Abstract
Conventionally, statins are used to treat high cholesterol levels. They exhibit pleiotropic effects, such as the prevention of cardiovascular disease and decreased cancer mortality. Gastric cancer (GC) is one of the most common cancers, ranking as the third leading global cause of cancer-related deaths, and is mainly attributed to chronic Helicobacter pylori infection. During their co-evolution with hosts, H. pylori has developed the ability to use the cellular components of the host to evade the immune system and multiply in intracellular niches. Certain H. pylori virulence factors, including cytotoxin-associated gene A (CagA), vacuolating cytotoxin A (VacA), and cholesterol-α-glucosyltransferase (CGT), have been shown to exploit host cholesterol during pathogenesis. Therefore, using statins to antagonize cholesterol synthesis might prove to be an ideal strategy for reducing the occurrence of H. pylori-related GC. This review discusses the current understanding of the interplay of H. pylori virulence factors with cholesterol and reactive oxygen species (ROS) production, which may prove to be novel therapeutic targets for the development of effective treatment strategies against H. pylori-associated GC. We also summarize the findings of several clinical studies on the association between statin therapy and the development of GC, especially in terms of cancer risk and mortality.
1. Introduction
Gastric cancer (GC) is a global health burden with more than a million new cases diagnosed in 2020, and the mortality rate is only surpassed by that of lung and liver cancers [1]. GC progresses in multiple stages including superficial gastritis, atrophic gastritis, intestinal metaplasia, dysplasia, and finally GC [2]. Lifestyle and behavior are exogenous risk factors closely associated with the development of GC [3]. Helicobacter pylori infection is one of the most important causative agents of GC and is associated with higher risk in at least 80% of cases [4]. H. pylori is a gram-negative, microaerophilic, spiral bacterium that generally colonizes the stomach. Usually acquired at a young age and without therapeutic means of eradication, H. pylori can colonize the host’s stomach for their entire lifetime [5]. Persistent H. pylori infection accumulates oxidative stress and prolongs the inflammation in gastric mucosa, resulting in the development of GC [6].
Surgical resection is a common curative treatment for GC, whereas endoscopic resection is an option for patients with small, well-differentiated early-stage tumors [7]. Adjuvant and neoadjuvant therapies, such as chemotherapy and radiotherapy are recommended in combination with surgery [8]. However, surgical treatments are invasive, and patients often suffer from surgical trauma, immunosuppression, postoperative morbidity, and mortality [9]. The management of metastatic GC includes cytotoxic therapy and chemotherapy. Although the former is associated with a high frequency of toxic effects, the latter appears to have a limited effect on overall mortality. As many patients with early-stage GC are asymptomatic, diagnosis is usually made when the tumor has metastasized and surgical resection no longer remains a curative treatment option. Therefore, preventive measures for GC in high-risk individuals are worth investigating.
Statins are inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, which is commonly prescribed for primary and secondary cardiovascular disease prevention, and they have attracted attention as a popular candidate for chemoprevention [10]. By reducing low-density lipoprotein cholesterol (LDLC) levels in blood circulation, statins effectively lower the risk of atherosclerotic cardiovascular diseases [11,12]. Mounting evidence has shown reduced cancer incidence and mortality in patients who were prescribed statins [13,14,15,16,17,18,19]. In terms of adverse effects, it has been suggested that statins are potentially diabetogenic (causing diabetes mellitus) and increase the incidence of hemorrhagic strokes [20,21]. Myopathy, a condition defined as muscle pain and tenderness, has also been reported as an adverse effect [20]. However, a study found that statin use was not correlated with myopathy [22], and did not increase the risk of hemorrhagic stroke in patients without prior incidence of stroke [23].
Although eradication of H. pylori using antibiotics is a viable method to reduce the risk of GC, it may have consequences such as an increase in antibiotic resistance rates, short-term alterations in gut microbiota, and increased risks of asthma and allergies [24,25]. Because statins primarily target virulence factors, they do not affect the extracellular survival of H. pylori, they only reduce the intracellular burden of infected cells [26]. In addition, statins have been reported to decrease GC risk independent of H. pylori status [27,28]. As such, statins cause little changes to the gut microbiota, making it a more ideal candidate for chemoprevention than eradication therapy of H. pylori. Overall, statins are not associated with clinically significant side effects, underscoring their potential as risk-reducing agents for GC.
By drawing links between H. pylori virulence factors and statin actions, we unravel the mechanisms by which statins may be employed to reduce GC incidence in patients with H. pylori infection. We also discuss previous database analyses to summarize the effects of statins on GC prevention in clinical settings, and review the prospects for statins as potential chemopreventive medications.
2. H. pylori Virulence Factors Usurp Cholesterol and Lead to GC Development
H. pylori infection induces sustained inflammation, an event that has long been linked to cancer development [29,30]. In addition to the host immune response, H. pylori virulence factors promote sustained inflammation, thus maintaining a microenvironment rich in cytokines/chemokines, reactive nitrogen species (RNS), and reactive oxygen species (ROS) that can destabilize normal cellular homeostasis [29]. Chronic inflammation exhausts resident gastric stem cells [31] and leads to the recruitment of bone marrow-derived cells that are predisposed to improper differentiation, resulting in metaplasia and dysplasia [30]. H. pylori virulence factors play an important role in inducing inflammatory responses and promoting pro-tumorigenic activities.
Vacuolating cytotoxin A (VacA) is a virulence factor well known for its ability to cause vacuoles that possess the hallmarks of late endosomes and early lysosomes in host cells [32]. Although the role of these vacuoles in GC remains unclear, it is posited that VacA-induced vacuoles disrupt normal membrane trafficking at or near late endosomes [33]. Subsequently, VacA induces autophagy and impairs transient receptor potential membrane channel mucolipin 1 (TRPML1), a key regulator of the endolysosomal pathway [34]. Consequently, the inhibition of lysosomal function or preventing the fusion of autophagosomes with lysosomes promotes bacterial resistance and multiplication of autophagosomes, which is crucial for persistent bacterial infection and induction of gastric carcinogenesis [35,36,37]. VacA triggers the production of proinflammatory cytokines, such as tumor necrosis factor (TNF) and interleukin (IL)-6, which promote the expression of cyclooxygenase-2 (COX-2) in T cells, macrophages, and neutrophils [38]. In addition to participating in proinflammatory events, COX-2 catalyzes a key step in the production of prostaglandin, a group of lipids known to play a role in tumorigenesis [29].
Another virulence factor is cytotoxin-associated gene A (CagA), which is encoded by the cag-pathogenicity island (cag-PAI) and is the most extensively studied virulence factor of H. pylori for its cancer-causing actions. H. pylori strains carrying cagA (referred to as cagA-positive strains) were significantly more correlated with the exacerbation of gastric conditions and gastric adenocarcinoma than cagA-negative strains [39]. The cag-PAI gene encodes a type IV secretion system (T4SS), a protein complex that is critical for the translocation of CagA across the membrane. Following its translocation into gastric epithelial cells, CagA is localized to the inner membrane and immediately phosphorylated by members of the Src family kinase [40]. Subsequently, CagA binds to the SH2 domain-containing tyrosine phosphatase SHP2, which potentiates a downstream cascade of Erk/MAPK activity and affects the regulation of cellular proliferation, growth, and morphology, leading to deregulation of phase progression from G1 to S [41,42] and to an increase in the expression of proto-oncogenes c-fos and c-jun [43]. In addition, CagA phosphorylation induces activation of the transcriptional factor nuclear factor-kappa B (NF-κB) and production of the cytokine IL-8 in gastric epithelial cells [44,45]. NF-κB regulates many genes whose products are involved in angiogenesis, anti-apoptotic pathways, metastasis, enhanced cell cycle progression, and cytokine production. IL-8 has been reported to have an angiogenic role in several types of cancer [29].
Cholesterol-α-glucosyltransferase (CGT), encoded by the type I capsular polysaccharide biosynthesis protein J gene (capJ) catalyzes the glucosylation of cellular cholesterol into cholesteryl glucosides [46]. Cholesterol glucosylation dampens H. pylori phagocytosis and T-cell activation, leading to bacterial immune evasion [47]. Furthermore, CGT increases autophagosome formation, which enhances H. pylori survival in host macrophages by providing an intracellular niche [48,49]. This process also reduces autophagosome-lysosome fusion, which is a key step in eliminating intracellular pathogens. Furthermore, cholesterol glucosylation improves H. pylori-host cell binding and reorganizes lipid raft membranes, thereby promoting T4SS functions such as CagA translocation/phosphorylation. This activates NF-κB to promote IL-8 production, thereby aggravating inflammation [50].
3. Interplay between H. pylori and ROS Production to Induce Gastric Carcinogenesis
H. pylori CagA translocated in the cells is crucial for inducing the production of a significant amount of ROS, which is involved in the enforcement of cell cycle progression and acceleration of cell proliferation [51]. In addition, the accumulation of ROS increases oxidative stress, which damages mitochondrial DNA (mtDNA) and nuclear DNA, leading to gastric carcinogenesis [52]. Apart from ROS, nitrosative stress is another key mediator of H. pylori infection. Nitric oxide (NO) derived from inducible nitric oxide synthase (iNOS) is responsible for bacteria-induced inflammatory responses [53]. H. pylori elicits NO production in macrophages and gastric epithelial cells, which convert l-arginine to l-citrulline using iNOS [54,55]. In GC patients with H. pylori infection, iNOS expression is higher than that in H. pylori-negative individuals [56]. iNOS deficiency lowered NO production by iNOS and markedly reduced H. pylori-associated GC in mice [57], indicating that overexpression of iNOS and sustainable NO levels contribute to H. pylori-induced stomach carcinogenesis.
VacA also plays a role in ROS generation. In a gastric epithelial model, H. pylori with CagA+/VacA+-induced ROS production and mtDNA mutations were significantly higher than those in a VacA-mutant strain [58]. Furthermore, infection with the VacA+ strain elevates SQSTM1/p62 aggregation and disrupts autophagy to increase ROS expression in gastric epithelial cells, which may accelerate carcinogenesis [35]. Biopsies from patients with Crohn’s disease showed that ATG16L1 with the T300A mutation increases susceptibility to H. pylori infection, indicating that the ATG16L1 genotype modulates autophagy responses to VacA [59]. Notably, autophagy and CagA can be degraded by VacA by reducing intracellular glutathione levels, leading to enhanced ROS accumulation and Akt phosphorylation, resulting in GC development [60]. The mechanism by which VacA and CagA decrease autophagy may provide a unique strategy for persistent H. pylori colonization in the stomach. These findings support the view that elevated ROS production due to H. pylori infection enhances DNA damage and prevents DNA repair mechanisms from functioning properly, thereby contributing to gastric carcinogenesis [6].
H. pylori has evolved to elicit detrimental effects in cells while dampening the host’s defenses using strategic mechanisms [61]. H. pylori arginase competes with cellular iNOS for the substrate l-arginine, which reduces NO production [62]. In addition, arginase II produced by macrophages suppresses H. pylori-induced NO production by inhibiting iNOS expression [63]. Reducing l-arginine availability decreases H. pylori-stimulated iNOS expression and NO levels [64]. In parallel, VacA inhibits the expression of integrin-linked kinase and endothelial nitric oxide synthase, thereby decreasing ROS production in macrophage/monocyte-lineages [65]. These findings indicate that H. pylori exploits host factors to orchestrate ROS generation, resulting in simultaneous damage to cells and immune evasion.
4. Statins Lower GC Risk by Reducing H. pylori Survival and Inhibition of Virulence Factor Actions
Statins are competitive inhibitors that block the conversion site of HMG-CoA reductase to prevent substrate access and effectively inhibit the conversion of HMG-CoA into mevalonic acid [66]. With a reduced level of mevalonic acid, the cholesterol synthesis pathway is interrupted in the liver [67]. This triggers the production of microsomal 3-hydroxy-3-methylglutaryl-CoA reductase and cell-surface low-density lipoprotein (LDL) receptors, which assist in lowering the level of circulating LDL in the bloodstream to 20–55% [68]. By inhibiting mevalonate synthesis, statins inhibit the production of mevalonate-derived intermediates, which are involved in the post-translational modification of proteins crucial for intracellular signaling, cell growth, and cellular differentiation [69,70].
Simvastatin is a class of statins that has been reported to reduce the level of cellular cholesterol in macrophages and gastric epithelial cells [13,26]. As both CagA translocation and phosphorylation depend on adequate cholesterol levels, treatment with simvastatin significantly reduces CagA translocation into gastric epithelial cells [13], which may in turn attenuate CagA-induced oncogenesis (Figure 1). In addition, treatment of cells with cholesterol-depleting agents reduces VacA internalization and cholesterol likely plays an important role in VacA entry into cells [71]. It is possible that statins may inhibit VacA internalization by gastric epithelial cells, subsequently inhibiting its pro-tumorigenic effects. Our recent study further demonstrated that the cholesterol-reducing effect of statins interrupts the cholesterol-dependent cellular evasion strategies of H. pylori mediated by CGT and reduces the bacterial load in macrophages [26]. Most importantly, several studies have reported a wide range of actions performed by statins that could reduce cancer incidence besides the inhibition of the internalization of H. pylori virulence factors. These include reduction in the plasma concentration of inflammatory cytokines, attenuation of proliferative response, and modulation of immune responses [69].
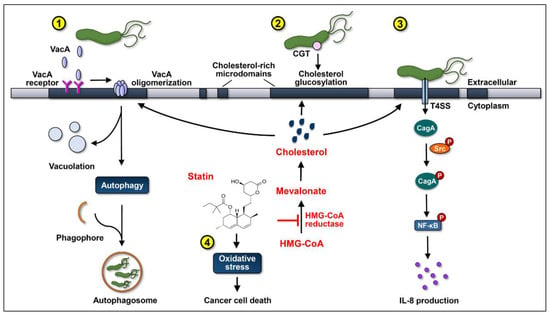
Figure 1.
Statins inhibit cholesterol synthesis by interrupting the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to mevalonate, consequently suppressing the actions of the virulence factors of Helicobacter pylori and increasing oxidative stress to promote gastric cancer cell death. (1) After cholesterol-dependent internalization, vacuolating cytotoxin A (VacA) induces vacuolation in the late endosomal compartments in host cells. Subsequently, VacA promotes autophagy and interferes with lysosomal functions, which sequesters H. pylori in intracellular niches to multiply. (2) Cholesterol-α-glucosyltransferase (CGT) catalyzes the glucosylation of cholesterol, contributing to immune evasion and intracellular survival of H. pylori. (3) Cytotoxin-associated gene A (CagA) translocation/phosphorylation depends on the coalescence of cholesterol-rich microdomains and glucosylated cholesterol, which in turn activates the nuclear factor-kappa B (NF-κB) and enhances the production of interleukin (IL)-8. The use of statins reduces the cellular cholesterol levels, inhibits the internalization of VacA, decreases the glucosylation of cholesterol, and attenuates the actions of CagA, thereby alleviating the H. pylori-induced pathogenesis. (4) Statins increase the production of reactive oxygen species (ROS) to enhance oxidative stress, thereby synergistically promoting their anti-cancer properties.
5. Statins Modulate MicroRNAs and Exosome Levels
MicroRNAs (miRNAs) are short, noncoding segments of RNA involved in the regulation of gene expression at the post-translational level [72]. Zambrano et al. investigated the effects of low dose short-term statin treatment and found that statin affects the expression of certain miRNAs [73]. Although simvastatin did not significantly impact the expression of the 86 miRNAs studied, it appeared to up-regulate several miRNAs that are involved in tumor progression in subjects who exhibited a high reduction in the level of LDLC. In addition, atorvastatin was associated with poor expression of some miRNAs studied, and it possibly downregulated the level of miRNA-33, a miRNA that reduces fatty acid metabolism and cholesterol transport [73,74].
Other than miRNAs, exosomes have been reported to lower cholesterol levels through the use of statins. Exosomes are extracellular vesicles that function in cell-to-cell signaling, and simvastatin was found to repress exosomal formation and secretion [75]. As cholesterol is an integral component of exosomal membrane, it is natural to attribute this outcome to its cholesterol-lowering effect; however, the results of the study revealed that alternative pathways may be at play instead. Because exosomes are thought to have proinflammatory activities, inhibition of exosome biogenesis and secretion may reduce inflammation, which suggests a possible chemopreventive mechanism of simvastatin.
In addition to regulating cholesterol synthesis, the virulence factors of H. pylori, and ROS responses, it is possible that statins reduce the risk of H. pylori-associated GC via the regulation of miRNAs and exosomes. For example, miRNA-146 and miRNA-155 are induced after H. pylori infection to regulate inflammation [76,77]. Another miRNA, let-7b is reduced in GC cells in a CagA-dependent manner, which may lead to the downregulation of TLR4 [78]. Additionally, miRNA-451, which inhibits the macrophage migration inhibitory factor (MIF) and functions as a tumor suppressor, is also downregulated after H. pylori infection in GC cells [79]. miRNA-29a is downregulated in GC cells to promote cell cycle progression and proliferation [80]. However, statins are known to reverse the functions of these miRNAs, suggesting that these drugs may be useful for treating GC caused by H. pylori. Meanwhile, simvastatin is known to suppress exosome formation [75], which allows cells to deliver various miRNAs and CagA for the pathogenesis of H. pylori.
6. Cholesterol-Independent Beneficial Effects of Statins in Cancer Therapy
In addition to their cholesterol-lowering function, statins exert pleiotropic therapeutic effects, which have been demonstrated to reduce the risk of several types of cancer. Statins possess anti-cancer properties, mainly by virtue of their high NO production, which is essential for tumor cytotoxicity [81]. For example, fluvastatin and simvastatin have been shown to be cytotoxic to human breast cancer cells by elevating iNOS activity and inhibiting geranylgeranylation [82]. Activation of iNOS increases NO levels, which arrests the cell cycle in the G1 phase and downregulates cyclin D1, leading to synergistically enhanced statin-induced cancer cell death [83]. In addition, lovastatin and simvastatin increase mitochondrial membrane potential (ΔΨ) and modulate mitochondrial metabolism in several cancer cells independent of cholesterol content [84]. Nonsteroidal anti-inflammatory drugs (NSAID) have been found to cause gastropathy, which was attributed to redox imbalance [85]. Notably, statins exert a protective effect against NSAID-induced lesions by increasing NO production and prostaglandin expression [86]. Overall, the NO-mediated proapoptotic, tumoricidal, and antiproliferative effects of statins confer anti-cancer properties.
Although ROS is activated as the cellular defense mechanism against bacterial invasion, H. pylori has evolved a variety of strategies, including antioxidant and DNA repair enzymes for facilitating its long-term survival within host cells [63,87]. However, persistent H. pylori infection enhances genetic instability and high mutations are generated during the repair processes, leading to gastric carcinogenesis [88]. In contrast, the excessive ROS induced by statins is irreversible, which leads to the accumulation of these ROS/RNS at toxic levels and results in profound cell death for therapeutic benefits [89,90].
7. Statin Use Reduces GC Risk
Statins were originally used to lower cholesterol levels to prevent cardiovascular disease. In addition to cholesterol restriction, statins are potential drugs for cancer therapy. To investigate the direct relationship between statins and anti-cancer activity, its effect on several types of cancers has been analyzed [91]. The results showed that statins induce cancer cell death by triggering the apoptotic pathway, not only in cell models but also in murine GC xenografts [92,93]. These findings suggest that statins may possess anti-cancer activity; however, very few clinical studies have reported that statins are anti-cancer drugs. This review further discussed the role of statin use in GC prevention and treatment (Table 1 and Table 2, respectively). Chiu et al. conducted a population-based case-control study in Taiwan, which showed a lower risk of GC in statin users than in non-users (OR = 0.68, 95% CI = 0.49–0.95) [18]. Notably, a dose-dependent effect was observed between statin use and GC risk. Lee et al. conducted a clinical study to examine the association between statin use and GC by analyzing patients with diabetes [27]. Their results showed that prescription for any statins exhibited a significant inverse association with GC. Additionally, the duration of statin use was positively correlated with a reduction in the risk of GC in patients with diabetes. These anti-GC effects warrant further research on the potential clinical use of statins.

Table 1.
The effects of statins on GC prevention.
Table 2.
The effects of statins on GC treatment.
H. pylori infection is closely associated with GC incidence [4,99,100]. Membrane cholesterol-rich microdomains provide specific regions for H. pylori virulence factor-induced pathogenesis and GC development [47,101,102]. Statins may reduce cholesterol levels and attenuate bacterial virulence factor actions, which may alleviate H. pylori-associated diseases, making them a plausible therapeutic option to treat H. pylori-induced GC. Our recent study reported that statin treatment reduced CagA translocation/phosphorylation levels and mitigated H. pylori-induced pathogenesis [13]. These results suggest that statins can decrease several pathogenic effects caused by H. pylori virulence factors. We then conducted population-based case-control studies by analyzing the Taiwan National Health Insurance Research Database and demonstrated that patients who received simvastatin exhibited a remarkably low incidence of GC [13]. GC risk reduction is especially significant in patients with H. pylori infection compared to that in statin non-users (adjusted OR = 0.25, 95% CI = 0.12–0.50). A similar trend was observed with the use of other types of statins. In addition to the type of statin prescribed, the defined daily dose is a factor related to the efficacy of GC risk reduction. These results demonstrate that statin use significantly reduces the incidence of GC, particularly in patients with H. pylori infection. However, the mechanism by which statins lower the risk of H. pylori-related GC requires further investigation.
Conversely, research by Toyoda’s group indicated that pitavastatin failed to suppress GC in murine models [94]. This study examined the relationship between pitavastatin and H. pylori-associated gastric carcinogenesis by adding pitavastatin to the diet of H. pylori-infected Mongolian gerbils. Compared to the control group, the incidence of H. pylori-associated gastric adenocarcinomas was not reduced in pitavastatin-treated mice. Serum total cholesterol also increased in the experimental groups treated with pitavastatin compared to that in the untreated controls. These results indicate that pitavastatin is ineffective in suppressing H. pylori-induced GC in murine models.
Although H. pylori infection is an important causative agent of GC, the possibility of developing GC still exists [103]. Statins interfere with H. pylori infection and suppress the delivery of virulence factors to the cells [13,26]. H. pylori infection could be a confounding factor that may cause bias in clinical studies. To eliminate the confounding effect of H. pylori status, Cheung et al. investigated the effect of statins in H. pylori-eradicated GC by analyzing the clinical data and reporting system in Hong Kong [96]. Competing risk regression with propensity score matching revealed that statin prescription was related to reducing the risk of GC in patients who had received H. pylori eradication therapy. The risk difference was 2.6 times lower in GC cases (95% CI = 1.56–3.12) per 10,000 person-years in statin users than in statin non-users. These results support the use of statins as chemopreventive agents for the treatment of GC in H. pylori-eradicated patients.
Spence et al. analyzed the relationship between statin use and GC mortality in England [14]. Two independent databases, the UK Clinical Practice Research Datalink in England and the Prescribing Information System in Scotland were investigated. These two databases recorded statin prescription and death information identified from the national mortality records. Combined cohorts and hazard ratio (HR) analysis showed that patients with GC who received statins exhibited a reduction in mortality (adjusted HR = 0.83, 95% CI = 0.74–0.92). Cancer-specific mortality was also reduced for patients who were prescribed statins before diagnosis with GC (adjusted HR = 0.91, 95% CI = 0.84–0.98). However, the doses of statins used in the treatment of patients with GC were not analyzed.
The association between statin use and lower GC risk remains elusive, and some discrepancies have emerged. A study by Cho et al. showed that statins were able to reduce mortality in GC but failed to decrease its incidence [98]. These findings are inconsistent with results from previous systematic reviews and meta-analyses, which may be because different study populations and settings were analyzed [17]. Although recent research has focused on using statins as an agent in GC treatment, whether statins can be used in the prevention of GC requires further supporting evidence. Further research that includes a large cohort with different populations should be conducted to clarify whether the clinical use of statins in the treatment of GC is feasible.
8. Conclusions and Perspectives
Most studies conducted in Western and Eastern countries to investigate the relationship between statin use and the development of GC have reported similar results, which suggest that the use of statins can reduce the risk of GC. However, most of these studies were still in the preclinical stage or were only conducted in the form of database analysis. The mechanisms by which statins inhibit H. pylori infection, especially the link between statin treatment and the manipulation of autophagy to eliminate H. pylori, remain unclear and need to be investigated further [26]. Moreover, the mechanisms by which statins increase ROS levels and regulate oxidative stress to promote GC cell death need to be elucidated. The current information from in vivo studies is insufficient, and further investigations should be conducted to determine whether statins can potentially be used for the treatment of GC.
Some studies have reported that the use of statins is associated with reducing the risk of other types of cancer. Data mining conducted using databases from the Food and Drug Administration (FDA) Adverse Event Reporting System and the Japan Medical Data Center, examined the association between the use of statins and different types of cancer, including colorectal, lung, pancreatic, gastric, esophageal, breast, and prostate cancers, as well as hematological malignancies and melanoma [104]. These results indicate that different statin categories target different types of cancer. For example, simvastatin exhibits the best efficacy in reducing the risk of GC, whereas it is positively correlated with the risk of pancreatic cancer. As conflicting results were obtained in various clinical observation studies, it is necessary to perform a large-scale prospective trial on the effects of statins in cancer therapy, which will assist physicians in determining the type of statin that is suitable for a specific type of cancer and in understanding the side effects of their use before administration to patients. In addition to further investigating the relationship between the use of statins and GC, the potential applications of statins for the treatment of other types of cancer are worth examining.
Author Contributions
Conception and design: C.-H.C., C.-J.K., and C.-H.L.; literature analysis and interpretation: T.-Y.L., W.-H.L., Y.-F.C., and C.-L.F.; writing the manuscript: T.-Y.L., W.-H.L., Y.-F.C., C.-L.F., C.-H.C., C.-J.K., and C.-H.L.; T.-Y.L., W.-H.L., and Y.-F.C. contributed equally to this work; final approval: all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Ministry of Science and Technology (109-2320-B-182-025-MY3, 109-2320-B-182-029-MY3, and 109-2622-B-182-002), Chang Gung Memorial Hospital (CMRPD1I0061-3, CMRPD1J0021-3, CMRPD1K0361, CMRPG3K0691, and BMRPE90), and Tomorrow Medical Foundation.
Acknowledgments
The authors would like to thank the editor and reviewers for the editorial assistance and their valuable comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Burkitt, M.D.; Duckworth, C.A.; Williams, J.M.; Pritchard, D.M. Helicobacter pylori-induced gastric pathology: Insights from in vivo and ex vivo models. Dis. Models Mech. 2017, 10, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Weber, A.; Graham, D.Y. Age, period, and cohort effects on gastric cancer mortality. Dig. Dis. Sci. 2015, 60, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y. Helicobacter pylori update: Gastric cancer, reliable therapy, and possible benefits. Gastroenterology 2015, 148, 719–731.e713. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; El-Serag, H.B. Burden of gastric cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 534–542. [Google Scholar] [CrossRef]
- Butcher, L.D.; den Hartog, G.; Ernst, P.B.; Crowe, S.E. Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 316–322. [Google Scholar] [CrossRef]
- Moehler, M.; Baltin, C.T.; Ebert, M.; Fischbach, W.; Gockel, I.; Grenacher, L.; Holscher, A.H.; Lordick, F.; Malfertheiner, P.; Messmann, H.; et al. International comparison of the german evidence-based s3-guidelines on the diagnosis and multimodal treatment of early and locally advanced gastric cancer, including adenocarcinoma of the lower esophagus. Gastric Cancer 2015, 18, 550–563. [Google Scholar] [CrossRef]
- Waddell, T.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; European Society for Medical Oncology (ESMO); European Society of Surgical Oncology (ESSO); European Society of Radiotherapy and Oncology (ESTRO). Gastric cancer: Esmo-esso-estro clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), 57–63. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Taylor, F.C.; Huffman, M.; Ebrahim, S. Statin therapy for primary prevention of cardiovascular disease. JAMA 2013, 310, 2451–2452. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Davis, A.M. Management of blood cholesterol. JAMA 2019, 321, 800–801. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Lin, C.J.; Liao, W.C.; Lin, H.J.; Hsu, Y.M.; Lin, C.L.; Chen, Y.A.; Feng, C.L.; Chen, C.J.; Kao, M.C.; Lai, C.H.; et al. Statins attenuate helicobacter pylori caga translocation and reduce incidence of gastric cancer: In vitro and population-based case-control studies. PLoS ONE 2016, 11, e0146432. [Google Scholar]
- Spence, A.D.; Busby, J.; Hughes, C.M.; Johnston, B.T.; Coleman, H.G.; Cardwell, C.R. Statin use and survival in patients with gastric cancer in two independent population-based cohorts. Pharmacoepidemiol. Drug Saf 2019, 28, 460–470. [Google Scholar] [CrossRef]
- Yang, P.R.; Tsai, Y.Y.; Chen, K.J.; Yang, Y.H.; Shih, W.T. Statin use improves overall survival of patients with gastric cancer after surgery and adjuvant chemotherapy in Taiwan: A nationwide matched cohort study. Cancers 2020, 12, 2055. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, W.; Jin, G.; Chu, P.; Li, H. Effect of statins on gastric cancer incidence: A meta-analysis of case control studies. J. Cancer Res. Ther. Oncol. 2014, 10, 859–865. [Google Scholar]
- Singh, P.P.; Singh, S. Statins are associated with reduced risk of gastric cancer: A systematic review and meta-analysis. Ann. Oncol. 2013, 24, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.F.; Ho, S.C.; Chang, C.C.; Wu, T.N.; Yang, C.Y. Statins are associated with a reduced risk of gastric cancer: A population-based case-control study. Am. J. Gastroenterol. Suppl. 2011, 106, 2098–2103. [Google Scholar] [CrossRef]
- Wu, X.D.; Zeng, K.; Xue, F.Q.; Chen, J.H.; Chen, Y.Q. Statins are associated with reduced risk of gastric cancer: A meta-analysis. Eur. J. Clin. Pharmacol. 2013, 69, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef]
- Brault, M.; Ray, J.; Gomez, Y.H.; Mantzoros, C.S.; Daskalopoulou, S.S. Statin treatment and new-onset diabetes: A review of proposed mechanisms. Metabolism 2014, 63, 735–745. [Google Scholar] [CrossRef]
- Naci, H.; Brugts, J.; Ades, T. Comparative tolerability and harms of individual statins: A study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. Adverse effects of statin therapy: Perception vs. The evidence-focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Heart J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.M.; Lee, Y.C.; El-Omar, E.M.; Wu, M.S. Efficacy and long-term safety of H. pylori eradication for gastric cancer prevention. Cancers 2019, 11, 593. [Google Scholar] [CrossRef]
- Zuo, Z.T.; Ma, Y.; Sun, Y.; Bai, C.Q.; Ling, C.H.; Yuan, F.L. The protective effects of helicobacter pylori infection on allergic asthma. Int. Arch. Allergy Immunol. 2021, 182, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Huang, M.Z.; Wang, M.L.; Lin, C.J.; Lu, T.L.; Lo, H.R.; Pan, Y.J.; Sun, Y.C.; Kao, M.C.; Lim, H.J.; et al. Statin decreases helicobacter pylori burden in macrophages by promoting autophagy. Front. Cell Infect. Microbiol. 2016, 6, 203. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.H.; Hur, K.Y.; Woo, S.Y.; Kim, S.W.; Kang, W.K. Statins and the risk of gastric cancer in diabetes patients. BMC Cancer 2012, 12, 596. [Google Scholar] [CrossRef]
- You, H.S.; You, N.; Lee, J.W.; Lim, H.J.; Kim, J.; Kang, H.T. Inverse association between statin use and stomach cancer incidence in individuals with hypercholesterolemia, from the 2002-2015 nhis-heals data. Int. J. Environ. Res. Public Health 2020, 17, 1054. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef]
- Stoicov, C.; Li, H.; Cerny, J.; Houghton, J.M. How the study of helicobacter infection can contribute to the understanding of carcinoma development. Clin. Microbiol. Infect. 2009, 15, 813–822. [Google Scholar] [CrossRef]
- Isomoto, H.; Moss, J.; Hirayama, T. Pleiotropic actions of helicobacter pylori vacuolating cytotoxin, vaca. Tohoku J. Exp. Med. 2010, 220, 3–14. [Google Scholar] [CrossRef]
- Montecucco, C.; de Bernard, M. Molecular and cellular mechanisms of action of the vacuolating cytotoxin (vaca) and neutrophil-activating protein (hp-nap) virulence factors of helicobacter pylori. Microbes Infect. 2003, 5, 715–721. [Google Scholar] [CrossRef]
- Capurro, M.I.; Greenfield, L.K.; Prashar, A.; Xia, S.; Abdullah, M.; Wong, H.; Zhong, X.Z.; Bertaux-Skeirik, N.; Chakrabarti, J.; Siddiqui, I.; et al. Vaca generates a protective intracellular reservoir for helicobacter pylori that is eliminated by activation of the lysosomal calcium channel trpml1. Nat. Microbiol. 2019, 4, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.; Hussey, S.; Ang, M.; Terebiznik, M.R.; Sibony, M.; Galindo-Mata, E.; Gupta, V.; Blanke, S.R.; Delgado, A.; Romero-Gallo, J.; et al. Vacuolating cytotoxin and variants in atg16l1 that disrupt autophagy promote helicobacter pylori infection in humans. Gastroenterology 2012, 142, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, L.K.; Jones, N.L. Modulation of autophagy by helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol. 2013, 21, 602–612. [Google Scholar] [CrossRef]
- Capurro, M.I.; Prashar, A.; Jones, N.L. Mcoln1/trpml1 inhibition-a novel strategy used by. Autophagy 2020, 16, 169–170. [Google Scholar] [CrossRef]
- Montecucco, C.; de Bernard, M. Immunosuppressive and proinflammatory activities of the vaca toxin of helicobacter pylori. J. Exp. Med. 2003, 198, 1767–1771. [Google Scholar] [CrossRef]
- Nomura, A.M.; Lee, J.; Stemmermann, G.N.; Nomura, R.Y.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori caga seropositivity and gastric carcinoma risk in a japanese american population. J. Infect. Dis. 2002, 186, 1138–1144. [Google Scholar] [CrossRef]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src is the kinase of the helicobacter pylori caga protein in vitro and in vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Higashi, H. Helicobacter pylori caga: A new paradigm for bacterial carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [CrossRef]
- Roovers, K.; Assoian, R.K. Integrating the map kinase signal into the g1 phase cell cycle machinery. Bioessays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Meyer-ter-Vehn, T.; Covacci, A.; Kist, M.; Pahl, H.L. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J. Biol. Chem. 2000, 275, 16064–16072. [Google Scholar] [CrossRef]
- Lamb, A.; Chen, L.F. Role of the helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef]
- Brandt, S.; Kwok, T.; Hartig, R.; Konig, W.; Backert, S. Nf-kappab activation and potentiation of proinflammatory responses by the Helicobacter pylori caga protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, A.H.; Wunder, C.; Hildebrand, J.; Churin, Y.; Zahringer, U.; Lindner, B.; Meyer, T.F.; Heinz, E.; Warnecke, D. Cloning of a cholesterol-alpha-glucosyltransferase from helicobacter pylori. J. Biol. Chem. 2006, 281, 27765–27772. [Google Scholar] [CrossRef] [PubMed]
- Wunder, C.; Churin, Y.; Winau, F.; Warnecke, D.; Vieth, M.; Lindner, B.; Zahringer, U.; Mollenkopf, H.J.; Heinz, E.; Meyer, T.F. Cholesterol glucosylation promotes immune evasion by helicobacter pylori. Nat. Med. 2006, 12, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Huang, J.C.; Cheng, H.H.; Wu, M.C.; Huang, M.Z.; Hsu, H.Y.; Chen, Y.A.; Hsu, C.Y.; Pan, Y.J.; Chu, Y.T.; et al. Helicobacter pylori cholesterol glucosylation modulates autophagy for increasing intracellular survival in macrophages. Cell. Microbiol. 2018, 20, e12947. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Y.; Wang, H.J.; Cheng, H.H.; Chen, S.D.; Wang, L.H.; Wang, W.C. Cholesterol glucosylation by helicobacter pylori delays internalization and arrests phagosome maturation in macrophages. J. Microbiol. Immunol. Infect. 2016, 49, 636–645. [Google Scholar] [CrossRef]
- Wang, H.J.; Cheng, W.C.; Cheng, H.H.; Lai, C.H.; Wang, W.C. Helicobacter pylori cholesteryl glucosides interfere with host membrane phase and affect type iv secretion system function during infection in ags cells. Mol. Microbiol. 2012, 83, 67–84. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Caga protein of helicobacter pylori: A hijacker of gastric epithelial cell signaling. Biochem. Pharmacol. 2007, 73, 1697–1702. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Redox biology and gastric carcinogenesis: The role of helicobacter pylori. Redox Rep. 2011, 16, 1–7. [Google Scholar] [CrossRef]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Wilson, K.T.; Ramanujam, K.S.; Mobley, H.L.; Musselman, R.F.; James, S.P.; Meltzer, S.J. Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 1996, 111, 1524–1533. [Google Scholar] [CrossRef]
- Fu, S.; Ramanujam, K.S.; Wong, A.; Fantry, G.T.; Drachenberg, C.B.; James, S.P.; Meltzer, S.J.; Wilson, K.T. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in helicobacter pylori gastritis. Gastroenterology 1999, 116, 1319–1329. [Google Scholar] [CrossRef]
- Goto, T.; Haruma, K.; Kitadai, Y.; Ito, M.; Yoshihara, M.; Sumii, K.; Hayakawa, N.; Kajiyama, G. Enhanced expression of inducible nitric oxide synthase and nitrotyrosine in gastric mucosa of gastric cancer patients. Clin. Cancer Res. 1999, 5, 1411–1415. [Google Scholar]
- Nam, K.T.; Oh, S.Y.; Ahn, B.; Kim, Y.B.; Jang, D.D.; Yang, K.H.; Hahm, K.B.; Kim, D.Y. Decreased helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut 2004, 53, 1250–1255. [Google Scholar] [CrossRef]
- Huang, X.W.; Luo, R.H.; Zhao, Q.; Shen, Z.Z.; Huang, L.L.; An, X.Y.; Zhao, L.J.; Wang, J.; Huang, Y.Z. Helicobacter pylori induces mitochondrial DNA mutation and reactive oxygen species level in ags cells. Int. J. Med. Sci. 2011, 8, 56–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raju, D.; Hussey, S.; Jones, N.L. Crohn disease atg16l1 polymorphism increases susceptibility to infection with helicobacter pylori in humans. Autophagy 2012, 8, 1387–1388. [Google Scholar] [CrossRef]
- Tsugawa, H.; Suzuki, H.; Saya, H.; Hatakeyama, M.; Hirayama, T.; Hirata, K.; Nagano, O.; Matsuzaki, J.; Hibi, T. Reactive oxygen species-induced autophagic degradation of helicobacter pylori caga is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012, 12, 764–777. [Google Scholar] [CrossRef]
- Lu, D.Y.; Tang, C.H.; Chang, C.H.; Maa, M.C.; Fang, S.H.; Hsu, Y.M.; Lin, Y.H.; Lin, C.J.; Lee, W.C.; Lin, H.J.; et al. Helicobacter pylori attenuates lipopolysaccharide-induced nitric oxide production by murine macrophages. Innate Immun. 2012, 18, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Gobert, A.P.; McGee, D.J.; Akhtar, M.; Mendz, G.L.; Newton, J.C.; Cheng, Y.; Mobley, H.L.; Wilson, K.T. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: A strategy for bacterial survival. Proc. Natl. Acad. Sci. USA 2001, 98, 13844–13849. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.D.; Asim, M.; Barry, D.P.; Singh, K.; de Sablet, T.; Boucher, J.L.; Gobert, A.P.; Chaturvedi, R.; Wilson, K.T. Arginase ii restricts host defense to helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J. Immunol. 2010, 184, 2572–2582. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Lewis, N.D.; Algood, H.M.; Cover, T.L.; Kim, P.Y.; Wilson, K.T. l-arginine availability regulates inducible nitric oxide synthase-dependent host defense against helicobacter pylori. J. Immunol. 2007, 75, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Li, P.; Tao, J.; Shi, X.; Hu, B.; Chen, H.; Guo, X.H. Pylori escape host immunoreaction through inhibiting ilk expression by vaca. Cell Mol. Immunol. 2009, 6, 191–197. [Google Scholar] [CrossRef]
- Istvan, E. Statin inhibition of hmg-coa reductase: A 3-dimensional view. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef]
- Lennernas, H.; Fager, G. Pharmacodynamics and pharmacokinetics of the hmg-coa reductase inhibitors. Similarities and differences. Clin. Pharmacokinet. 1997, 32, 403–425. [Google Scholar] [CrossRef]
- McKenney, J.M.; Jones, P.H.; Adamczyk, M.A.; Cain, V.A.; Bryzinski, B.S.; Blasetto, J.W.; Group, S.S. Comparison of the efficacy of rosuvastatin versus atorvastatin, simvastatin, and pravastatin in achieving lipid goals: Results from the stellar trial. Curr. Med. Res. Opin. 2003, 19, 689–698. [Google Scholar] [CrossRef]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Patel, H.K.; Willhite, D.C.; Patel, R.M.; Ye, D.; Williams, C.L.; Torres, E.M.; Marty, K.B.; MacDonald, R.A.; Blanke, S.R. Plasma membrane cholesterol modulates cellular vacuolation induced by the helicobacter pylori vacuolating cytotoxin. Infect. Immun. 2002, 70, 4112–4123. [Google Scholar] [CrossRef]
- Mohajeri, M.; Banach, M.; Atkin, S.L.; Butler, A.E.; Ruscica, M.; Watts, G.F.; Sahebkar, A. Micrornas: Novel molecular targets and response modulators of statin therapy. Trends Pharmacol. Sci. 2018, 39, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, T.; Hirata, R.D.C.; Hirata, M.H.; Cerda, Á.; Salazar, L.A. Statins differentially modulate micrornas expression in peripheral cells of hyperlipidemic subjects: A pilot study. Eur. J. Pharm. Sci. 2018, 117, 55–61. [Google Scholar] [CrossRef]
- Moore, K.J.; Rayner, K.J.; Suárez, Y.; Fernández-Hernando, C. The role of micrornas in cholesterol efflux and hepatic lipid metabolism. Annu. Rev. Nutr. 2011, 31, 49–63. [Google Scholar] [CrossRef]
- Kulshreshtha, A.; Singh, S.; Ahmad, M.; Khanna, K.; Ahmad, T.; Agrawal, A.; Ghosh, B. Simvastatin mediates inhibition of exosome synthesis, localization and secretion via multicomponent interventions. Sci. Rep. 2019, 9, 16373. [Google Scholar] [CrossRef] [PubMed]
- Hoces de la Guardia, A.; Staedel, C.; Kaafarany, I.; Clement, A.; Roubaud Baudron, C.; Megraud, F.; Lehours, P. Inflammatory cytokine and microrna responses of primary human dendritic cells cultured with helicobacter pylori strains. Front. Microbiol. 2013, 4, 236. [Google Scholar] [CrossRef]
- Sasaran, M.O.; Melit, L.E.; Dobru, E.D. Microrna modulation of host immune response and inflammation triggered by helicobacter pylori. Int. J. Mol. Sci. 2021, 22, 1406. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.G.; Wang, W.H.; Dai, Y.; Wang, S.J.; Chu, Y.X.; Li, J. Let-7b is involved in the inflammation and immune responses associated with helicobacter pylori infection by targeting toll-like receptor 4. PLoS ONE 2013, 8, e56709. [Google Scholar] [CrossRef] [PubMed]
- Bandres, E.; Bitarte, N.; Arias, F.; Agorreta, J.; Fortes, P.; Agirre, X.; Zarate, R.; Diaz-Gonzalez, J.A.; Ramirez, N.; Sola, J.J.; et al. Microrna-451 regulates macrophage migration inhibitory factor production and proliferation of gastrointestinal cancer cells. Clin. Cancer Res. 2009, 15, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Su, W.Y.; Xing, J.; Wang, Y.C.; Wang, P.; Chen, X.Y.; Shen, Z.Y.; Cao, H.; Lu, Y.Y.; Fang, J.Y. Mir-29a inhibits cell proliferation and induces cell cycle arrest through the downregulation of p42.3 in human gastric cancer. PLoS ONE 2011, 6, e25872. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed]
- Kotamraju, S.; Williams, C.L.; Kalyanaraman, B. Statin-induced breast cancer cell death: Role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007, 67, 7386–7394. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (mda-mb-231): Potential role of cyclin d1. Proc. Natl. Acad. Sci. USA 2001, 98, 3583–3588. [Google Scholar] [CrossRef]
- Christie, C.F.; Fang, D.; Hunt, E.G.; Morris, M.E.; Rovini, A.; Heslop, K.A.; Beeson, G.C.; Beeson, C.C.; Maldonado, E.N. Statin-dependent modulation of mitochondrial metabolism in cancer cells is independent of cholesterol content. FASEB J. 2019, 33, 8186–8201. [Google Scholar] [CrossRef]
- Cherkas, A.; Zarkovic, N. 4-hydroxynonenal in redox homeostasis of gastrointestinal mucosa: Implications for the stomach in health and diseases. Antioxidants 2018, 7, 118. [Google Scholar]
- Ghaisas, M.M.; Dandawate, P.R.; Zawar, S.A.; Ahire, Y.S.; Gandhi, S.P. Antioxidant, antinociceptive and anti-inflammatory activities of atorvastatin and rosuvastatin in various experimental models. Inflammopharmacology 2010, 18, 169–177. [Google Scholar] [CrossRef]
- Stent, A.; Every, A.L.; Sutton, P. Helicobacter pylori defense against oxidative attack. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G579–G587. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Chiba, T.; Marusawa, H. Helicobacter pylori-mediated genetic instability and gastric carcinogenesis. In Molecular Pathogenesis and Signal Transduction by Helicobacter Pylori; Tegtmeyer, N.M., Backert, S., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 305–323. [Google Scholar]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- McGregor, G.H.; Campbell, A.D.; Fey, S.K.; Tumanov, S.; Sumpton, D.; Blanco, G.R.; Mackay, G.; Nixon, C.; Vazquez, A.; Sansom, O.J.; et al. Targeting the metabolic response to statin-mediated oxidative stress produces a synergistic antitumor response. Cancer Res. 2020, 80, 175–188. [Google Scholar] [CrossRef]
- Corcos, L.; Le Jossic-Corcos, C. Statins: Perspectives in cancer therapeutics. Dig. Liver Dis 2013, 45, 795–802. [Google Scholar] [CrossRef]
- Gibot, L.; Follet, J.; Metges, J.P.; Auvray, P.; Simon, B.; Corcos, L.; Le Jossic-Corcos, C. Human caspase 7 is positively controlled by srebp-1 and srebp-2. Biochem. J. 2009, 420, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Follet, J.; Remy, L.; Hesry, V.; Simon, B.; Gillet, D.; Auvray, P.; Corcos, L.; Le Jossic-Corcos, C. Adaptation to statins restricts human tumour growth in nude mice. BMC Cancer 2011, 11, 491. [Google Scholar] [CrossRef]
- Toyoda, T.; Tsukamoto, T.; Takasu, S.; Hirano, N.; Ban, H.; Shi, L.; Kumagai, T.; Tanaka, T.; Tatematsu, M. Pitavastatin fails to lower serum lipid levels or inhibit gastric carcinogenesis in helicobacter pylori-infected rodent models. Cancer Prev. Res. 2009, 2, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.W.; Liao, K.F.; Lin, C.Y.; Lin, C.L.; Sung, F.C. Statins on the risk of gastric cancer: A population-based observation in taiwan. Am. J. Gastroenterol. 2013, 108, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Chan, E.W.; Wong, A.Y.S.; Chen, L.; Seto, W.K.; Wong, I.C.K.; Leung, W.K. Statins were associated with a reduced gastric cancer risk in patients with eradicated helicobacter pylori infection: A territory-wide propensity score matched study. Cancer Epidemiol. Biomark. Prev. 2020, 29, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Lee, H.; Park, J.C.; Shin, S.K.; Lee, S.K.; Hyung, W.J.; Lee, Y.C.; Kang, M.W.; Noh, S.H. Long-term statin therapy improves oncological outcome after radical gastrectomy for stage ii and iii gastric cancer. Anticancer Res. 2014, 34, 355–361. [Google Scholar]
- Cho, M.H.; Yoo, T.G.; Jeong, S.M.; Shin, D.W. Association of aspirin, metformin, and statin use with gastric cancer incidence and mortality: A nationwide cohort study. Cancer Prev. Res. 2021, 14, 95–104. [Google Scholar] [CrossRef]
- Megraud, F.; Bessede, E.; Varon, C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. 2015, 21, 984–990. [Google Scholar] [CrossRef]
- Hartgrink, H.H.; Jansen, E.P.; van Grieken, N.C.; van de Velde, C.J. Gastric cancer. Lancet 2009, 374, 477–490. [Google Scholar] [CrossRef]
- Schraw, W.; Li, Y.; McClain, M.S.; van der Goot, F.G.; Cover, T.L. Association of helicobacter pylori vacuolating toxin (vaca) with lipid rafts. J. Biol. Chem. 2002, 277, 34642–34650. [Google Scholar] [CrossRef]
- Lai, C.H.; Chang, Y.C.; Du, S.Y.; Wang, H.J.; Kuo, C.H.; Fang, S.H.; Fu, H.W.; Lin, H.H.; Chiang, A.S.; Wang, W.C. Cholesterol depletion reduces helicobacter pylori caga translocation and caga-induced responses in ags cells. Infect. Immun. 2008, 76, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Piscione, M.; Mazzone, M.; Di Marcantonio, M.C.; Muraro, R.; Mincione, G. Eradication of helicobacter pylori and gastric cancer: A controversial relationship. Front. Microbiol. 2021, 12, 630852. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Higuchi, T.; Hosomi, K.; Takada, M. Association between statin use and cancer: Data mining of a spontaneous reporting database and a claims database. Int. J. Med. Sci. 2015, 12, 223–233. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).