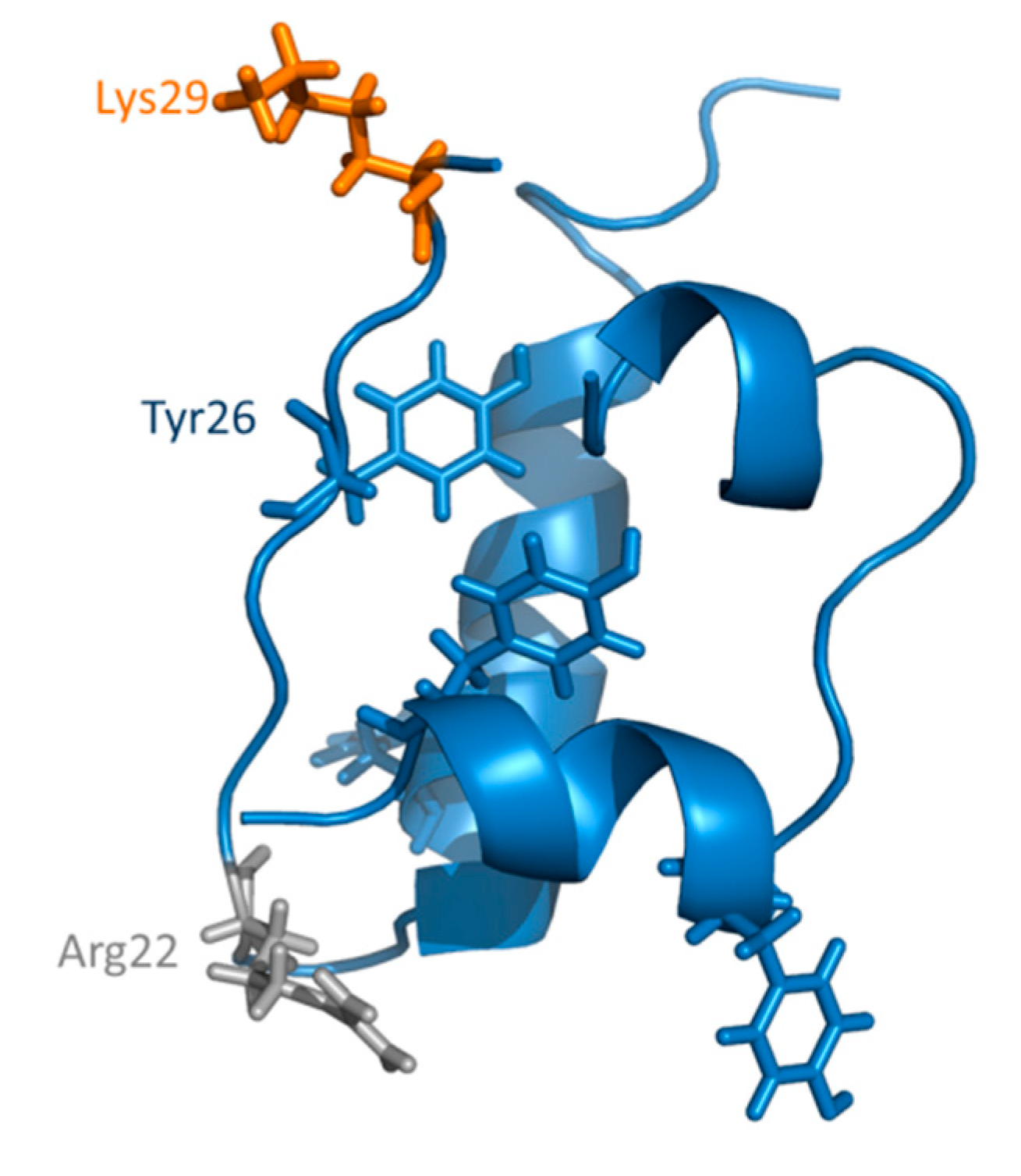
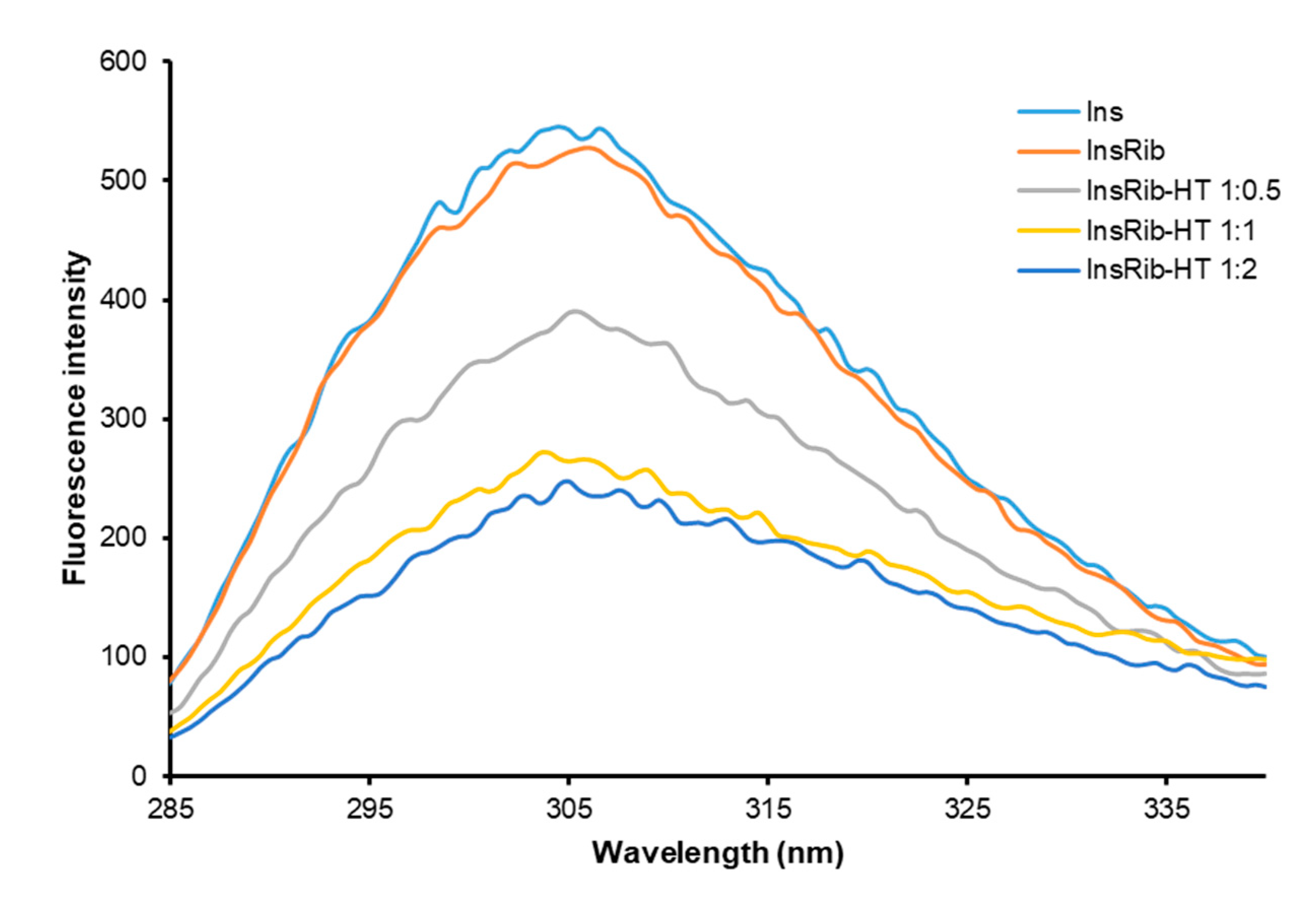
3.2. HT-Insulin Interaction in AGE Formation
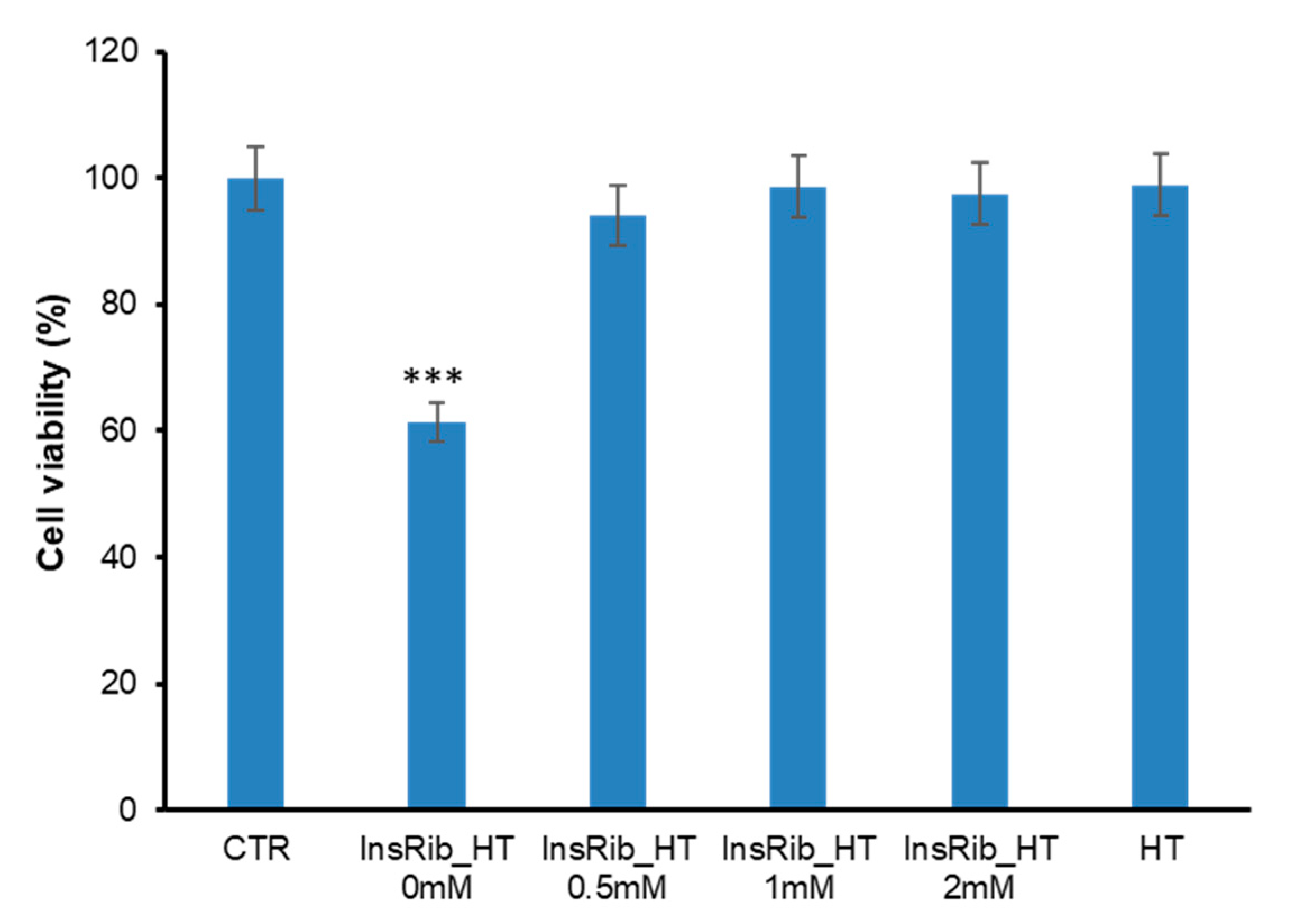
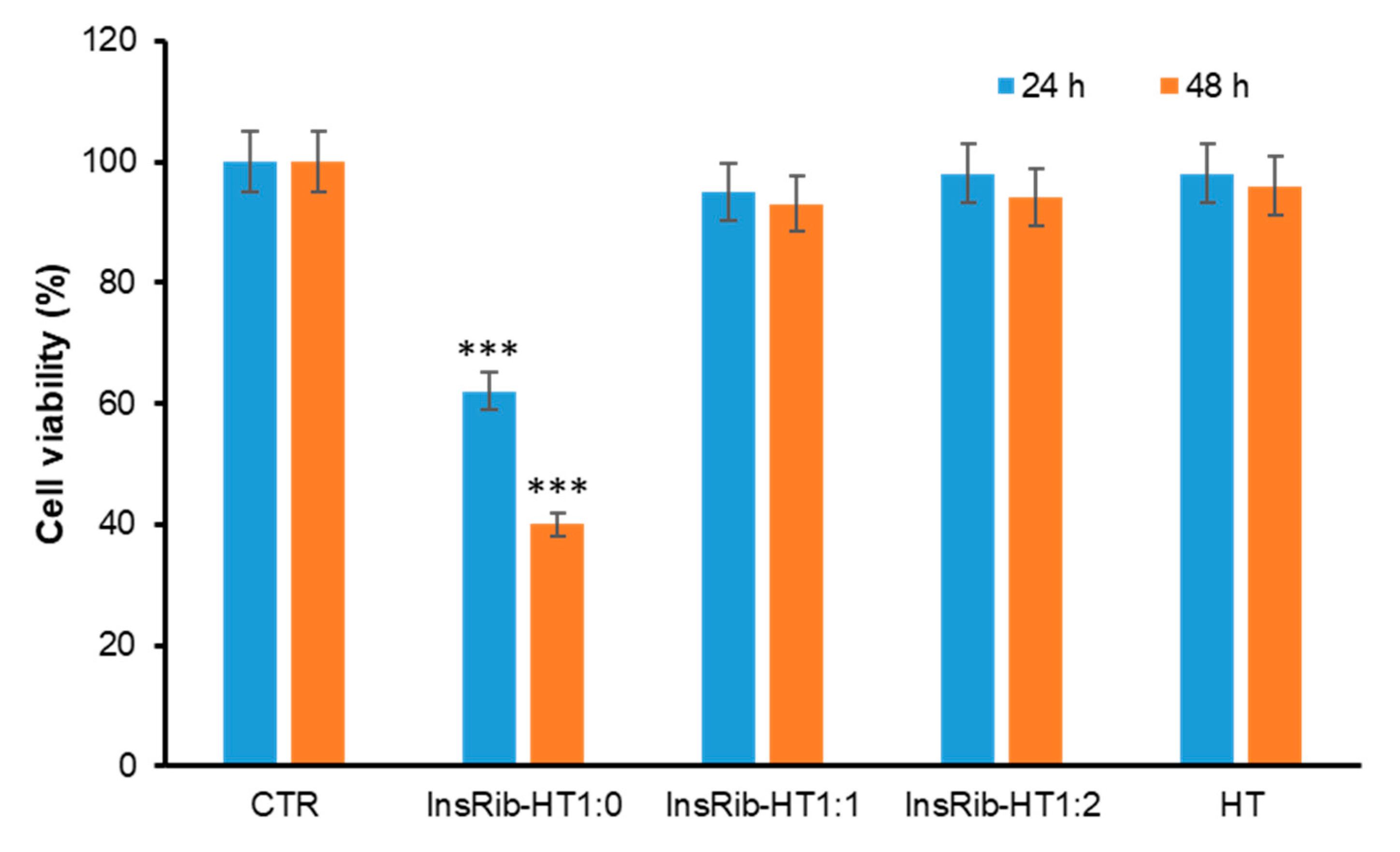
3.3. HT Protective Effect in AGE-Induced Toxicity
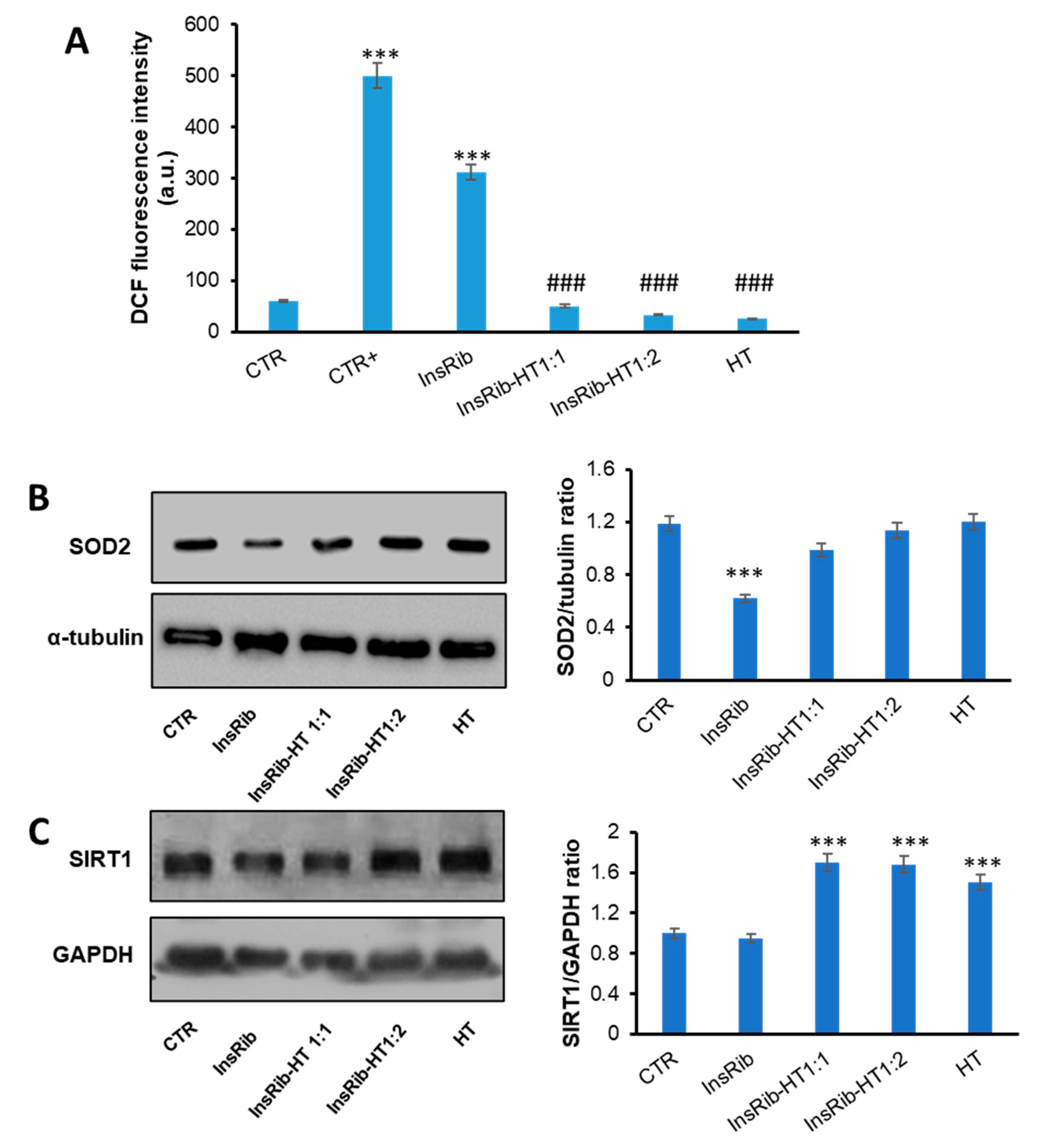
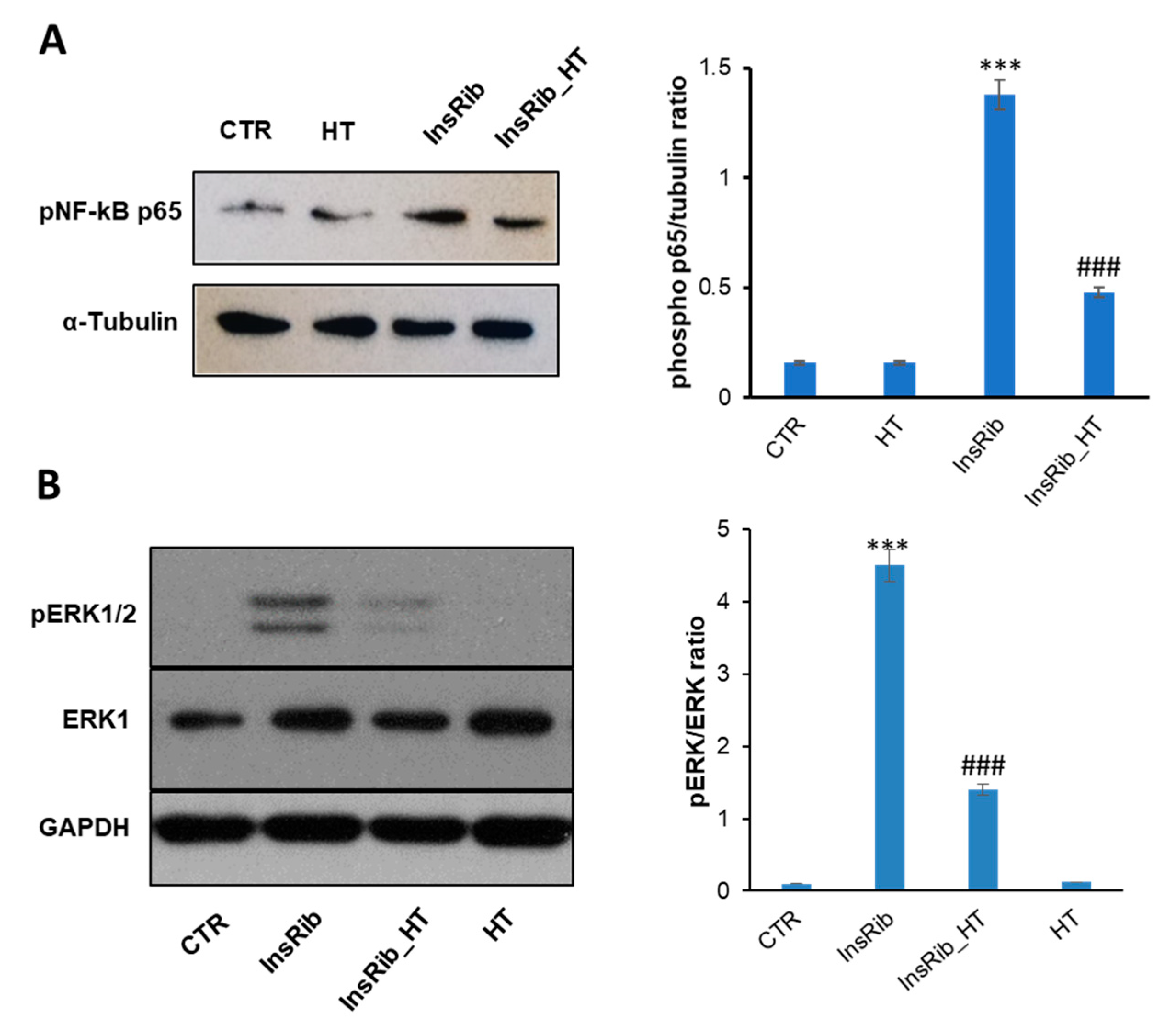
3.4. HT Protective Effect on AGE-Induced Oxidative Stress and Inflammatory Pathways
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef] [Green Version]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Hashimoto, K.; Kunikata, H.; Yasuda, M.; Ito, A.; Aizawa, N.; Sawada, S.; Kondo, K.; Satake, C.; Takano, Y.; Nishiguchi, K.M.; et al. The relationship between advanced glycation end products and ocular circulation in type 2 diabetes. J. Diabetes Compl. 2016, 30, 1371–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grillo, M.A.; Colombatto, S. Advanced glycation end-products (AGEs): Involvement in aging and in neurodegenerative diseases. Amino Acids 2007, 35, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Farhan, M. Impact of Non-Enzymatic Glycation in Neurodegenerative Diseases: Role of Natural Products in Prevention. Adv. Neurobiol. 2016, 12, 125–151. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, P.; Rabbani, G.; Khan, R.H. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19, 407–437. [Google Scholar] [CrossRef]
- Hrelia, P.; Sita, G.; Ziche, M.; Ristori, E.; Marino, A.; Cordaro, M.; Molteni, R.; Spero, V.; Malaguti, M.; Morroni, F.; et al. Common Protective Strategies in Neurodegenerative Disease: Focusing on Risk Factors to Target the Cellular Redox System. Oxidative Med. Cell. Longev. 2020, 2020, 8363245. [Google Scholar] [CrossRef]
- Takeuchi, M. Possible Involvement of Advanced Glycation End-Products (AGEs) in the Pathogenesis of Alzheimers Disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Katare, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10, S144–S149. [Google Scholar] [CrossRef] [PubMed]
- Batkulwar, K.; Godbole, R.K.; Banarjee, R.M.; Kassaar, O.M.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef]
- Jash, K.; Gondaliya, P.; Kirave, P.; Kulkarni, B.; Sunkaria, A.; Kalia, K. Cognitive dysfunction: A growing link between diabetes and Alzheimer’s disease. Drug Dev. Res. 2020, 81, 144–164. [Google Scholar] [CrossRef]
- Chou, P.-S.; Wu, M.-N.; Yang, C.-C.; Shen, C.-T.; Yang, Y.-H. Effect of Advanced Glycation End Products on the Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: The Receptor for Advanced Glycation End Products (RAGE). Front. Aging Neurosci. 2020, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Nass, N.; Bartling, B.; Navarrete Santos, A.; Scheubel, R.J.; Börgermann, J.; Silber, R.E.; Simm, A. Advanced glycation end products, diabetes and ageing. Z. Gerontol. Geriatr. 2007, 40, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Perkins, R.K.; Miranda, E.R.; Karstoft, K.; Beisswenger, P.J.; Solomon, T.P.J.; Haus, J.M. Experimental Hyperglycemia Alters Circulating Concentrations and Renal Clearance of Oxidative and Advanced Glycation End Products in Healthy Obese Humans. Nutrients 2019, 11, 532. [Google Scholar] [CrossRef] [Green Version]
- Wautier, J.-L.; Schmidt, A.M. Protein Glycation: A firm link to endothelial cell dysfunction. Circ. Res. 2004, 95, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. Differential effects of glycation on protein aggregation and amyloid formation. Front. Mol. Biosci. 2014, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [Green Version]
- Münch, G.; Mayer, S.; Michaelis, J.; Hipkiss, A.R.; Riederer, P.; Müller, R.; Neumann, A.; Schinzel, R.; Cunningham, A.M. Influence of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of β-amyloid peptide. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1997, 1360, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Ogata, A.; Shinpo, K.; Moriwaka, F.; Fujii, J.; Taniguchi, N.; Tashiro, K. Detection of an Amadori product, 1-hexitol-lysine, in the anterior horn of the amyotrophic lateral sclerosis and spinobulbar muscular atrophy spinal cord: Evidence for early involvement of glycation in motoneuron diseases. Acta Neuropathol. 2000, 99, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Carafa, V.; Altucci, L.; Irace, G.; Borriello, M.; Vinciguerra, R.; Sirangelo, I. Glycation of Wild-Type Apomyoglobin Induces Formation of Highly Cytotoxic Oligomeric Species. J. Cell. Physiol. 2015, 230, 2807–2820. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Vella, F.M.; Irace, G.; Manco, G.; Iannuzzi, C. Glycation in Demetalated Superoxide Dismutase 1 Prevents Amyloid Aggregation and Produces Cytotoxic Ages Adducts. Front. Mol. Biosci. 2016, 3, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soman, S.; Raju, R.; Sandhya, V.K.; Advani, J.; Khan, A.A.; Harsha, H.C.; Prasad, T.S.K.; Sudhakaran, P.R.; Pandey, A.; Adishesha, P.K. A multicellular signal transduction network of AGE/RAGE signaling. J. Cell Commun. Signal. 2012, 7, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Prasad, K.; Tiwari, S. Therapeutic Interventions for Advanced Glycation-End Products and its Receptor- Mediated Cardiovascular Disease. Curr. Pharm. Des. 2017, 23, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Knani, I.; Bouzidi, H.; Zrour, S.; Bergaoui, N.; Hammami, M.; Kerkeni, M. Increased serum concentrations of Nɛ-carboxymethyllysine are related to the presence and the severity of rheumatoid arthritis. Ann. Clin. Biochem. Int. J. Lab. Med. 2017, 55, 430–436. [Google Scholar] [CrossRef]
- Prasad, K. AGE–RAGE Stress in the Pathophysiology of Atrial Fibrillation and Its Treatment. Int. J. Angiol. 2020, 29, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Waghela, B.N.; Vaidya, F.U.; Ranjan, K.; Chhipa, A.S.; Tiwari, B.S.; Pathak, C. AGE-RAGE synergy influences programmed cell death signaling to promote cancer. Mol. Cell. Biochem. 2021, 476, 585–598. [Google Scholar] [CrossRef]
- Juranek, J.; Ray, R.; Banach, M.; Rai, V. Receptor for advanced glycation end-products in neurodegenerative diseases. Rev. Neurosci. 2015, 26, 691–698. [Google Scholar] [CrossRef]
- Abate, G.; Marziano, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Nutrition and AGE-ing: Focusing on Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buelna-Chontal, M.; Zazueta, C. Redox activation of Nrf2 & NF-κB: A double end sword? Cell. Signal. 2013, 25, 2548–2557. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for Advanced Glycation End Products and Its Involvement in Inflammatory Diseases. Int. J. Inflamm. 2013, 2013, 403460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Hernandez, M.M.A. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, W.; Zhou, Q.; Jie, J.; Chen, X.; Wang, F.; Gong, X. Retracted: Advanced glycation end-products induce oxidative stress through the Sirt1/Nrf2 axis by interacting with the receptor of AGEs under diabetic conditions. J. Cell. Biochem. 2019, 120, 2159–2170. [Google Scholar] [CrossRef]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef]
- Shen, C.-Y.; Lu, C.-H.; Wu, C.-H.; Li, K.-J.; Kuo, Y.-M.; Hsieh, S.-C.; Yu, C.-L. The Development of Maillard Reaction, and Advanced Glycation End Product (AGE)-Receptor for AGE (RAGE) Signaling Inhibitors as Novel Therapeutic Strategies for Patients with AGE-Related Diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Rupasinghe, H.P.V. Polyphenols: Multipotent Therapeutic Agents in Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2013, 2013, 891748. [Google Scholar] [CrossRef] [Green Version]
- Sadowska-Bartosz, I.; Bartosz, G. Prevention of Protein Glycation by Natural Compounds. Molecules 2015, 20, 3309–3334. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Irace, G.; Cammarota, M.; Di Maro, A.; Sirangelo, I. Vanillin Affects Amyloid Aggregation and Non-Enzymatic Glycation in Human Insulin. Sci. Rep. 2017, 7, 15086. [Google Scholar] [CrossRef] [Green Version]
- Yeh, W.-J.; Hsia, S.-M.; Lee, W.-H.; Wu, C.-H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef] [PubMed]
- González, I.; Morales, M.A.; Rojas, A. Polyphenols and AGEs/RAGE axis. Trends and challenges. Food Res. Int. 2020, 129, 108843. [Google Scholar] [CrossRef]
- Peng, X.; Ma, J.; Chen, F.; Wang, M. Naturally occurring inhibitors against the formation of advanced glycation end-products. Food Funct. 2011, 2, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; He, X.-W.; Jiang, J.-G.; Xu, X.-L. Hydroxytyrosol and Its Potential Therapeutic Effects. J. Agric. Food Chem. 2014, 62, 1449–1455. [Google Scholar] [CrossRef]
- Rodríguez-Morató, J.; Xicota, L.; Fitó, M.; Farré, M.; Dierssen, M.; De La Torre, R. Potential Role of Olive Oil Phenolic Compounds in the Prevention of Neurodegenerative Diseases. Molecules 2015, 20, 4655–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles-Almazan, M.; Pulido-Moran, M.; Moreno-Fernandez, J.; Ramirez-Tortosa, C.; Rodriguez-Garcia, C.; Quiles, J.L.; Ramirez-Tortosa, M. Hydroxytyrosol: Bioavailability, toxicity, and clinical applications. Food Res. Int. 2018, 105, 654–667. [Google Scholar] [CrossRef]
- Karković Marković, A.; Torić, J.; Barbarić, M.; Jakobušić Brala, C. Hydroxytyrosol, Tyrosol and Derivatives and Their Potential Effects on Human Health. Molecules 2019, 24, 2001. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, M.; Kiani, A.K.; Paolacci, S.; Manara, E.; Kurti, D.; Dhuli, K.; Bushati, V.; Miertus, J.; Pangallo, D.; Baglivo, M.; et al. Hydroxytyrosol: A natural compound with promising pharmacological activities. J. Biotechnol. 2020, 309, 29–33. [Google Scholar] [CrossRef]
- Navarro, M.; Morales, F.J. Effect of hydroxytyrosol and olive leaf extract on 1,2-dicarbonyl compounds, hydroxymethylfurfural and advanced glycation endproducts in a biscuit model. Food Chem. 2017, 217, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal Modification of Protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, J.R.; McKillop, A.M.; Mooney, M.H.; O’Harte, F.P.M.; Bell, P.M.; Flatt, P. Demonstration of increased concentrations of circulating glycated insulin in human Type 2 diabetes using a novel and specific radioimmunoassay. Diabetologia 2003, 46, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.J.; Boyd, A.C.; O’Harte, F.; McKillop, A.M.; Wiggam, M.I.; Mooney, M.H.; McCluskey, J.T.; Lindsay, J.R.; Ennis, C.N.; Gamble, R.; et al. Demonstration of Glycated Insulin in Human Diabetic Plasma and Decreased Biological Activity Assessed by Euglycemic-Hyperinsulinemic Clamp Technique in Humans. Diabetes 2003, 52, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.; Olson, D.J.H.; Ross, A.R.S.; Wu, L. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 2006, 20, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.M.; Lages, A.; Gomes, R.A.; Neves, H.; Família, C.; Coelho, A.V.; Quintas, A. Insulin glycation by methylglyoxal results in native-like aggregation and inhibition of fibril formation. BMC Biochem. 2011, 12, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalkwijk, C.G. Vascular AGE-ing by methylglyoxal: The past, the present and the future. Diabetologia 2015, 58, 1715–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, C.; Borriello, M.; Carafa, V.; Altucci, L.; Vitiello, M.; Balestrieri, M.L.; Ricci, G.; Irace, G.; Sirangelo, I. D-ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, L.H.C.; Birch, D.J.S.; Vyshemirsky, V.; Ryadnov, M.G.; Rolinski, O.J. Tracking Insulin Glycation in Real Time by Time-Resolved Emission Spectroscopy. J. Phys. Chem. B 2019, 123, 7812–7817. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Portaccio, M.; Irace, G.; Sirangelo, I. Insights into Insulin Fibril Assembly at Physiological and Acidic pH and Related Amyloid Intrinsic Fluorescence. Int. J. Mol. Sci. 2017, 18, 2551. [Google Scholar] [CrossRef] [Green Version]
- Matiacevich, S.B.; Buera, M.P. A critical evaluation of fluorescence as a potential marker for the Maillard reaction. Food Chem. 2005, 95, 423–430. [Google Scholar] [CrossRef]
- Borriello, M.; Iannuzzi, C.; Sirangelo, I. Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin. Cells 2019, 8, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirangelo, I.; Borriello, M.; Vilasi, S.; Iannuzzi, C. Hydroxytyrosol Inhibits Protein Oligomerization and Amyloid Aggregation in Human Insulin. Int. J. Mol. Sci. 2020, 21, 4636. [Google Scholar] [CrossRef] [PubMed]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; D’Agostino, A.; Cimini, D.; Schiraldi, C.; Sirangelo, I. Protective effect of extractive and biotechnological chondroitin in insulin amyloid and advanced glycation end product-induced toxicity. J. Cell. Physiol. 2019, 234, 3814–3828. [Google Scholar] [CrossRef]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [Green Version]
- De Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858. [Google Scholar] [CrossRef]
- Chhipa, A.S.; Borse, S.P.; Baksi, R.; Lalotra, S.; Nivsarkar, M. Targeting receptors of advanced glycation end products (RAGE): Preventing diabetes induced cancer and diabetic complications. Pathol. Res. Pract. 2019, 215, 152643. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxidative Med. Cell. Longev. 2020, 2020, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Morales, F.J. Mechanism of reactive carbonyl species trapping by hydroxytyrosol under simulated physiological conditions. Food Chem. 2015, 175, 92–99. [Google Scholar] [CrossRef]
- Navarro, M.; Morales, F.J. In vitro investigation on the antiglycative and carbonyl trapping activities of hydroxytyrosol. Eur. Food Res. Technol. 2016, 242, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Kim, Y.S. The Role of Advanced Glycation End Products in Diabetic Vascular Complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar] [CrossRef]
- Wang, W.; Jing, T.; Yang, X.; He, Y.; Wang, B.; Xiao, Y.; Shang, C.; Zhang, J.; Lin, R. Hydroxytyrosol regulates the autophagy of vascular adventitial fibroblasts through the SIRT1-mediated signaling pathway. Can. J. Physiol. Pharmacol. 2018, 96, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jing, T.; Li, Y.; He, Y.; Zhang, W.; Wang, B.; Xiao, Y.; Wang, W.; Zhang, J.; Wei, J.; et al. Hydroxytyrosol Attenuates LPS-Induced Acute Lung Injury in Mice by Regulating Autophagy and Sirtuin Expression. Curr. Mol. Med. 2017, 17, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Shi, Y.; Chen, H.; Le, R.; Gong, X.; Xu, K.; Zhu, Q.; Shen, F.; Chen, Z.; Gu, X.; et al. Isoliquiritigenin prevents hyperglycemia-induced renal injuries by inhibiting inflammation and oxidative stress via SIRT1-dependent mechanism. Cell Death Dis. 2020, 11, 1040. [Google Scholar] [CrossRef]
- Huang, K.; Chen, C.; Hao, J.; Huang, J.; Wang, S.; Liu, P.; Huang, H. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating Sirt1 to resist AGEs-induced upregulation of fibronetin and transforming growth factor-β1 in rat glomerular messangial cells. Mol. Cell. Endocrinol. 2015, 399, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Cabrerizo, S.; De La Cruz, J.P.; López-Villodres, J.A.; Muñoz-Marín, J.; Guerrero, A.; Reyes, J.J.; Labajos, M.T.; Gonzalez-Correa, J.A. Role of the inhibition of oxidative stress and inflammatory mediators in the neuroprotective effects of hydroxytyrosol in rat brain slices subjected to hypoxia reoxygenation. J. Nutr. Biochem. 2013, 24, 2152–2157. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirangelo, I.; Borriello, M.; Liccardo, M.; Scafuro, M.; Russo, P.; Iannuzzi, C. Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity. Antioxidants 2021, 10, 1127. https://doi.org/10.3390/antiox10071127
Sirangelo I, Borriello M, Liccardo M, Scafuro M, Russo P, Iannuzzi C. Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity. Antioxidants. 2021; 10(7):1127. https://doi.org/10.3390/antiox10071127
Chicago/Turabian StyleSirangelo, Ivana, Margherita Borriello, Maria Liccardo, Marika Scafuro, Paola Russo, and Clara Iannuzzi. 2021. "Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity" Antioxidants 10, no. 7: 1127. https://doi.org/10.3390/antiox10071127
APA StyleSirangelo, I., Borriello, M., Liccardo, M., Scafuro, M., Russo, P., & Iannuzzi, C. (2021). Hydroxytyrosol Selectively Affects Non-Enzymatic Glycation in Human Insulin and Protects by AGEs Cytotoxicity. Antioxidants, 10(7), 1127. https://doi.org/10.3390/antiox10071127