
NADPH-Oxidase, Rho-Kinase and Autophagy Mediate the (Pro)renin-Induced Pro-Inflammatory Microglial Response and Enhancement of Dopaminergic Neuron Death

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
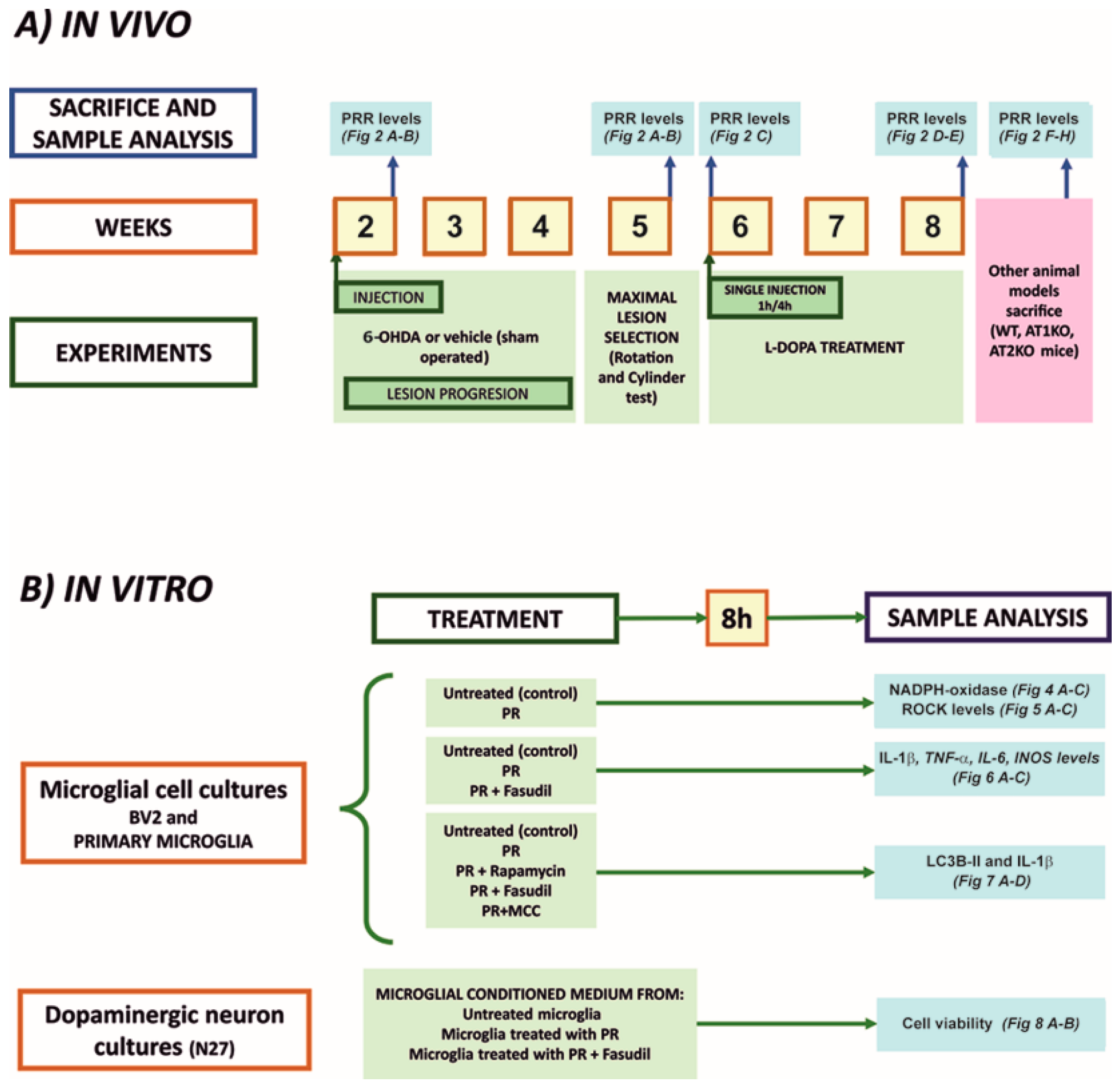
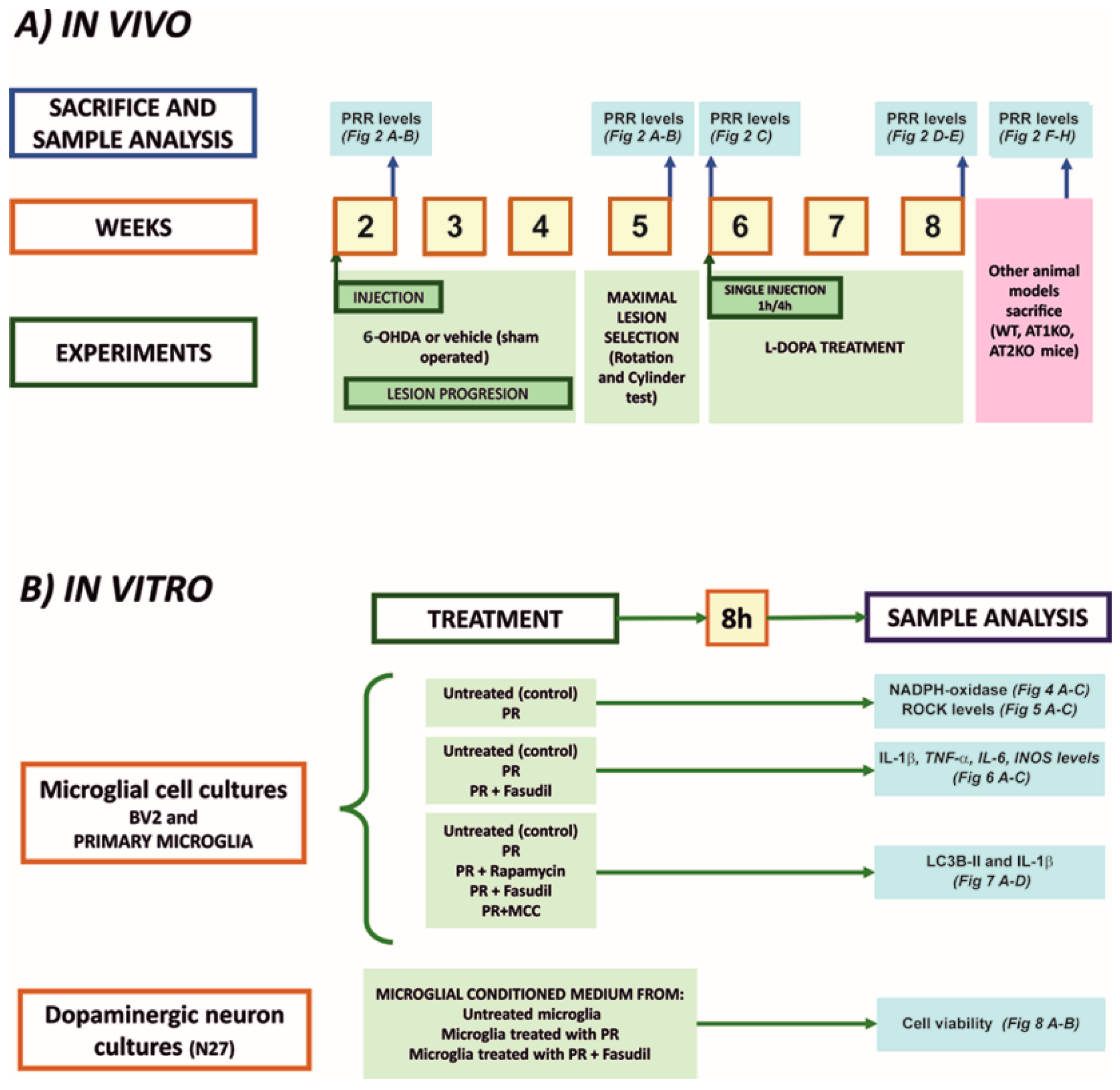
2.1. Experimental Design
2.2. Animal Models
2.3. Isolation of Microglia from Adult Brains
2.4. Laser Capture Microdissection of Mouse Nigral Neurons
2.5. Microglial and Neuronal Cell Cultures
2.6. Treatment of Cultures
2.7. Immunofluorescent Labeling
2.8. NADPH-Oxidase and ROCK Activities
2.9. RNA Extraction and Real-Time Quantitative RT-PCR
2.10. Western Blot Analysis and Autophagy Assay
2.11. MTT Viability Assay
2.12. Statistical Analysis
3. Results
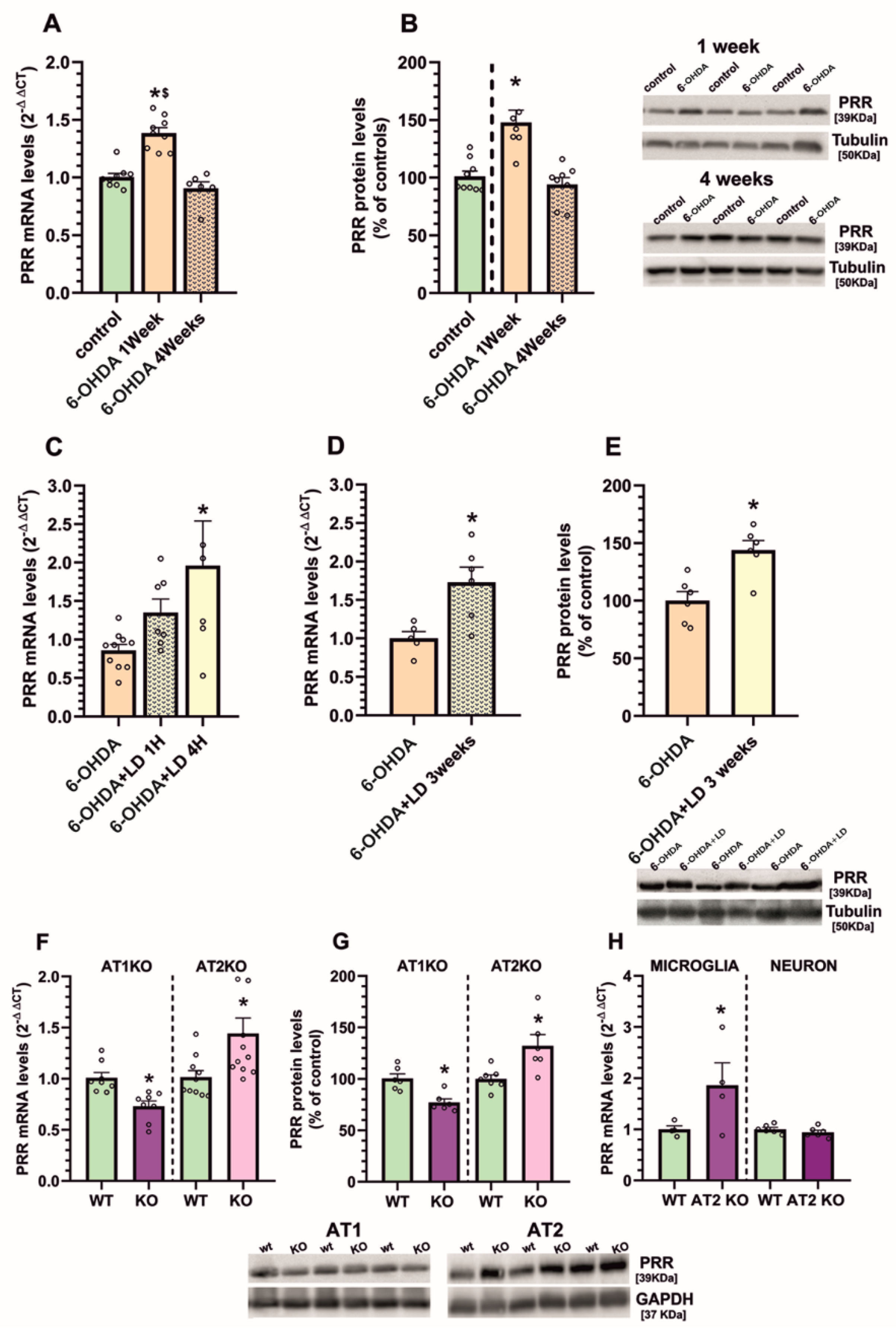
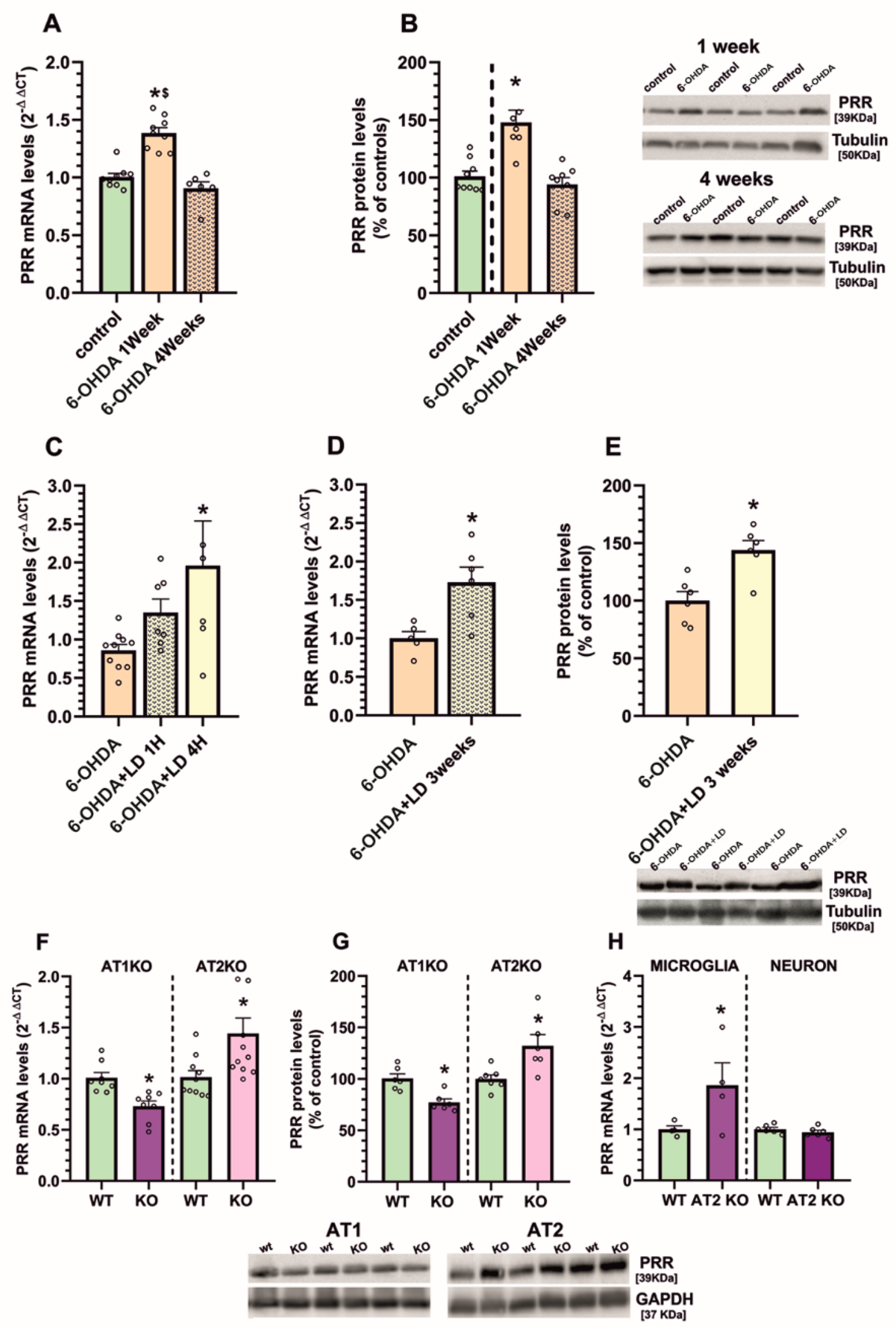
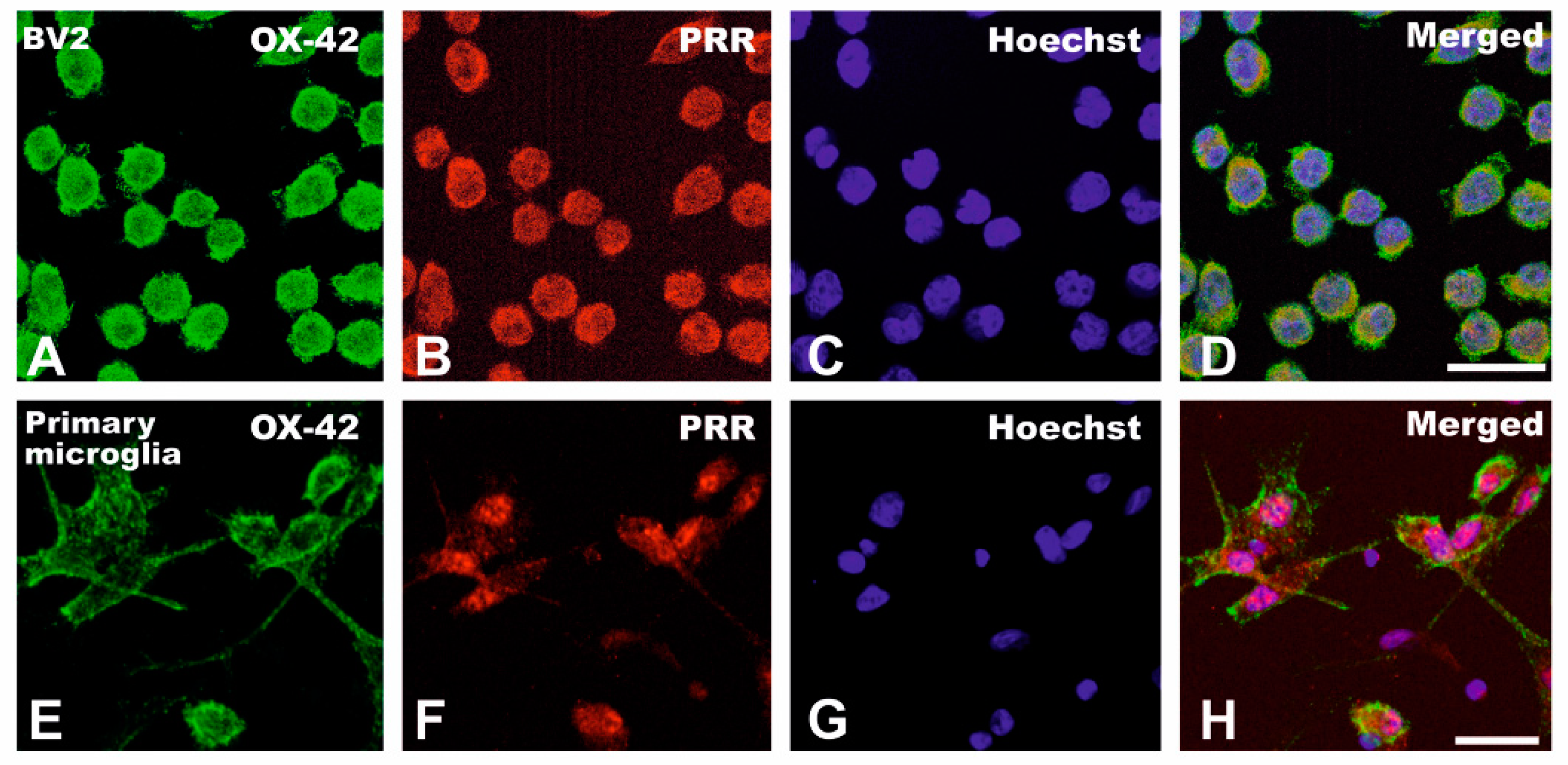
3.1. Upregulation of (Pro)Renin Receptors in Nigral Inflammatory Processes. Upregulation in Microglia
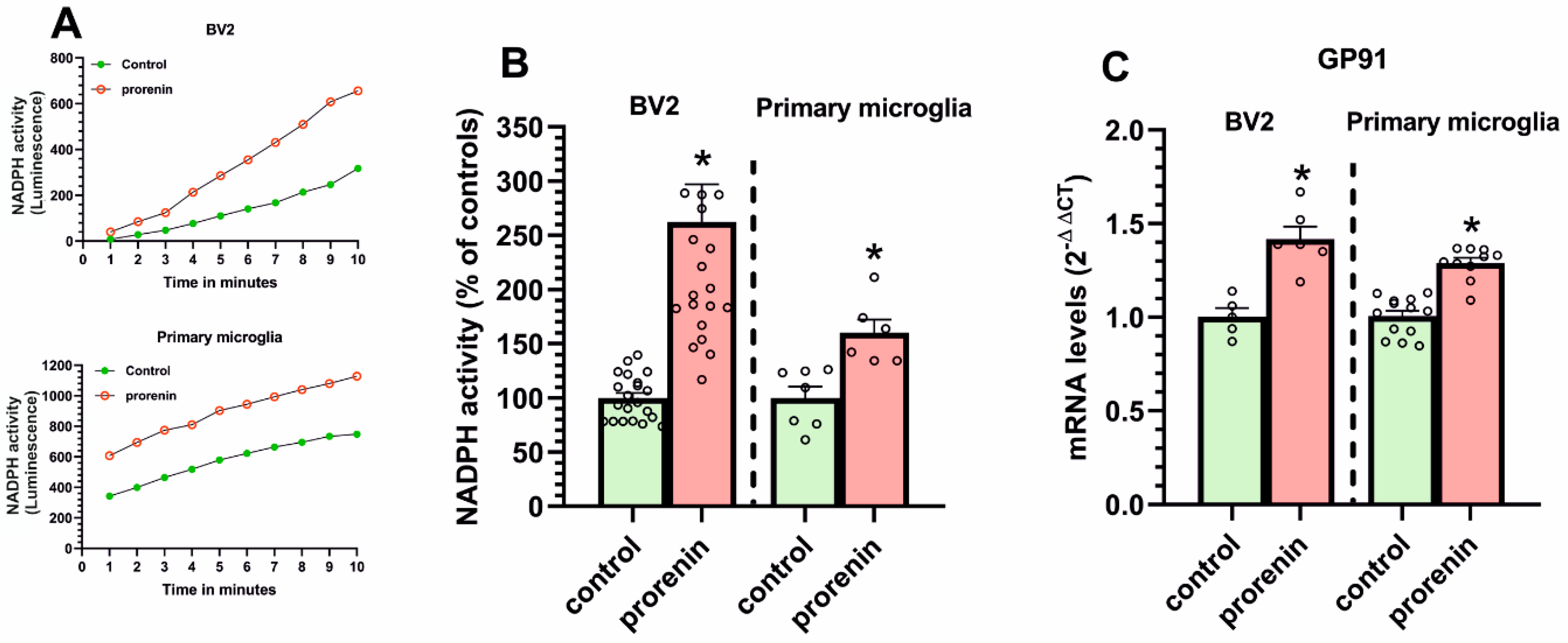
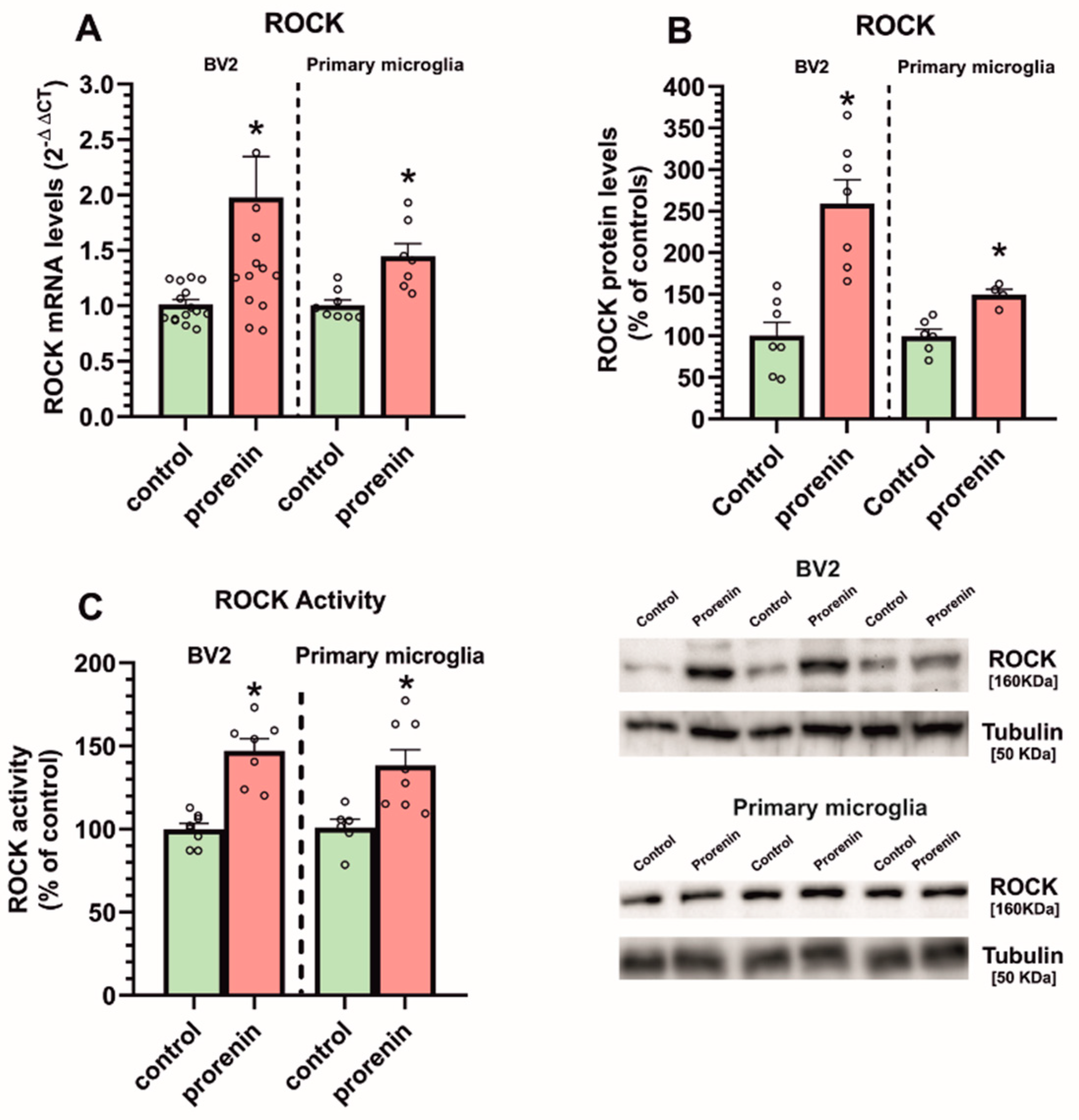
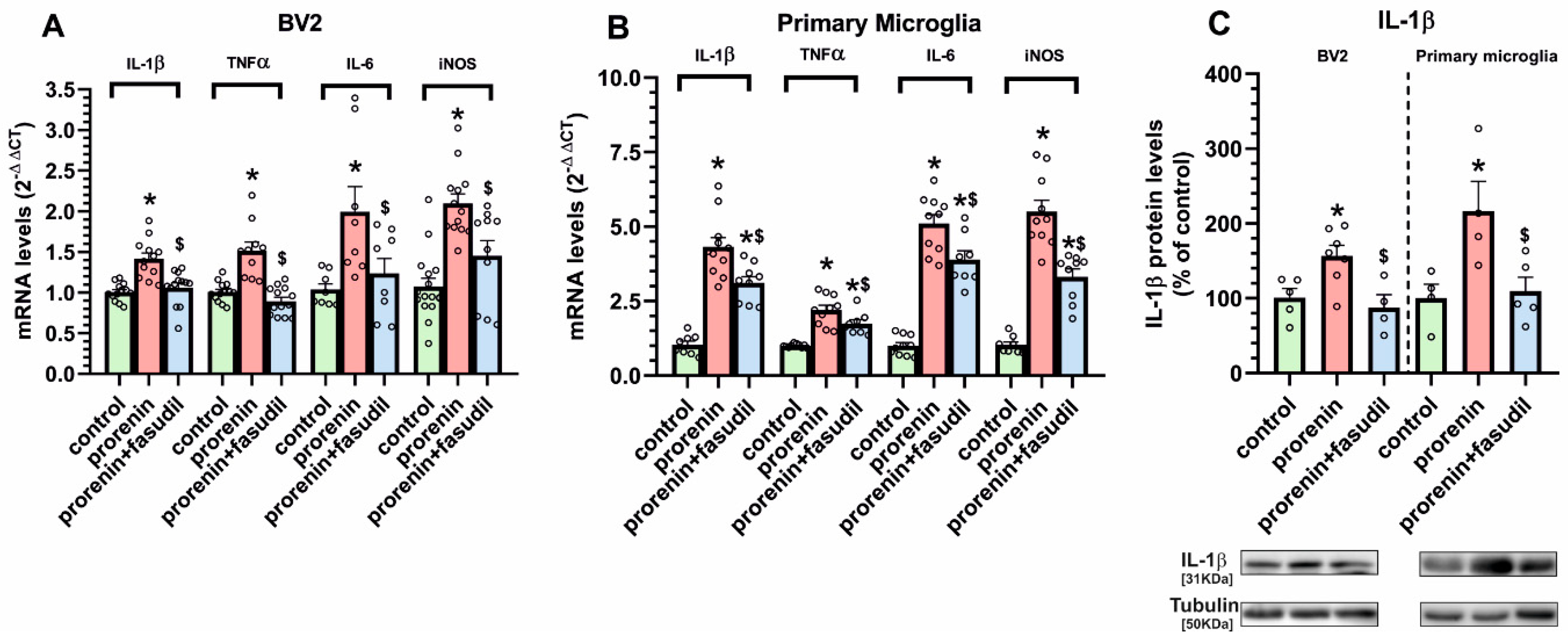
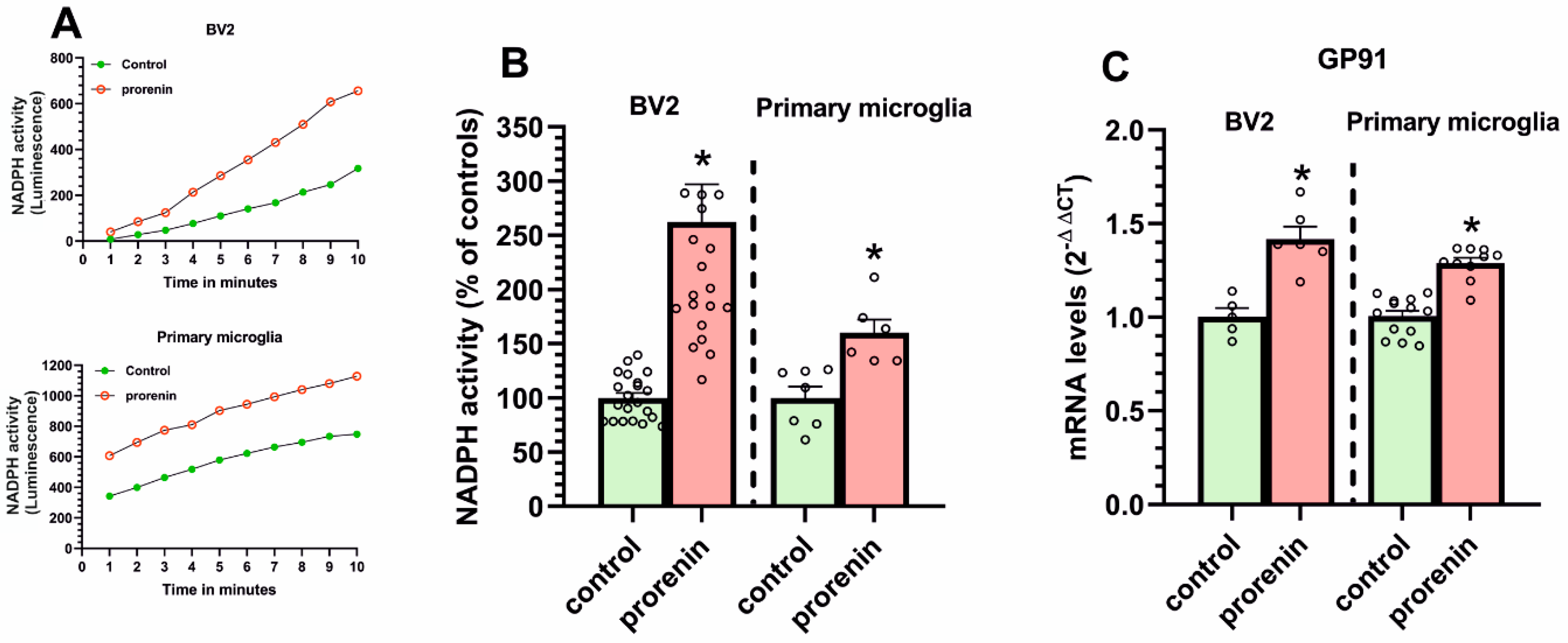
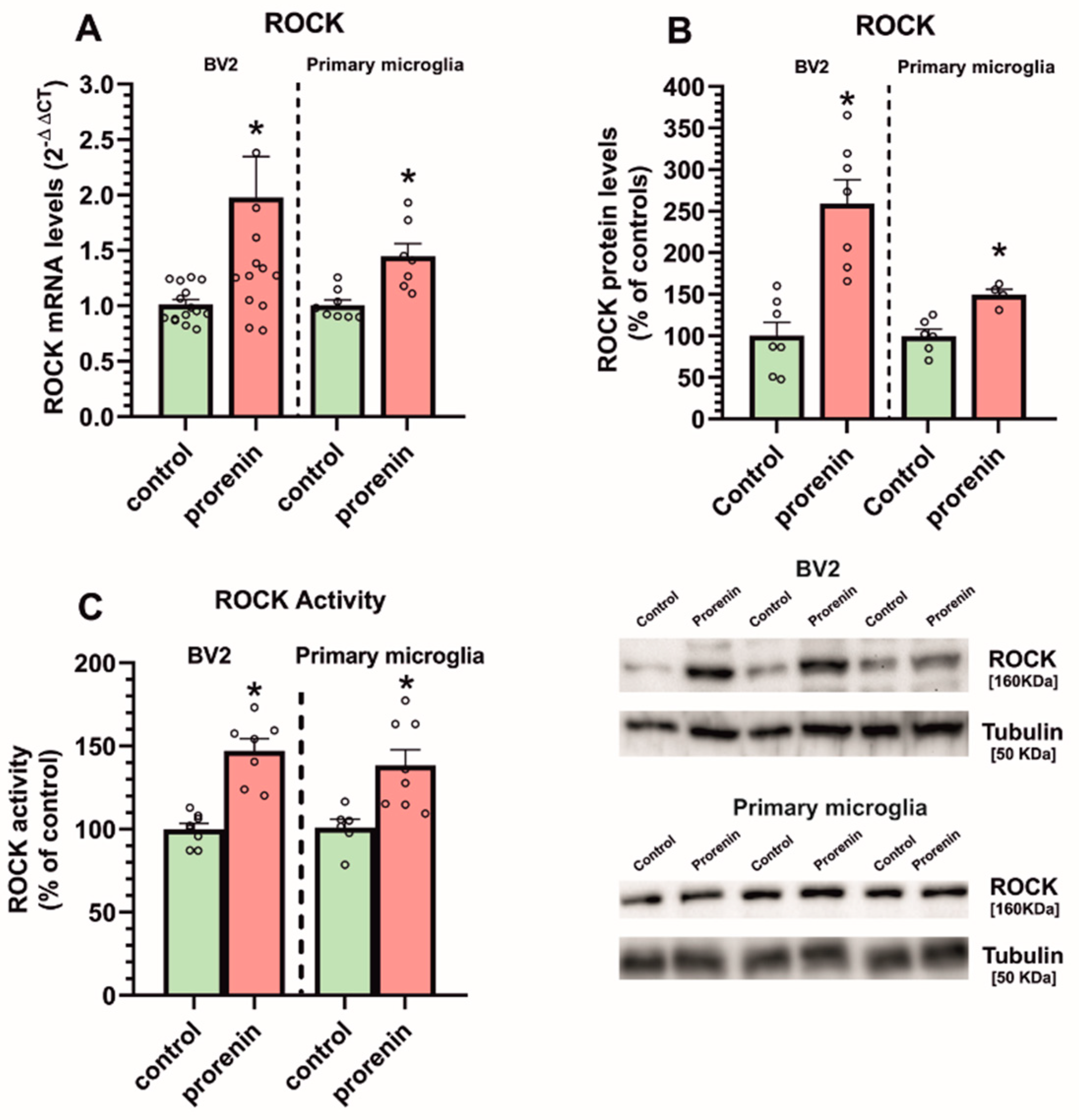
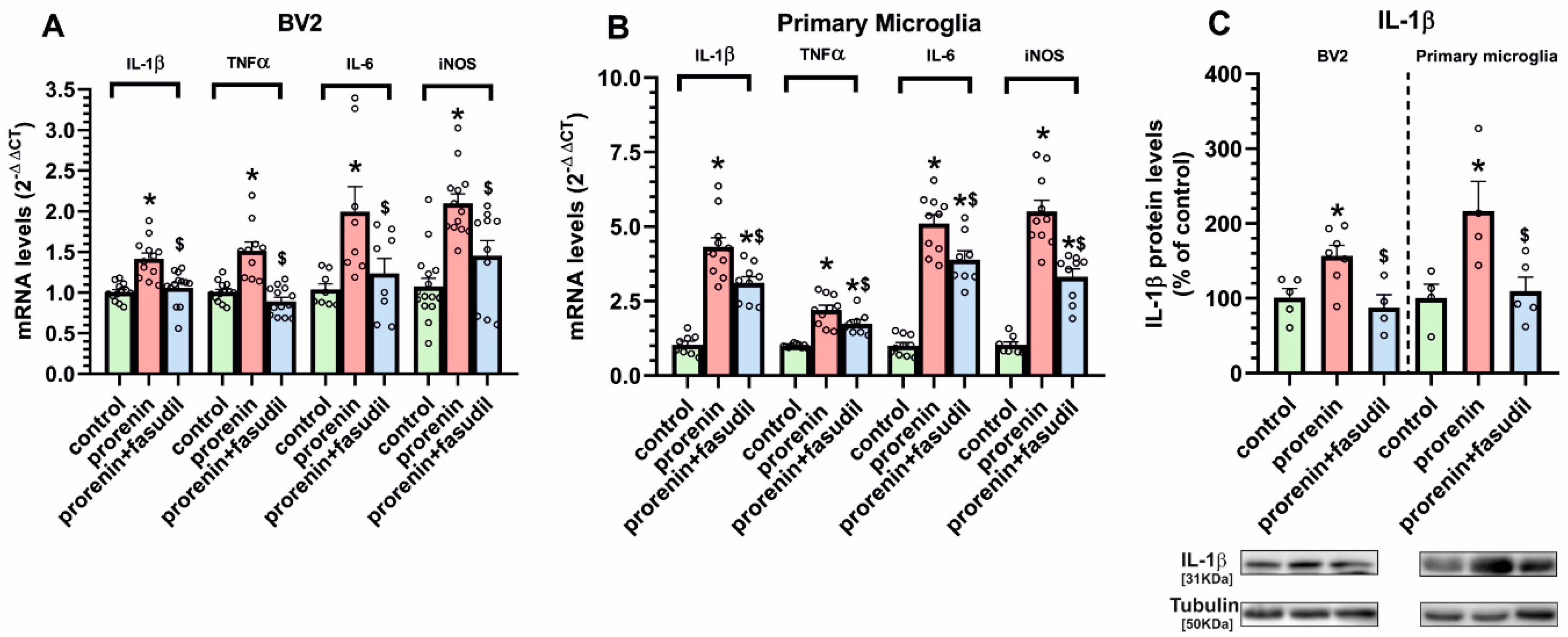
3.2. Cultured Microglia Express (Pro)Renin Receptors That Activated by (Pro)Renin Upregulate Major Components of the Microglial Pro-Inflammatory Response
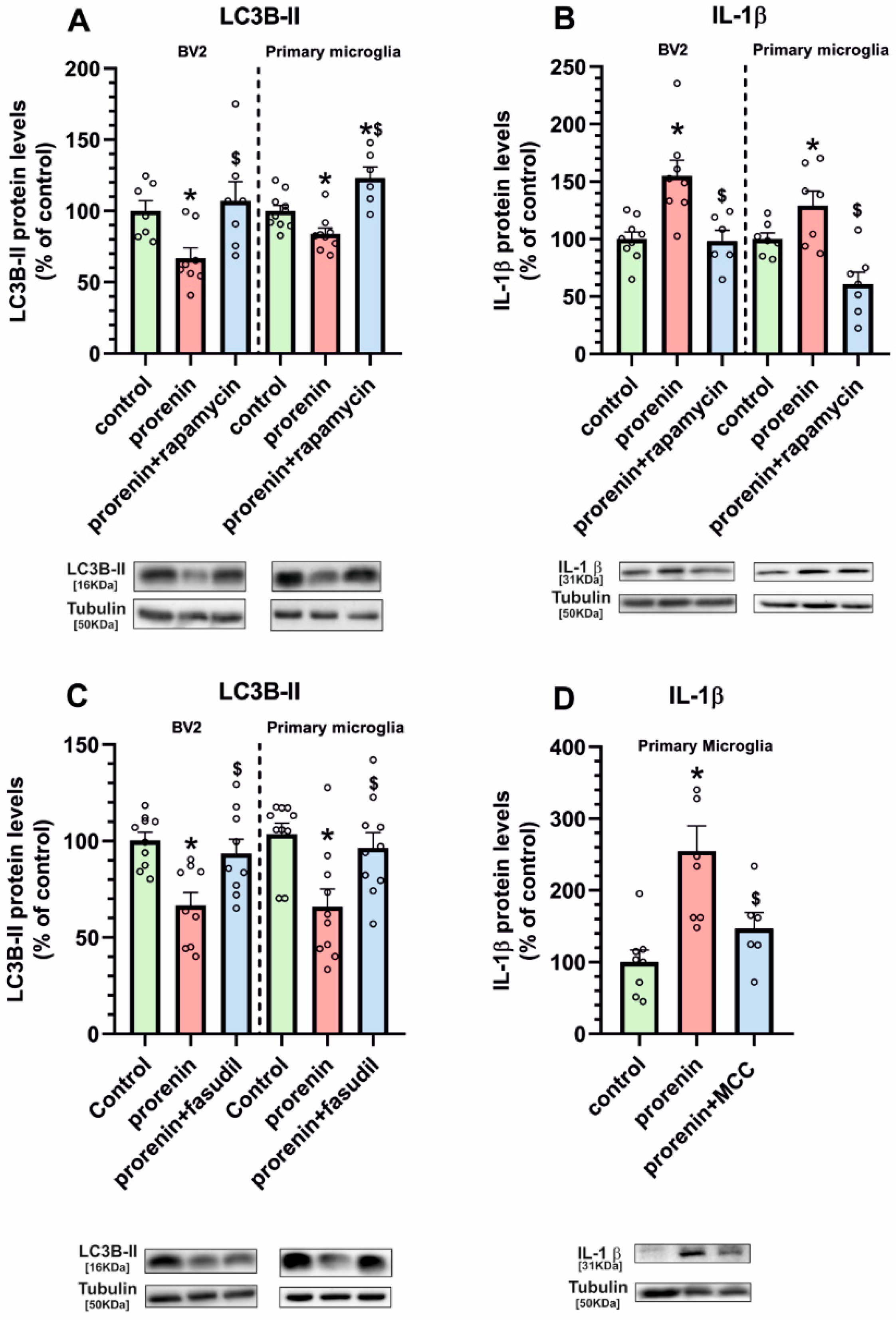
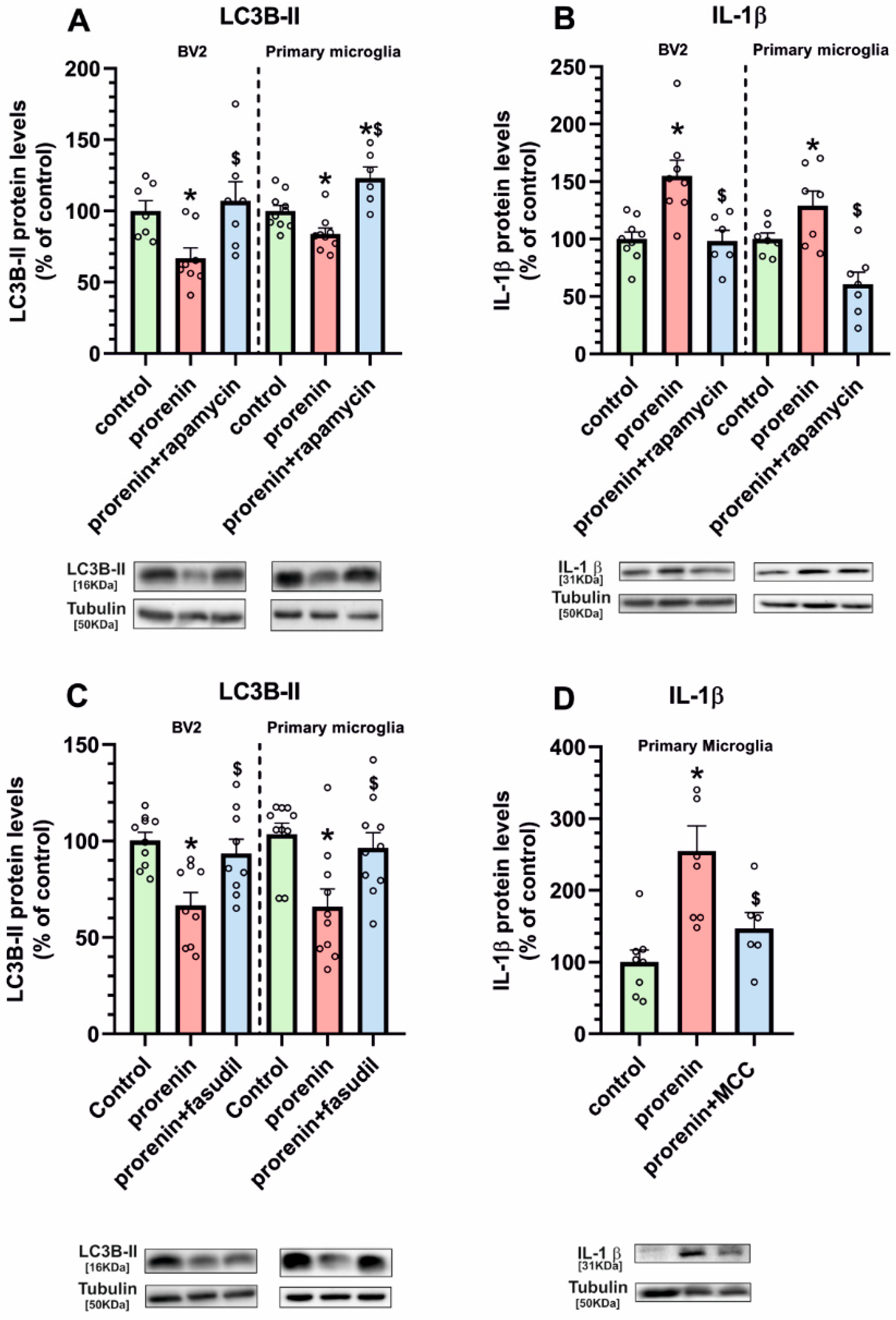
3.3. Microglial Autophagy and Inflammasome Are Involved in the (Pro)Renin-Induced Microglial Pro-Inflammatory Response
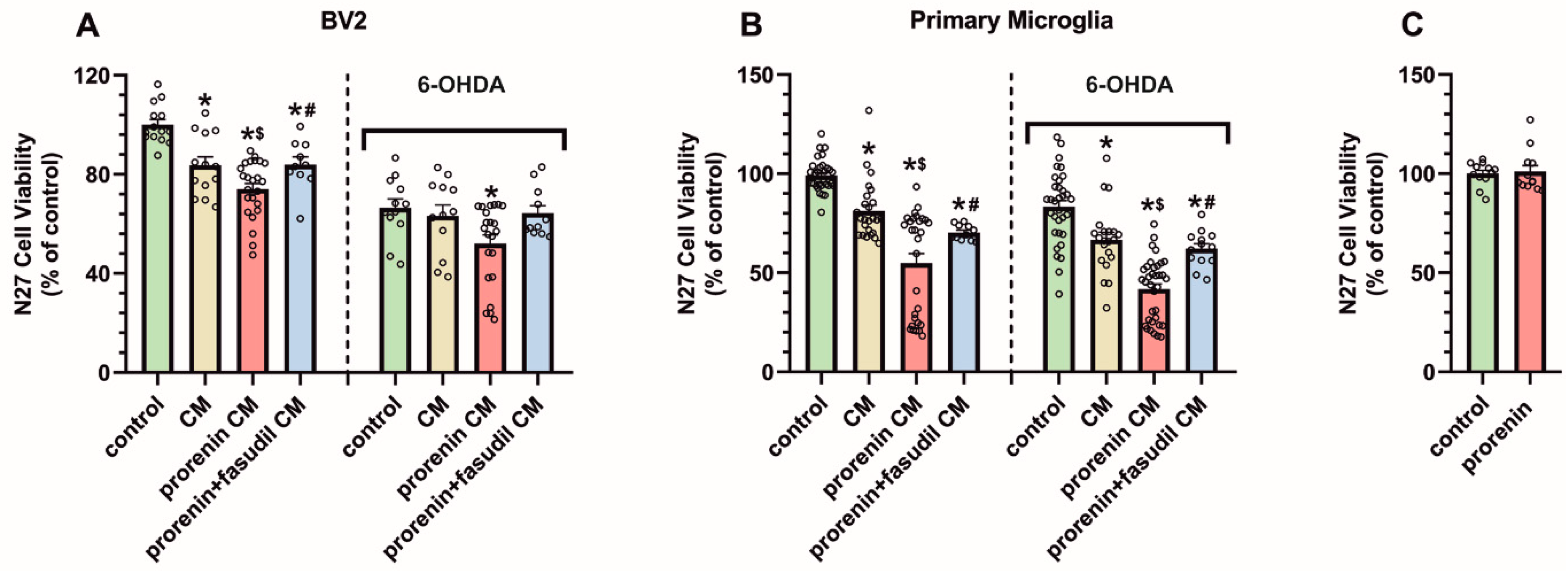
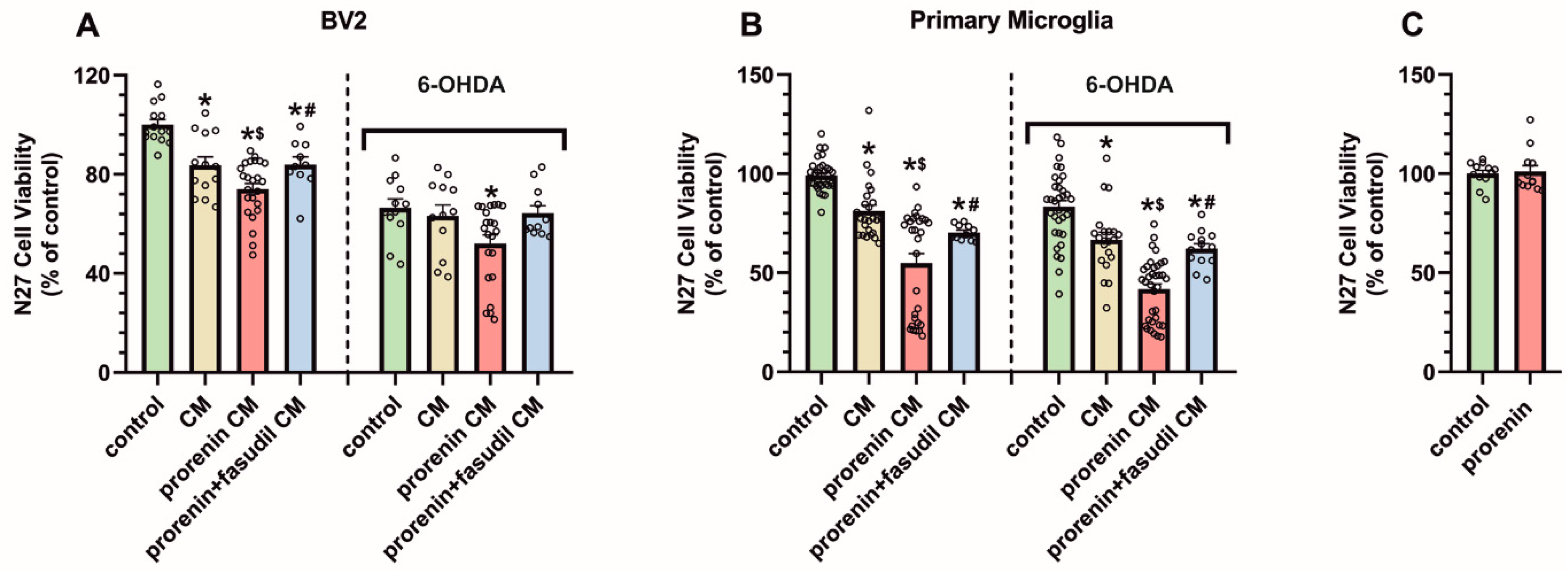
3.4. PRR Activation Significantly Increases the Deleterious Effect of Microglia on Dopaminergic Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| ACE | angiotensin converting enzyme |
| ACE2 | angiotensin converting enzyme 2 |
| Ang | angiotensin |
| AT1 | angiotensin type 1 receptors |
| AT2 | angiotensin type 2 receptors |
| CM | conditioned medium |
| DMEM | Dulbecco’s modified Eagles medium |
| dNTP | deoxynucleotide triphosphate |
| DPBS | Dulbecco’s phosphate buffered saline |
| HRP | horseradish peroxidase |
| IL-1β | interleukin1β |
| IL-6 | interleukin 6 |
| iNOS | inducible nitric oxide synthases |
| LC3B | microtubule associated protein 1 light chain 3 beta |
| LCM | laser capture microdissection |
| LD | levodopa |
| MasR | Mas receptors |
| MCC | N-[[(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)amino]carbonyl]-4-(1-hydroxy-1-methylethyl)-2-furansulfonamide] |
| MCM | conditioned medium from microglia |
| MMLV | Moloney murine leukemia virus reverse transcriptase |
| MTT | 3-(45-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide |
| mRNA | messenger ribonucleic acid |
| NADPH oxidase | nicotinamide adenine dinucleotide phosphate oxidase |
| NLRP | NOD-like receptor family pyrin domain-containing 3 |
| PD | Parkinson´s disease |
| PMSF | phenylmethanesulfonyl fluoride |
| PR | (pro)renin |
| PRR | (pro) renin receptor |
| RAS | renin–angiotensin system |
| RIPA | radio immunoprecipitation assay |
| ROCK | Rho-kinase |
| RPMI | Roswell Park Memorial Institute |
| RT-PCR | reverse transcriptase polymerase chain reaction |
| TNF-α | tumor necrosis factor alpha. |
References
- Jackson-Cowan, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [Green Version]
- Ranjbar, R.; Shafiee, M.; Hesari, A.; Ferns, G.A.; Ghasemi, F.; Avan, A. The potential therapeutic use of renin–angiotensin system inhibitors in the treatment of inflammatory diseases. J. Cell. Physiol. 2018, 234, 2277–2295. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Perez, A.I.R.; Valenzuela, R.; Villar-Cheda, B.; Guerra, M.J.; Labandeira-Garcia, J.L. Dopaminergic neuroprotection of hormonal replacement therapy in young and aged menopausal rats: Role of the brain angiotensin system. Brain 2012, 135, 124–138. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Labandeira-Garcia, J.L.; Perez, A.I.R.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Lanciego, J.L.; Guerra, M.J. Brain Renin-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Front. Aging Neurosci. 2017, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Sarzani, R.; Giulietti, F.; Di Pentima, C.; Giordano, P.; Spannella, F. Disequilibrium between the classic renin-angiotensin system and its opposing arm in SARS-CoV-2-related lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L325–L336. [Google Scholar] [CrossRef]
- Nguyen, G.; Delarue, F.; Burckle, C.; Bouzhir, L.; Giller, T.; Sraer, J.-D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef]
- Ichihara, A.; Yatabe, M.S. The (pro)renin receptor in health and disease. Nat. Rev. Nephrol. 2019, 15, 693–712. [Google Scholar] [CrossRef]
- Schefe, J.H.; Menk, M.; Reinemund, J.; Effertz, K.; Hobbs, R.M.; Pandolfi, P.P.; Ruiz, P.; Unger, T.; Funke-Kaiser, H. A Novel Signal Transduction Cascade Involving Direct Physical Interaction of the Renin/Prorenin Receptor With the Transcription Factor Promyelocytic Zinc Finger Protein. Circ. Res. 2006, 99, 1355–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Z.; Cuadra, A.E.; Sumners, C.; Raizada, M.K. Characterization of a functional (pro)renin receptor in rat brain neurons. Exp. Physiol. 2008, 93, 701–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labandeira-Garcia, J.L.; Rodriguez-Pallares, J.; Meijide, A.D.; Valenzuela, R.; Villar-Cheda, B.; Perez, A.I.R. Dopamine-Angiotensin interactions in the basal ganglia and their relevance for Parkinson’s disease. Mov. Disord. 2013, 28, 1337–1342. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. Contributions by the Brain Renin-Angiotensin System to Memory, Cognition, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pallares, J.; Rey, P.; Parga, J.; Muñoz, A.; Guerra, M.; Labandeira-Garcia, J. Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol. Dis. 2008, 31, 58–73. [Google Scholar] [CrossRef]
- Joglar, B.; Rodriguez-Pallares, J.; Perez, A.I.R.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: Relevance to progression of the disease. J. Neurochem. 2009, 109, 656–669. [Google Scholar] [CrossRef]
- Valenzuela, R.; Barroso-Chinea, P.; Villar-Cheda, B.; Joglar, B.; Muñoz, A.; Lanciego, J.L.; Labandeira-Garcia, J.L. Location of Prorenin Receptors in Primate Substantia Nigra: Effects on Dopaminergic Cell Death. J. Neuropathol. Exp. Neurol. 2010, 69, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gil, P.; Valenzuela, R.; Villar-Cheda, B.; Lanciego, J.L.; Labandeira-Garcia, J.L. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: An intracellular renin–angiotensin system in the nigra. Brain Struct. Funct. 2013, 218, 373–388. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Gil, P.; Perez, A.I.R.; Fernandez-Rodriguez, P.; Lanciego, J.L.; Labandeira-Garcia, J.L. Expression of angiotensinogen and receptors for angiotensin and prorenin in the rat and monkey striatal neurons and glial cells. Brain Struct. Funct. 2017, 222, 2559–2571. [Google Scholar] [CrossRef]
- Shi, P.; Grobe, J.; Desland, F.A.; Zhou, G.; Shen, X.Z.; Shan, Z.; Liu, M.; Raizada, M.K.; Sumners, C. Direct Pro-Inflammatory Effects of Prorenin on Microglia. PLoS ONE 2014, 9, e92937. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Miller, A.G.; Deliyanti, D.; Berka, D.R.; Agrotis, A.; Campbell, D.J.; Wilkinson-Berka, J. Prorenin stimulates a pro-angiogenic and pro-inflammatory response in retinal endothelial cells and an M1 phenotype in retinal microglia. Clin. Exp. Pharmacol. Physiol. 2015, 42, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, A.; Perez, A.I.R.; Diaz-Ruiz, C.; Guerra, M.J.; Labandeira-Garcia, J.L. Microglial TNF-α mediates enhancement of dopaminergic degeneration by brain angiotensin. Glia 2014, 62, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, A.; Perez, A.I.R.; Villar-Cheda, B.; Guerra, M.J.; Labandeira-Garcia, J.L. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology 2014, 85, 1–8. [Google Scholar] [CrossRef]
- Perez, A.I.R.; Borrajo, A.; Rodriguez-Pallares, J.; Guerra, M.J.; Labandeira-Garcia, J.L. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia 2015, 63, 466–482. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rozas, G.; Martín, M.E.L.; Liste, I.; Guerra, M.J. Time course of striatal changes induced by 6-hydroxydopamine lesion of the nigrostriatal pathway, as studied by combined evaluation of rotational behaviour and striatal Fos expression. Exp. Brain Res. 1996, 108, 69–84. [Google Scholar] [CrossRef]
- Walsh, S.; Finn, D.; Dowd, E. Time-course of nigrostriatal neurodegeneration and neuroinflammation in the 6-hydroxydopamine-induced axonal and terminal lesion models of Parkinson’s disease in the rat. Neuroscience 2011, 175, 251–261. [Google Scholar] [CrossRef]
- Barnum, C.; Eskow, K.; Dupre, K.; Blandino, P.; Deak, T.; Bishop, C. Exogenous corticosterone reduces l-DOPA-induced dyskinesia in the hemi-parkinsonian rat: Role for interleukin-1beta. Neuroscience 2008, 156, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Mulas, G.; Espa, E.; Fenu, S.; Spiga, S.; Cossu, G.; Pillai, E.; Carboni, E.; Simbula, G.; Jadžić, D.; Angius, F.; et al. Differential induction of dyskinesia and neuroinflammation by pulsatile versus continuous l -DOPA delivery in the 6-OHDA model of Parkinson’s disease. Exp. Neurol. 2016, 286, 83–92. [Google Scholar] [CrossRef]
- Lopez-Lopez, A.; Labandeira, C.M.; Labandeira-Garcia, J.L.; Muñoz, A. Rho kinase inhibitor fasudil reduces l -DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Br. J. Pharmacol. 2020, 177, 5622–5641. [Google Scholar] [CrossRef]
- Benigni, A.; Corna, D.; Zoja, C.; Sonzogni, A.; Latini, R.; Salio, M.; Conti, S.; Rottoli, D.; Longaretti, L.; Cassis, P.; et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Investig. 2009, 119, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gil, P.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of PPAR-γ in the neuroprotective and anti-inflammatory effects of angiotensin type 1 receptor inhibition: Effects of the receptor antagonist telmisartan and receptor deletion in a mouse MPTP model of Parkinson’s disease. J. Neuroinflamm. 2012, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Perez, A.I.; Garrido-Gil, P.; Pedrosa, M.A.; Garrote, M.G.; Valenzuela, R.; Navarro, G.; Franco, R.; Labandeira-Garcia, J.L. Angiotensin type 2 receptors: Role in aging and neuroinflammation in the substantia nigra. Brain Behav. Immun. 2020, 87, 256–271. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 7th ed.; Academic Press: New York, NY, USA, 2013. [Google Scholar]
- Winklerab, C.; Kirika, D.; Björklund, A.; Cenci, M. l-DOPA-Induced Dyskinesia in the Intrastriatal 6-Hydroxydopamine Model of Parkinson’s Disease: Relation to Motor and Cellular Parameters of Nigrostriatal Function. Neurobiol. Dis. 2002, 10, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Schallert, T.; Tillerson, J.L. Intervention Strategies for Degeneration of Dopamine Neurons in Parkinsonism: Optimising Behavioural Assessment of Outcome; Humana Press: Clifton, NJ, USA, 1999. [Google Scholar]
- Cenci, M.A.; Lee, C.S.; Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase Mrna. Eur. J. Neurosci. 1998, 10, 2694–2706. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gil, P.; Fernandez-Rodríguez, P.; Rodríguez-Pallares, J.; Labandeira-Garcia, J.L. Laser capture microdissection protocol for gene expression analysis in the brain. Histochem. Cell Biol. 2017, 148, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Rodriguez-Pallares, J.; Valenzuela, R.; Muñoz, A.; Guerra, M.J.; Baltatu, O.C.; Labandeira-Garcia, J.L. Nigral and striatal regulation of angiotensin receptor expression by dopamine and angiotensin in rodents: Implications for progression of Parkinson’s disease. Eur. J. Neurosci. 2010, 32, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Höing, S.; Rudhard, Y.; Reinhardt, P.; Glatza, M.; Stehling, M.; Wu, G.; Peiker, C.; Böcker, A.; Parga, J.A.; Bunk, E.; et al. Discovery of Inhibitors of Microglial Neurotoxicity Acting Through Multiple Mechanisms Using a Stem-Cell-Based Phenotypic Assay. Cell Stem Cell 2012, 11, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-Y.; Tang, R.-H.; Qi, R.-Q. Interleukin-4 affects microglial autophagic flux. Neural Regen. Res. 2019, 14, 1594–1602. [Google Scholar] [CrossRef]
- Liu, C.; Ma, H.; Slitt, A.L.; Seeram, N.P. Inhibitory Effect of Cannabidiol on the Activation of NLRP3 Inflammasome Is Associated with Its Modulation of the P2X7 Receptor in Human Monocytes. J. Nat. Prod. 2020, 83, 2025–2029. [Google Scholar] [CrossRef]
- Villar-Cheda, B.; Meijide, A.D.; Joglar, B.; Perez, A.I.R.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of microglial RhoA/Rho-Kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012, 47, 268–279. [Google Scholar] [CrossRef]
- Li, C.; Siragy, H.M. (Pro)renin receptor regulates autophagy and apoptosis in podocytes exposed to high glucose. Am. J. Physiol. Metab. 2015, 309, E302–E310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parga, J.A.; Rodriguez-Perez, A.I.; Garrote, M.G.; Rodriguez-Pallares, J.; Labandeira-Garcia, J.L. Angiotensin II induces oxidative stress and upregulates neuroprotective signaling from the NRF2 and KLF9 pathway in dopaminergic cells. Free Radic. Biol. Med. 2018, 129, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Matavelli, L.C.; Huang, J.; Siragy, H.M. (Pro)renin receptor contributes to diabetic nephropathy by enhancing renal inflammation. Clin. Exp. Pharmacol. Physiol. 2010, 37, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadri, S.S.; Culver, S.; Siragy, H.M. Prorenin receptor mediates inflammation in renal ischemia. Clin. Exp. Pharmacol. Physiol. 2018, 45, 133–139. [Google Scholar] [CrossRef]
- Dong, X.; Yu, S.; Wang, Y.; Yang, M.; Xiong, J.; Hei, N.; Dong, B.; Su, Q.; Chen, J. (Pro)renin receptor-mediated myocardial injury, apoptosis, and inflammatory response in rats with diabetic cardiomyopathy. J. Biol. Chem. 2019, 294, 8218–8226. [Google Scholar] [CrossRef] [Green Version]
- Cooper, S.G.; Trivedi, D.P.; Yamamoto, R.; Worker, C.J.; Feng, C.-Y.; Sorensen, J.T.; Yang, W.; Xiong, Z.; Feng, Y. Increased (pro)renin receptor expression in the subfornical organ of hypertensive humans. Am. J. Physiol. Circ. Physiol. 2018, 314, H796–H804. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, M.; Souza, L.A.C.; Aliabadi, S.; Worker, C.J.; Cooper, S.G.; Afrin, S.; Murata, Y.; Xiong, Z.; Earley, Y.F. Increased (Pro)renin Receptor Expression in the Hypertensive Human Brain. Front. Physiol. 2020, 11, 606811. [Google Scholar] [CrossRef]
- Wang, G.; Anrather, J.; Huang, J.; Speth, R.C.; Pickel, V.M.; Iadecola, C. NADPH Oxidase Contributes to Angiotensin II Signaling in the Nucleus Tractus Solitarius. J. Neurosci. 2004, 24, 5516–5524. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, F.; Annese, V.; Sauvage, M.A.C.-D.; Ros-Bernal, F.; Gómez, A.; Yuste, J.E.; Campuzano, C.M.; De Pablos, V.; Fernandez-Villalba, E.; et al. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci. Rep. 2012, 2, 809. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhou, X.; Guo, J.-J.; Mao, L.; Wang, Y.-J.; Sun, J.; Sun, L.-X.; Zhang, L.-Y.; Zhou, X.-F.; Liao, H. Nogo-66 inhibits adhesion and migration of microglia via GTPase Rho pathway in vitro. J. Neurochem. 2011, 120, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xu, Y.; Zhu, L.; Rao, P.; Wen, J.; Sang, Y.; Shang, F.; Liu, Y. Fasudil inhibits LPS-induced migration of retinal microglial cells via regulating p38-MAPK signaling pathway. Mol. Vis. 2016, 22, 836–846. [Google Scholar]
- Tönges, L.; Frank, T.; Tatenhorst, L.; Saal, K.A.; Koch, J.C.; Szegő, É.M.; Bähr, M.; Weishaupt, J.H.; Lingor, P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain 2012, 135, 3355–3370. [Google Scholar] [CrossRef] [PubMed]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Da Fonseca, T.L.; Koch, J.C.; Becker, S.; Tönges, L.; Bähr, M.; et al. Fasudil attenuates aggregation of α-synuclein in models of Parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labandeira-Garcia, J.L.; Perez, A.I.R.; Villar-Cheda, B.; Borrajo, A.; Meijide, A.D.; Guerra, M.J. Rho Kinase and Dopaminergic Degeneration. Neuroscientist 2015, 21, 616–629. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, Y.-F.; Xi, J.-Y.; Yu, W.-B.; Xiao, B.-G. Rho kinase II interference by small hairpin RNA ameliorates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Mol. Med. Rep. 2016, 14, 4947–4956. [Google Scholar] [CrossRef] [Green Version]
- Han, H.E.; Kim, T.K.; Son, H.J.; Park, W.J.; Han, P.L. Activation of Autophagy Pathway Suppresses the Expression of iNOS, IL6 and Cell Death of LPS-Stimulated Microglia Cells. Biomol. Ther. Seoul. 2013, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zhou, T.; Chen, Y.; Lin, D.; Jing, X.; Peng, S.; Zheng, D.; Zeng, Z.; Lei, M.; Wu, X.; et al. Rifampicin inhibits rotenone-induced microglial inflammation via enhancement of autophagy. Neurotoxicology 2017, 63, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tönges, L.; Barski, E.; Michel, U.; Bahr, M.; Lingor, P. ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell Death Dis. 2014, 5, 1225. [Google Scholar] [CrossRef] [Green Version]
- Bauer, P.; Wong, H.-K.; Oyama, F.; Goswami, A.; Okuno, M.; Kino, Y.; Miyazaki, H.; Nukina, N. Inhibition of Rho Kinases Enhances the Degradation of Mutant Huntingtin. J. Biol. Chem. 2009, 284, 13153–13164. [Google Scholar] [CrossRef] [Green Version]
- Gentry, E.G.; Henderson, B.W.; Arrant, A.E.; Gearing, M.; Feng, Y.; Riddle, N.C.; Herskowitz, J.H. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J. Neurosci. 2016, 36, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.-T.; Yang, Y.-J.; Wu, J.-J.; Li, S.; Tang, Y.-L.; Zhao, J.; Liu, Z.-Y.; Xiao, B.-G.; Zuo, J.; Liu, W.; et al. Fasudil, a Rho kinase inhibitor, promotes the autophagic degradation of A53T α-synuclein by activating the JNK 1/Bcl-2/beclin 1 pathway. Brain Res. 2016, 1632, 9–18. [Google Scholar] [CrossRef]
- Riediger, F.; Quack, I.; Qadri, F.; Hartleben, B.; Park, J.-K.; Potthoff, S.A.; Sohn, D.; Sihn, G.; Rousselle, A.; Fokuhl, V.; et al. Prorenin Receptor Is Essential for Podocyte Autophagy and Survival. J. Am. Soc. Nephrol. 2011, 22, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Binger, K.J.; Muller, D.N. Autophagy and the (Pro)renin Receptor. Front. Endocrinol. 2013, 4, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.-G.; Zhou, X.-G.; Qiao, G.; Yu, L.; Tang, Y.; Yan, L.; Qiu, W.-Q.; Pan, R.; Yu, C.-L.; Law, B.Y.-K.; et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 2021, 65, 101202. [Google Scholar] [CrossRef]
- Haque, E.; Akther, M.; Jakaria, K.I.; Azam, S.; Choi, D. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson’s Disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) | |
|---|---|---|---|
| Rat/Mouse | Actβ | TCGTGCGTGACATTAAAGAG | TGCCACAGGATTCCATACC |
| Rat | Gp91 | ATCTTGCTGCCAGTGTGTCG | AATGGTGTGAATGGCCGTGTG |
| IL-1β | GGCAACTGTCCCTGAACTCA | TGTCGAGATGCTGCTGTGAGA | |
| IL-6 | GGATACCACCCACAACAGACC | AGTGCATCATCGCGTTCATACACA | |
| iNOS | CAGGCTTGGGTCTTGTTAGCC | GCCATGTCTGTGACTTTGTGCTT | |
| PRR | TGGTGGGAATGCAGTGGTAGAG | GGGACTTTGGGTGTTCTCTTGTT | |
| ROCKII | GTTCAGTTGGTTCGTCATAAGGCA | TGAACCACCCACGGACTGTT | |
| Tnf-α | CACGTCGTAGCAAACCACCA | GGTTGTCTTTGAGATCCATGCCA | |
| Mouse | Gp91 | GGAGTTCCAAGATGCCTGGA | CCACTAACATCACCACCTCATAGC |
| IL-1β | GCTATGGCAACTGTTCCTGA | TGATGTGCTGCTGCGAGA | |
| IL-6 | GACTGATGCTGGTGACAAC | GAGTGGTATCCTCTGTGAA | |
| iNOS | TGGTGAAGGGACTGAGCTGTTA | CAGGGGCAAGCCATGTCTGAG | |
| PRR | GTTTGTTGTCTCGTCATAAGC | ACTCTACCACTGCGTTCC | |
| ROCKII | CGAATAGAACTCCAGATGACC | GCACAGGCAATGACAACC | |
| Tnf-α | TGTGCTCAGAGCTTTCAACAA | CTTGATGGTGGTGCATGAGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Lopez, A.; Villar-Cheda, B.; Quijano, A.; Garrido-Gil, P.; Garcia-Garrote, M.; Díaz-Ruiz, C.; Muñoz, A.; Labandeira-Garcia, J.L. NADPH-Oxidase, Rho-Kinase and Autophagy Mediate the (Pro)renin-Induced Pro-Inflammatory Microglial Response and Enhancement of Dopaminergic Neuron Death. Antioxidants 2021, 10, 1340. https://doi.org/10.3390/antiox10091340
Lopez-Lopez A, Villar-Cheda B, Quijano A, Garrido-Gil P, Garcia-Garrote M, Díaz-Ruiz C, Muñoz A, Labandeira-Garcia JL. NADPH-Oxidase, Rho-Kinase and Autophagy Mediate the (Pro)renin-Induced Pro-Inflammatory Microglial Response and Enhancement of Dopaminergic Neuron Death. Antioxidants. 2021; 10(9):1340. https://doi.org/10.3390/antiox10091340
Chicago/Turabian StyleLopez-Lopez, Andrea, Begoña Villar-Cheda, Aloia Quijano, Pablo Garrido-Gil, María Garcia-Garrote, Carmen Díaz-Ruiz, Ana Muñoz, and José L. Labandeira-Garcia. 2021. "NADPH-Oxidase, Rho-Kinase and Autophagy Mediate the (Pro)renin-Induced Pro-Inflammatory Microglial Response and Enhancement of Dopaminergic Neuron Death" Antioxidants 10, no. 9: 1340. https://doi.org/10.3390/antiox10091340
APA StyleLopez-Lopez, A., Villar-Cheda, B., Quijano, A., Garrido-Gil, P., Garcia-Garrote, M., Díaz-Ruiz, C., Muñoz, A., & Labandeira-Garcia, J. L. (2021). NADPH-Oxidase, Rho-Kinase and Autophagy Mediate the (Pro)renin-Induced Pro-Inflammatory Microglial Response and Enhancement of Dopaminergic Neuron Death. Antioxidants, 10(9), 1340. https://doi.org/10.3390/antiox10091340