
Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.1.1. Model of Pressure Overload-Induced Heart Failure
2.1.2. Model of Volume Overload-Induced Heart Failure
2.1.3. Experimental Groups
- Sham-T (n = 12): rats undergoing sham operation as controls of TAC and followed for 14 weeks;
- TAC (n = 12): rats undergoing TAC and followed for 14 weeks;
- Sham-A (n = 12): rats undergoing sham operation as controls of ACF and followed for 24 weeks;
- ACF (n = 12): rats undergoing ACF operation and followed for 24 weeks.
2.2. Echocardiographic Measurements
2.3. Left Ventricular Pressure-Volume Analysis
2.4. RNA Isolation and Polymerase Chain Reaction
2.5. Protein Isolation and Western Blotting
2.6. Histology and Immunohistochemistry
2.7. Statistical Analysis
3. Results
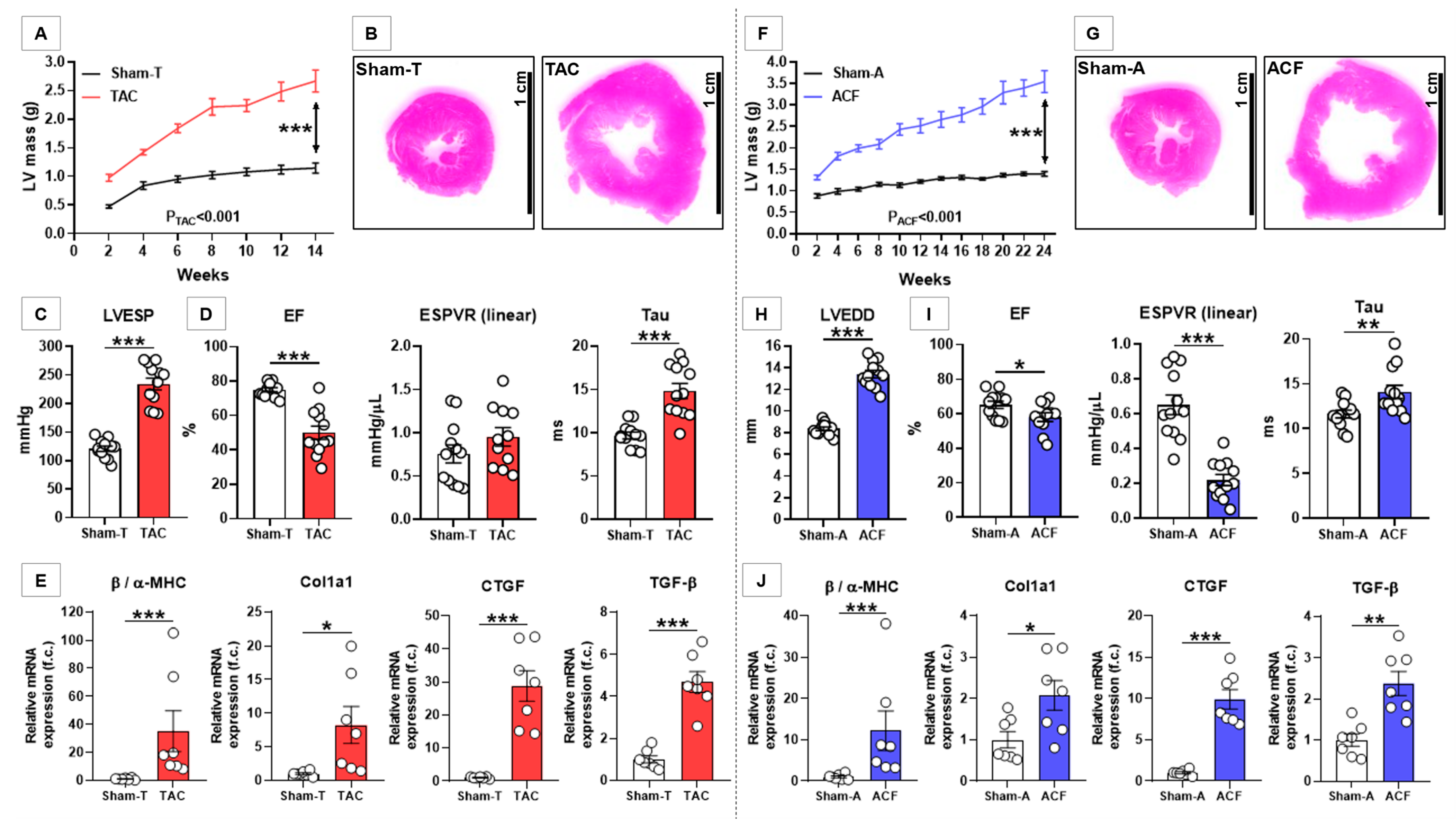
3.1. TAC Induced Characteristic LV Structural and Functional Alterations
3.2. ACF Induced Characteristic LV Structural and Functional Alterations
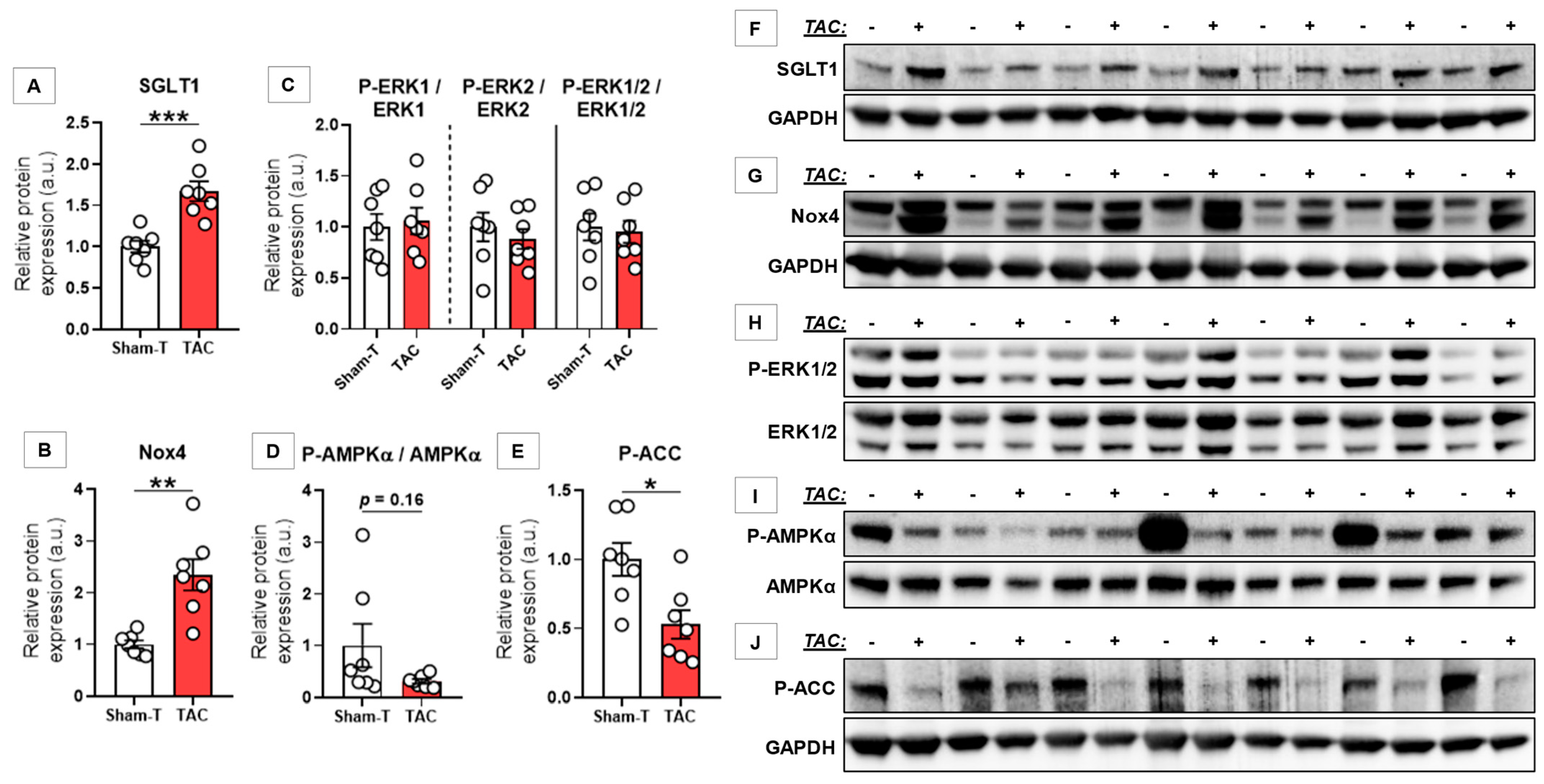
3.3. SGLT1 Protein Expression Was Upregulated Regardless of Type of HF
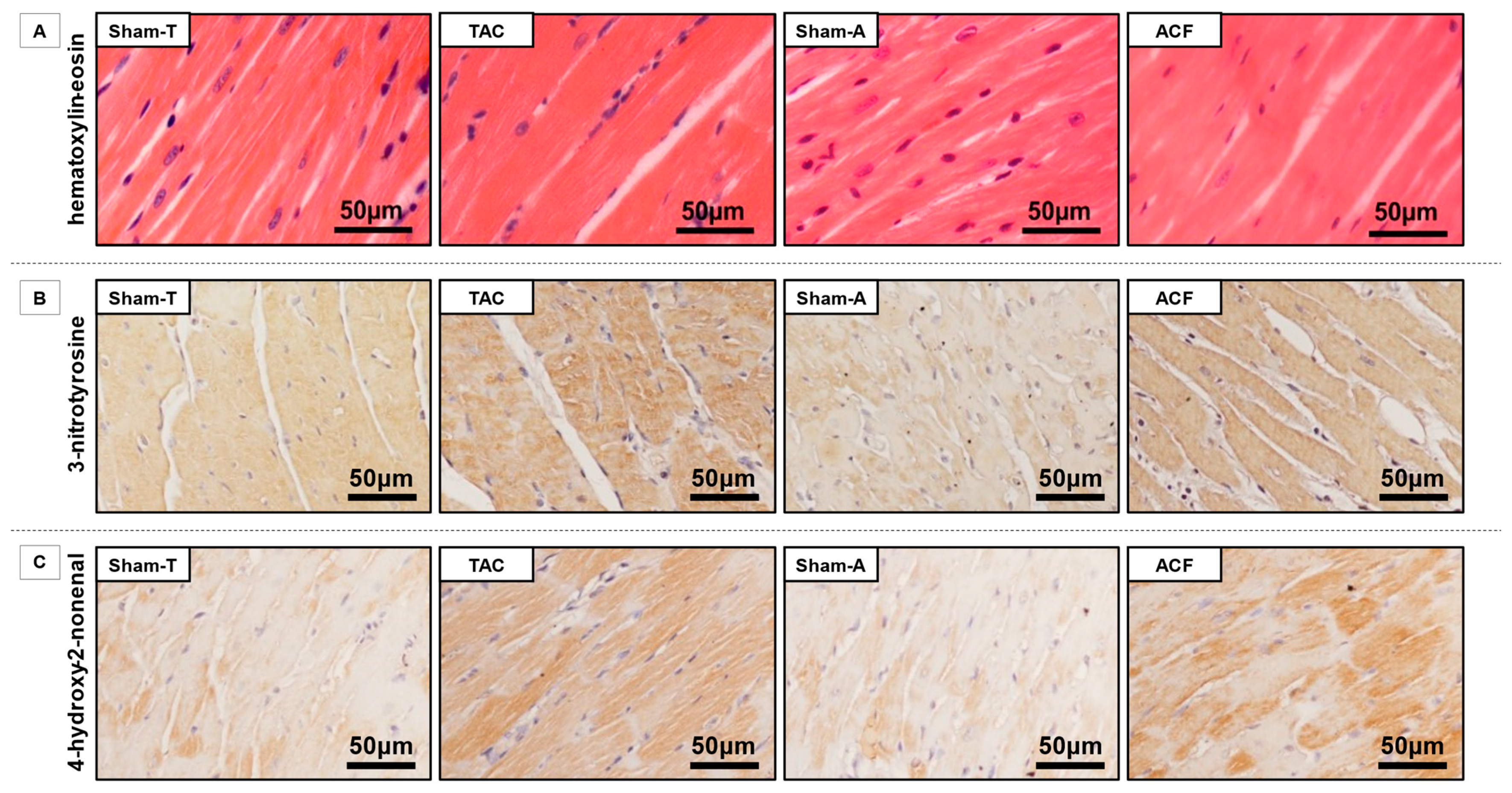
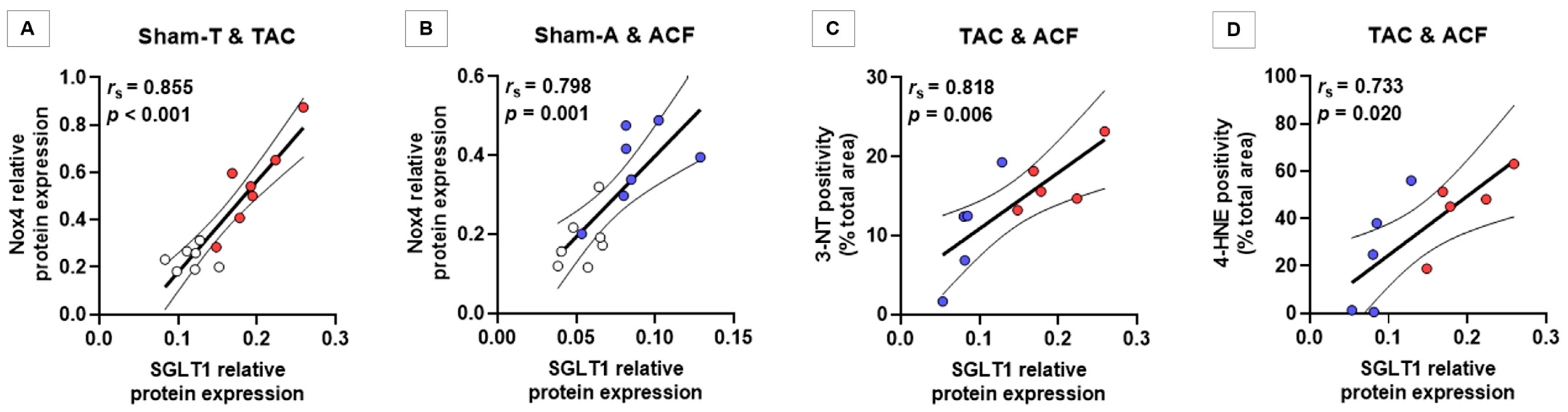
3.4. SGLT1 Protein Expression Correlates with Nox4 Protein Expression and with the Extent of Myocardial Nitro-Oxidative Stress
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, S.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayour, A.A.; Oláh, A.; Ruppert, M.; Barta, B.A.; Horváth, E.M.; Benke, K.; Pólos, M.; Hartyánszky, I.; Merkely, B.; Radovits, T. Characterization of left ventricular myocardial sodium-glucose cotransporter 1 expression in patients with end-stage heart failure. Cardiovasc. Diabetol. 2020, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, N.; Ishida, N.; Ibi, M.; Saito, M.; Sanbe, A.; Shimojo, H.; Suzuki, S.; Koepsell, H.; Takeishi, Y.; Morino, Y.; et al. Chronic pressure overload induces cardiac hypertrophy and fibrosis via increases in SGLT1 and IL-18 gene expression in mice. Int. Heart J. 2018, 59, 1123–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Guan, L.; Meng, L.; Uzui, H.; Guo, H. SGLT1 knockdown attenuates cardiac fibroblast activation in diabetic cardiac fibrosis. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Sun, Z.; Chai, Q.; Zhang, Z.; Lu, D.; Meng, Z.; Wu, W. Inhibition of SGLT1 protects against glycemic variability-induced cardiac damage and pyroptosis of cardiomyocytes in diabetic mice. Life Sci. 2021, 271, 119116. [Google Scholar] [CrossRef]
- Seidelmann, S.B.; Feofanova, E.; Yu, B.; Franceschini, N.; Claggett, B.; Kuokkanen, M.; Puolijoki, H.; Ebeling, T.; Perola, M.; Salomaa, V.; et al. Genetic variants in SGLT1, glucose tolerance, and cardiometabolic risk. J. Am. Coll. Cardiol. 2018, 72, 1763–1773. [Google Scholar] [CrossRef]
- Li, Z.; Agrawal, V.; Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Sincoular, A.; Jakubiak, M.; Music, M.L.; Kutschke, W.J.; Huang, X.N.; et al. Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc. Res. 2019, 115, 1646–1658. [Google Scholar] [CrossRef]
- Banerjee, S.; Wang, D.W.; Alzamora, R.; Huang, X.N.; Pastor-Soler, N.M.; Hallows, K.R.; McGaffin, K.R.; Ahmad, F. SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 49, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Balteau, M.; Tajeddine, N.; De Meester, C.; Ginion, A.; Rosiers, C.D.; Brady, N.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.-L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 92, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, M.; Lakatos, B.K.; Braun, S.; Tokodi, M.; Karime, C.; Oláh, A.; Sayour, A.A.; Hizoh, I.; Barta, B.A.; Merkely, B.; et al. Longitudinal strain reflects ventriculoarterial coupling rather than mere contractility in rat models of hemodynamic overload-induced heart failure. J. Am. Soc. Echocardiogr. 2020, 33, 1264–1275.e4. [Google Scholar] [CrossRef]
- Lakatos, B.K.; Ruppert, M.; Tokodi, M.; Oláh, A.; Braun, S.; Karime, C.; Ladányi, Z.; Sayour, A.A.; Barta, B.A.; Merkely, B.; et al. Myocardial work index: A marker of left ventricular contractility in pressure- or volume overload-induced heart failure. ESC Heart Fail. 2021, 8, 2220–2231. [Google Scholar] [CrossRef]
- Devereux, R.B.; Alonso, D.R.; Lutas, E.M.; Gottlieb, G.J.; Campo, E.; Sachs, I.; Reichek, N. Echocardiographic assessment of left ventricular hypertrophy: Comparison to necropsy findings. Am. J. Cardiol. 1986, 57, 450–458. [Google Scholar] [CrossRef]
- Teichholz, L.E.; Kreulen, T.; Herman, M.V.; Gorlin, R. Problems in echocardiographic volume determinations: Echocardiographic-angiographic correlations in the presence or absence of asynergy. Am. J. Cardiol. 1976, 37, 7–11. [Google Scholar] [CrossRef]
- Sayour, A.A.; Korkmaz-Icöz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Oláh, A.; Benke, K.; Brune, M.; Benkő, R.; Horváth, E.M.; et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J. Transl. Med. 2019, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, M.; Bódi, B.; Korkmaz-Icöz, S.; Loganathan, S.; Jiang, W.; Lehmann, L.; Oláh, A.; Barta, B.A.; Sayour, A.A.; Merkely, B.; et al. Myofilament Ca2+ sensitivity correlates with left ventricular contractility during the progression of pressure overload-induced left ventricular myocardial hypertrophy in rats. J. Mol. Cell. Cardiol. 2019, 129, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Van Steenbergen, A.; Balteau, M.; Ginion, A.; Ferté, L.; Battault, S.; Ravenstein, C.D.M.D.; Balligand, J.-L.; Daskalopoulos, E.-P.; Gilon, P.; Despa, F.; et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci. Rep. 2017, 7, 41166. [Google Scholar] [CrossRef]
- Von Lewinski, D.; Gasser, R.; Rainer, P.P.; Huber, M.-S.; Wilhelm, B.; Roessl, U.; Haas, T.; Wasler, A.; Grimm, M.; Bisping, E.; et al. Functional effects of glucose transporters in human ventricular myocardium. Eur. J. Heart Fail. 2010, 12, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Vrhovac, I.; Eror, D.B.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radović, N.; Jadrijević, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na+-d-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflügers Arch. Eur. J. Physiol. 2014, 467, 1881–1898. [Google Scholar] [CrossRef]
- Bell, R.M.; Yellon, D.M. SGLT2 inhibitors: Hypotheses on the mechanism of cardiovascular protection. Lancet Diabetes Endocrinol. 2018, 6, 435–437. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.; Fitchett, D.H.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.; Cahn, A.; Trimarco, B.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in patients with diabetes and chronic kidney disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Sattar, N.; Brueckmann, M.; Jamal, W.; Cotton, D.; et al. Empagliflozin in patients with heart failure, reduced ejection fraction, and volume overload. J. Am. Coll. Cardiol. 2021, 77, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Lee, S.J.; Wang, D.; Huang, X.Y.N.; Ahmad, F. Transgenic knockdown of cardiac sodium/glucose cotransporter 1 (SGLT1) attenuates PRKAG2 cardiomyopathy, whereas transgenic overexpression of cardiac SGLT1 causes pathologic hypertrophy and dysfunction in mice. J. Am. Heart Assoc. 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, R.; Srodulski, S.; Peng, X.; Margulies, K.B.; Despa, F.; Despa, S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J. Am. Heart Assoc. 2015, 4, e002183. [Google Scholar] [CrossRef] [Green Version]
- Sawa, Y.; Saito, M.; Ishida, N.; Ibi, M.; Matsushita, N.; Morino, Y.; Taira, E.; Hirose, M. Pretreatment with KGA-2727, a selective SGLT1 inhibitor, is protective against myocardial infarction-induced ventricular remodeling and heart failure in mice. J. Pharmacol. Sci. 2020, 142, 16–25. [Google Scholar] [CrossRef]
- Toischer, K.; Rokita, A.G.; Unsöld, B.; Zhu, W.; Kararigas, G.; Sossalla, S.; Reuter, S.P.; Becker, A.; Teucher, N.; Seidler, T.; et al. Differential cardiac remodeling in preload versus afterload. Circulation 2010, 122, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-M.; Ma, Y.-T.; Yang, Y.-N.; Liu, F.; Chen, B.-D.; Han, W.; Zhang, J.-F.; Gao, X.-M. Downregulation of survival signalling pathways and increased apoptosis in the transition of pressure overload-induced cardiac hypertrophy to heart failure. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1054–1061. [Google Scholar] [CrossRef]
- Schnelle, M.; Sawyer, I.; Anilkumar, N.; Mohamed, B.A.; Richards, D.A.; Toischer, K.; Zhang, M.; Catibog, N.; Sawyer, G.; Mongue-Din, H.; et al. NADPH oxidase-4 promotes eccentric cardiac hypertrophy in response to volume overload. Cardiovasc. Res. 2019, 117, 178–187. [Google Scholar] [CrossRef]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2011, 108, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Park, S.H.; Lee, Y.J. Signaling cascade of ANG II-induced inhibition of α-MG uptake in renal proximal tubule cells. Am. J. Physiol. Physiol. 2004, 286, F634–F642. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Park, J.Y.; Lee, Y.J.; Taub, M. Epidermal growth factor inhibits 14C-α-methyl-d-glucopyranoside uptake in renal proximal tubule cells: Involvement of PLC/PKC, p44/42 MAPK, and cPLA2. J. Cell. Physiol. 2004, 199, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.-Z.; Wu, Y.; Ni, Y.-J.; Liu, J.-H.; Gong, M.; Wang, X.-H.; Wei, F.; Wang, T.-Z.; Yuan, Z.; Ma, A.-Q.; et al. Isoproterenol instigates cardiomyocyte apoptosis and heart failure via AMPK inactivation-mediated endoplasmic reticulum stress. Apoptosis 2013, 18, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Asanuma, H.; Fujita, M.; Takahama, H.; Wakeno, M.; Ito, S.; Ogai, A.; Asakura, M.; Kim, J.; Minamino, T.; et al. Metformin prevents progression of heart failure in dogs. Circulation 2009, 119, 2568–2577. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Dyck, J.R. Is AMPK the savior of the failing heart? Trends Endocrinol. Metab. 2015, 26, 40–48. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Uzui, H.; Guo, H.; Tada, H. Role of SGLT1 in high glucose level-induced MMP-2 expression in human cardiac fibroblasts. Mol. Med. Rep. 2018, 17, 6887–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham-T | TAC | p Value | Sham-A | ACF | p Value | |
|---|---|---|---|---|---|---|
| Body weight, BW (g) | 520 ± 17 | 439 ± 17 | 0.002 | 642 ± 16 | 702 ± 22 | 0.042 |
| Tibial length, TL (cm) | 4.39 ± 0.04 | 4.21 ± 0.04 | 0.005 | 4.63 ± 0.03 | 4.67 ± 0.05 | 0.51 |
| Heart weight, HW (g) | 1.32 ± 0.05 | 2.72 ± 0.14 | <0.001 | 1.56 ± 0.04 | 3.41 ± 0.18 | <0.001 |
| HW/TL (g/cm) | 0.30 ± 0.01 | 0.65 ± 0.03 | <0.001 | 0.34 ± 0.01 | 0.73 ± 0.04 | <0.001 |
| Lung weight, LW (g) | 1.94 ± 0.07 | 4.06 ± 0.33 | <0.001 | 2.06 ± 0.08 | 3.25 ± 0.18 | <0.001 |
| LW/TL (g/cm) | 0.44 ± 0.01 | 0.97 ± 0.08 | <0.001 | 0.44 ± 0.02 | 0.70 ± 0.04 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayour, A.A.; Ruppert, M.; Oláh, A.; Benke, K.; Barta, B.A.; Zsáry, E.; Ke, H.; Horváth, E.M.; Merkely, B.; Radovits, T. Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure. Antioxidants 2021, 10, 1190. https://doi.org/10.3390/antiox10081190
Sayour AA, Ruppert M, Oláh A, Benke K, Barta BA, Zsáry E, Ke H, Horváth EM, Merkely B, Radovits T. Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure. Antioxidants. 2021; 10(8):1190. https://doi.org/10.3390/antiox10081190
Chicago/Turabian StyleSayour, Alex Ali, Mihály Ruppert, Attila Oláh, Kálmán Benke, Bálint András Barta, Eszter Zsáry, Haoran Ke, Eszter Mária Horváth, Béla Merkely, and Tamás Radovits. 2021. "Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure" Antioxidants 10, no. 8: 1190. https://doi.org/10.3390/antiox10081190
APA StyleSayour, A. A., Ruppert, M., Oláh, A., Benke, K., Barta, B. A., Zsáry, E., Ke, H., Horváth, E. M., Merkely, B., & Radovits, T. (2021). Left Ventricular SGLT1 Protein Expression Correlates with the Extent of Myocardial Nitro-Oxidative Stress in Rats with Pressure and Volume Overload-Induced Heart Failure. Antioxidants, 10(8), 1190. https://doi.org/10.3390/antiox10081190