
Agaricales Mushroom Lignin Peroxidase: From Structure–Function to Degradative Capabilities

 ,
,  , ,
, , 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Synthesis and Site-Directed Mutagenesis
2.2. Heterologous Expression and In Vitro Activation
2.3. Protein Purification and Quantification
2.4. ApeLiP Crystallization, Data Collection, and Refinement
2.5. pH and Temperature Stability
2.6. Steady-State Kinetic Constants
2.7. Native and Acetylated Softwood and Hardwood Lignins
2.8. Estimation of Transient-State Kinetic Constants with Lignosulfonates
2.9. Estimation of CI/RS, CI/CII and CII/RS Reduction Potentials
2.10. Steady-State Treatment of Lignin
2.11. 2D-NMR Analyses
3. Results
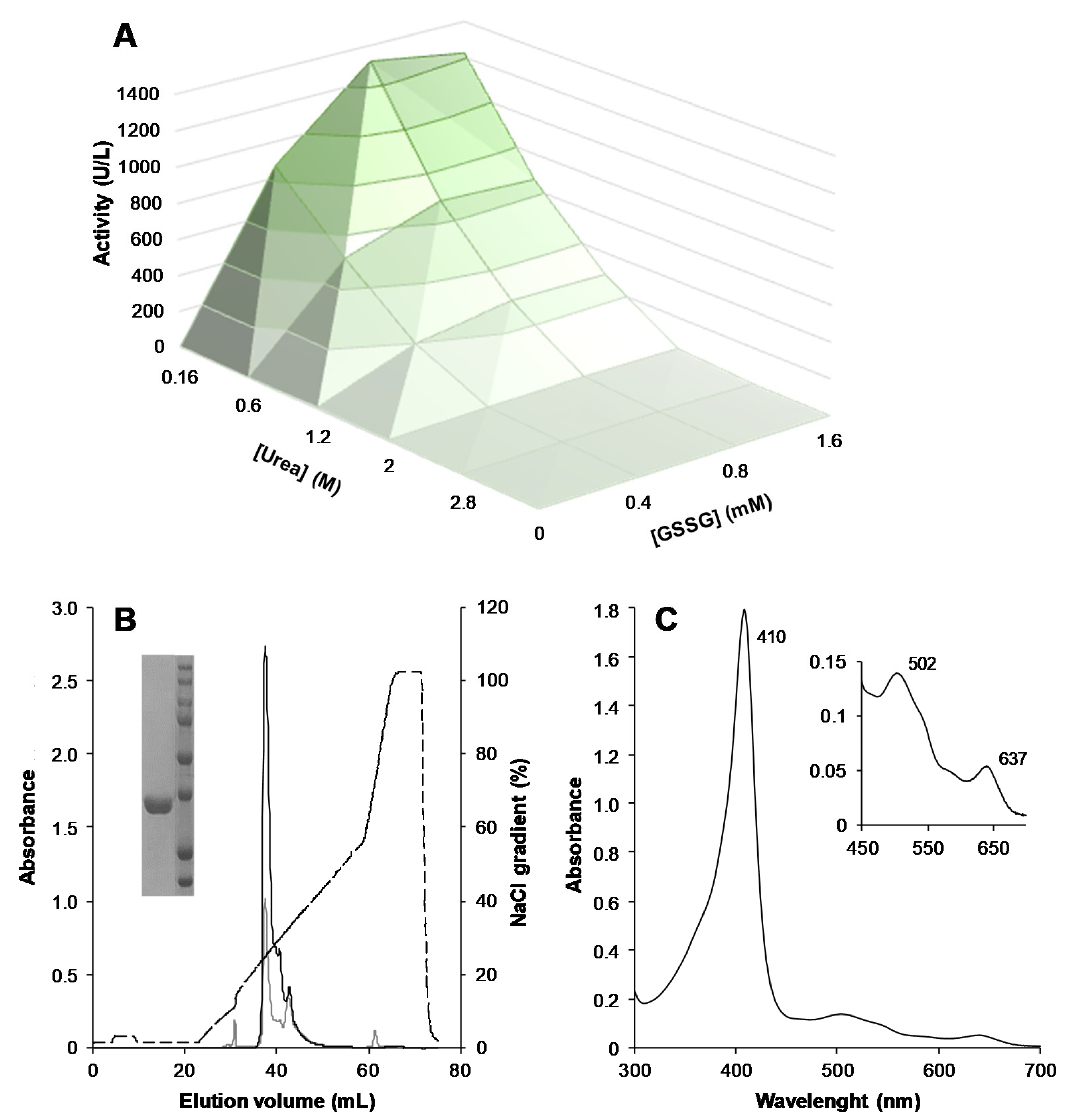
3.1. Heterologous Expression, Protein Activation, and Purification of ApeLiP
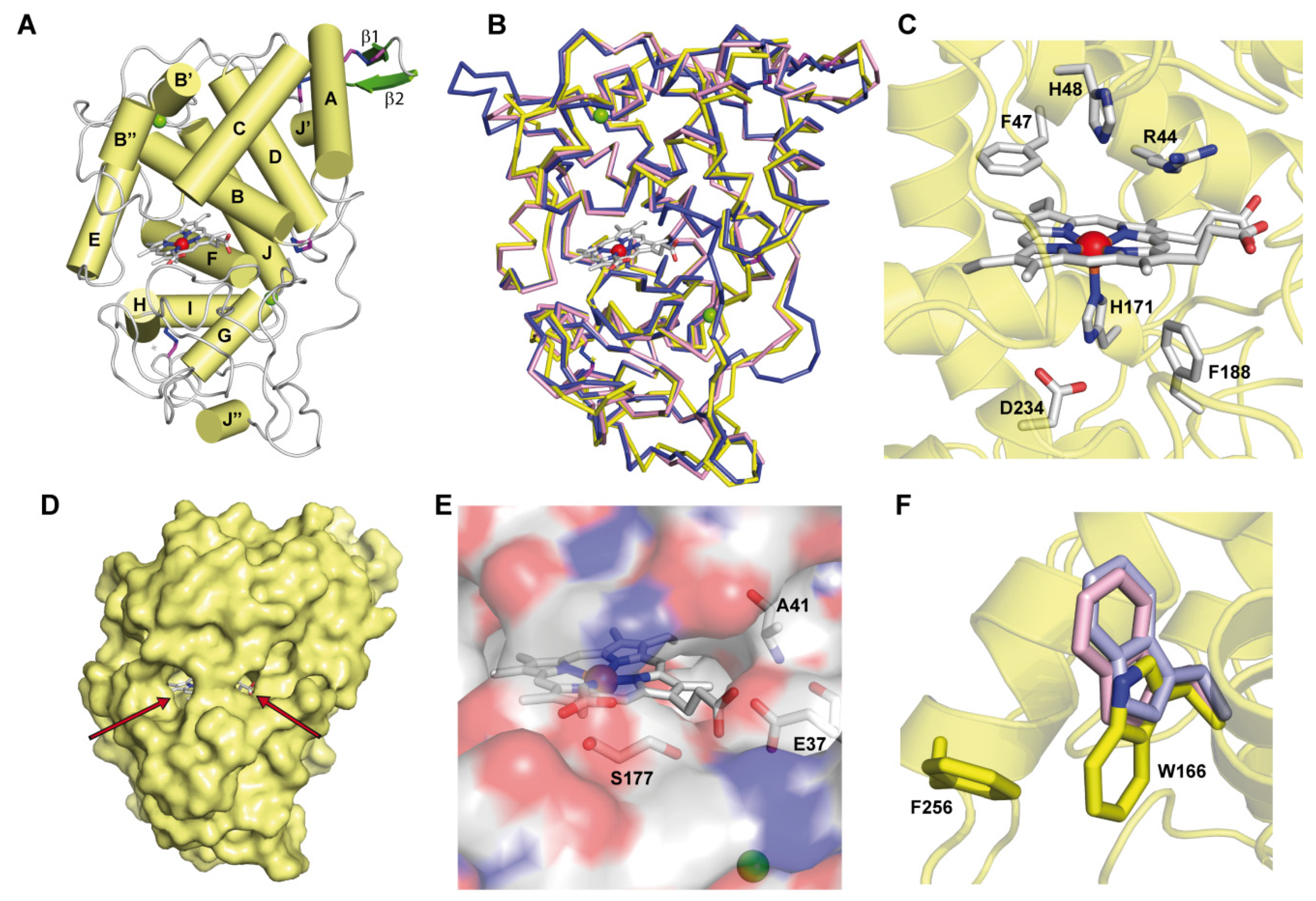
3.2. ApeLiP Crystallographic Studies
3.3. Temperature and pH Stability of Recombinant ApeLiP
3.4. Steady-State Kinetic Properties of ApeLiP
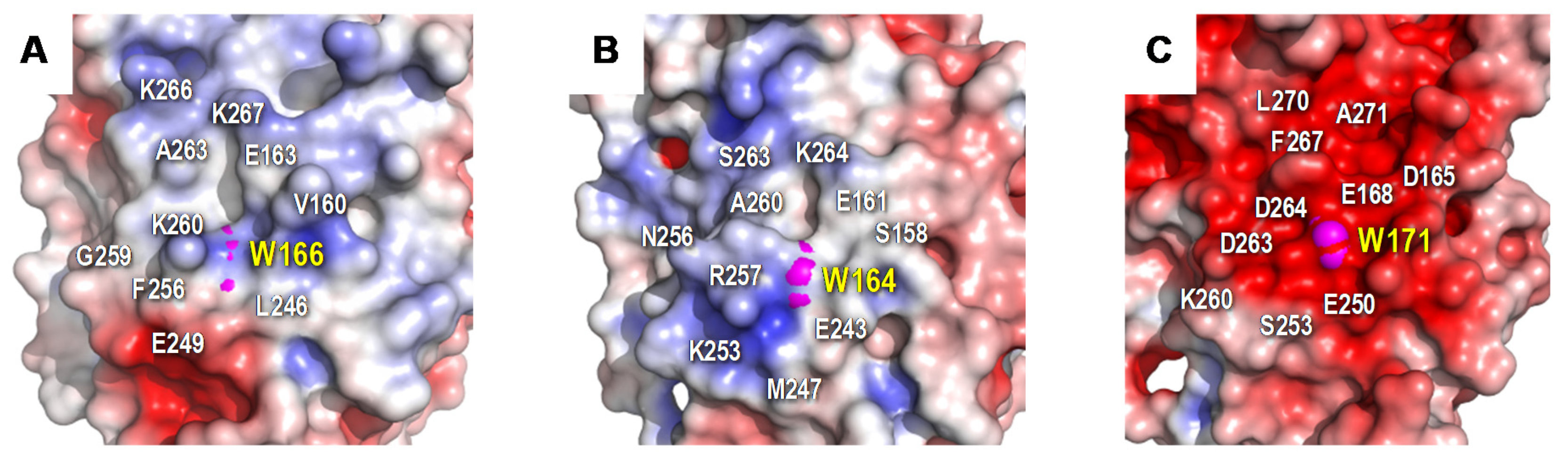
3.5. Identification of Trp166 as a Catalytic Residue
3.6. Catalytic Cycle and ApeLiP Reduction Potentials
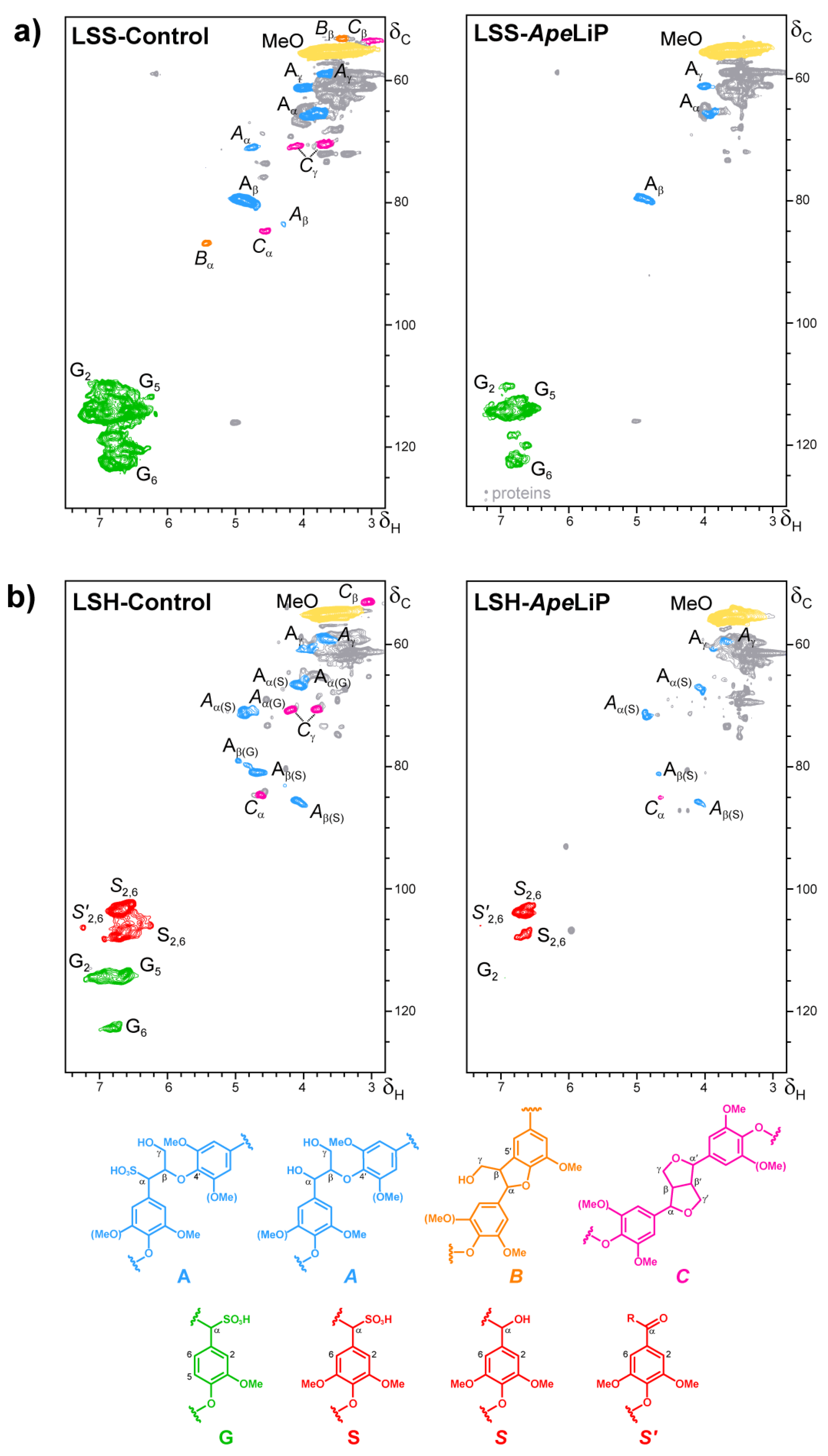
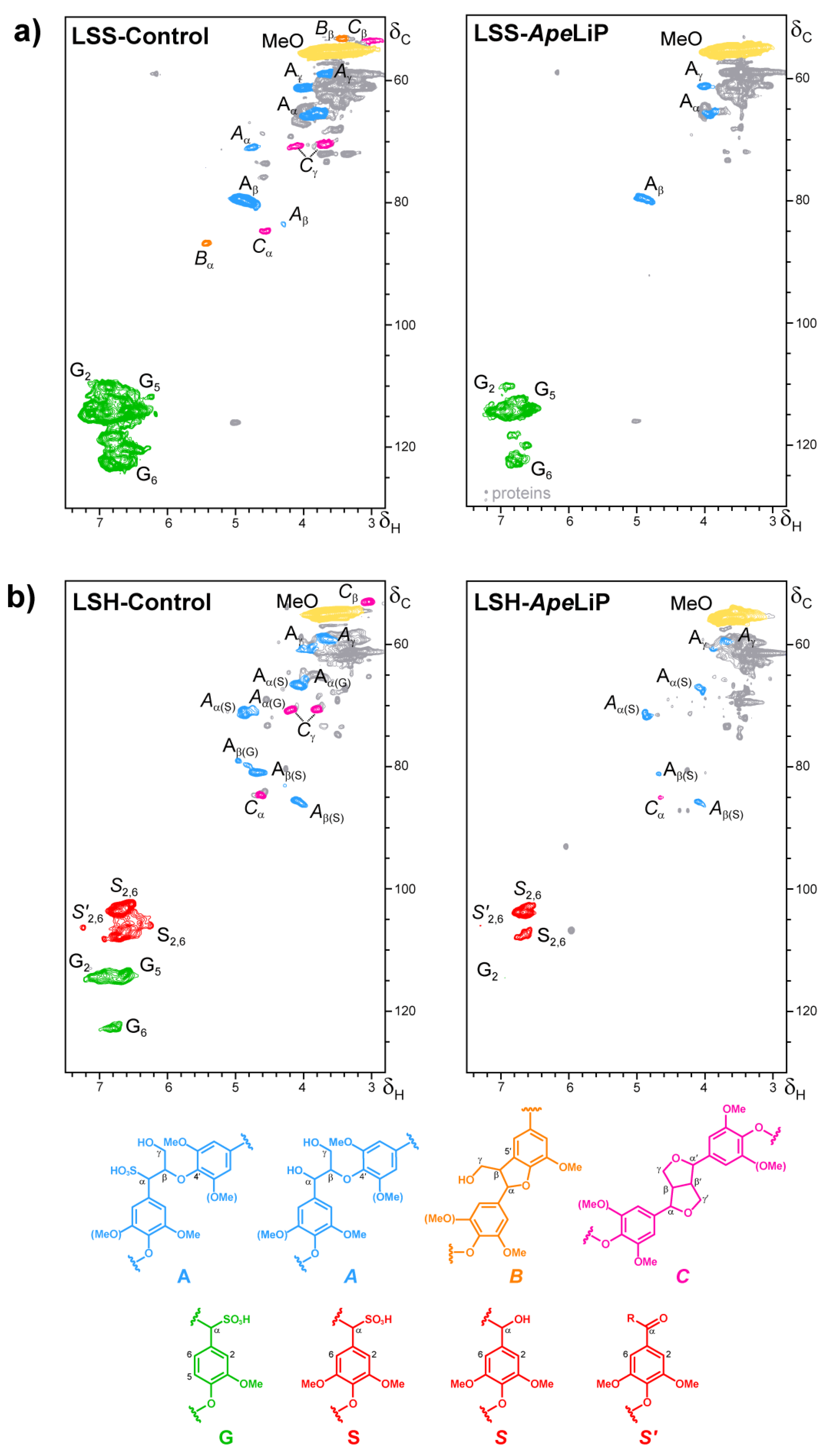
3.7. Lignin Oxidation by ApeLiP: Transient-State Kinetic Data and NMR Analyses
4. Discussion
4.1. Lignin Peroxidase in Agaricales Genomes
4.2. ApeLiP Activation and LRET Oxidation of Lignin Models
4.3. Heme-Channel Oxidation Site
4.4. Intriguing Catalytic Cycle and Lignin Decay Abilities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamm, B.; Gruber, P.R.; Kamm, M. Biorefineries-Industrial Processes and Products: Status Quo and Future Directions; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2010. [Google Scholar]
- Wagemann, K.; Tippkötter, N. Biorefineries; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Fengel, D.; Wegener, G. Wood: Chemistry, Ultrastructure, Reactions; De Gruyter: Berlin, Germany, 1984. [Google Scholar]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Ralph, J.; Lapierre, C.; Boerjan, W. Lignin structure and its engineering. Curr. Opin. Biotechnol. 2019, 56, 240–249. [Google Scholar] [CrossRef]
- Camarero, S.; Martínez, M.J.; Martínez, A.T. Understanding lignin biodegradation for the improved utilization of plant biomass in modern biorefineries. Biofuels Bioprod. Biorefining 2014, 8, 615–625. [Google Scholar] [CrossRef]
- Himmel, M.E.; Ding, S.Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef] [Green Version]
- Beckham, G.T.; Johnson, C.W.; Karp, E.M.; Salvachúa, D.; Vardon, D.R. Opportunities and challenges in biological lignin valorization. Curr. Opin. Biotechnol. 2016, 42, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, A.T.; Camarero, S.; Ruiz-Dueñas, F.J.; Martínez, M.J. Biological lignin degradation. In Lignin Valorization: Emerging Approaches; Beckham, G.T., Ed.; RSC: Cambridge, UK, 2018; pp. 199–225. [Google Scholar]
- Kamimura, N.; Sakamoto, S.; Mitsuda, N.; Masai, E.; Kajita, S. Advances in microbial lignin degradation and its applications. Curr. Opin. Biotechnol. 2019, 56, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Limura, Y.; Abe, H.; Otsuka, Y.; Sato, Y.; Habe, H. Bacterial community coexisting with white-rot fungi in decayed wood in Nature. Curr. Microbiol. 2021, 78, 3112–3217. [Google Scholar]
- Higuchi, Y.; Takahashi, K.; Kamimura, N.; Masai, E. Bacterial Enzymes for the Cleavage of Lignin b-Aryl Ether Bonds: Properties and Applications. In Lignin Valorization: Emerging Approaches; Beckham, G.T., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2018; Chapter 9; pp. 226–251. [Google Scholar]
- Martínez, A.T.; Speranza, M.; Ruiz-Dueñas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; del Río, J.C. Biodegradation of lignocellulosics: Microbiological, chemical and enzymatic aspects of fungal attack to lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar]
- Kirk, T.K.; Farrell, R.L. Enzymatic “combustion”: The microbial degradation of lignin. Annu. Rev. Microbiol. 1987, 41, 465–505. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.-E.L.; Blanchette, R.A.; Ander, P. Microbial and Enzymatic Degradation of Wood Components; Springer: Berlin, Germany, 1990. [Google Scholar]
- Hammel, K.E.; Cullen, D. Role of fungal peroxidases in biological ligninolysis. Curr. Opin. Plant Biol. 2008, 11, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Zámocký, M.; Hofbauer, S.; Schaffner, I.; Gasselhuber, B.; Nicolussi, A.; Soudi, M.; Pirker, K.F.; Furtmüller, P.G.; Obinger, C. Independent evolution of four heme peroxidase superfamilies. Arch. Biochem. Biophys. 2015, 574, 108–119. [Google Scholar] [CrossRef]
- Gold, M.H.; Youngs, H.L.; Gelpke, M.D. Manganese peroxidase. Met. Ions Biol. Syst. 2000, 37, 559–586. [Google Scholar]
- Ayuso-Fernández, I.; Martínez, A.T.; Ruiz-Dueñas, F.J. Experimental recreation of the evolution of lignin degrading enzymes from the Jurassic to date. Biotechnol. Biofuels 2017, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, W.A.; Blodig, W.; Veitch, N.C.; Piontek, K.; Smith, A.T. Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry 1998, 37, 15097–15105. [Google Scholar] [CrossRef] [PubMed]
- Mester, T.; Ambert-Balay, K.; Ciofi-Baffoni, S.; Banci, L.; Jones, A.D.; Tien, M. Oxidation of a tetrameric nonphenolic lignin model compound by lignin peroxidase. J. Biol. Chem. 2001, 276, 22985–22990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Dueñas, F.J.; Morales, M.; García, E.; Miki, Y.; Martínez, M.J.; Martínez, A.T. Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J. Exp. Bot. 2009, 60, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Ayuso-Fernández, I.; Ruiz-Dueñas, F.J.; Martínez, A.T. Evolutionary convergence in lignin degrading enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 6428–6433. [Google Scholar] [CrossRef] [Green Version]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Gelpke, M.D.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef]
- Johjima, T.; Itoh, H.; Kabuto, M.; Tokimura, F.; Nakagawa, T.; Wariishi, H.; Tanaka, H. Direct interaction of lignin and lignin peroxidase from Phanerochaete chrysosporium. Proc. Natl. Acad. Sci. USA 1999, 96, 1989–1994. [Google Scholar] [CrossRef] [Green Version]
- Sáez-Jiménez, V.; Baratto, M.C.; Pogni, R.; Rencoret, J.; Gutiérrez, A.; Santos, J.I.; Martínez, A.T.; Ruiz-Dueñas, F.J. Demonstration of lignin-to-peroxidase direct electron transfer: A transient-state kinetics, directed mutagenesis, EPR and NMR study. J. Biol. Chem. 2015, 290, 23201–23213. [Google Scholar] [CrossRef] [Green Version]
- Sáez-Jiménez, V.; Rencoret, J.; Rodríguez-Carvajal, M.A.; Gutiérrez, A.; Ruiz-Dueñas, F.J.; Martínez, A.T. Role of surface tryptophan for peroxidase oxidation of nonphenolic lignin. Biotechnol. Biofuels 2016, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Tien, M.; Kirk, T.K. Lignin-degrading enzyme from Phanerochaete chrysosporium: Purification, characterization, and catalytic properties of a unique H2O2-requiring oxygenase. Proc. Natl. Acad. Sci. USA 1984, 81, 2280–2284. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Dueñas, F.J.; Barrasa, J.M.; Sánchez-García, M.; Camarero, S.; Miyauchi, S.; Serrano, A.; Linde, D.; Babiker, R.; Drula, E.; Ayuso-Fernández, I.; et al. Genomic analysis enlightens Agaricales lifestyle evolution and increasing peroxidase diversity. Mol. Biol. Evol. 2021, 38, 1428–1446. [Google Scholar] [CrossRef]
- Puigbò, P.; Guzmán, E.; Romeu, A.; Garcia-Vallvé, S. OPTIMIZER: A web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007, 35, W126–W131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning, 3rd ed.; CSHL Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Pérez-Boada, M.; Doyle, W.A.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Martínez, A.T.; Smith, A.T. Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzym. Microb. Technol. 2002, 30, 518–524. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Ferreira, P.; Martínez, M.J.; Martínez, A.T. In vitro activation, purification, and characterization of Escherichia coli expressed aryl-alcohol oxidase, a unique H2O2-producing enzyme. Protein Express. Purif. 2006, 45, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Barr, I.; Guo, F. Pyridine hemochromagen assay for determining the concentration of heme in purified protein solutions. Bio Protoc. 2015, 5, e1594. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Read, R. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. Sect. D Struct. Biol. 2001, 57, 1373–1382. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Struct. Biol. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Morales, M.; Mate, M.J.; Romero, A.; Martínez, M.J.; Martínez, A.T.; Ruiz-Dueñas, F.J. Two oxidation sites for low redox-potential substrates: A directed mutagenesis, kinetic and crystallographic study on Pleurotus eryngii versatile peroxidase. J. Biol. Chem. 2012, 287, 41053–41067. [Google Scholar] [CrossRef] [Green Version]
- Kishi, K.; Kusters-van Someren, M.; Mayfield, M.B.; Sun, J.; Loehr, T.M.; Gold, M.H. Characterization of manganese(II) binding site mutants of manganese peroxidase. Biochemistry 1996, 35, 8986–8994. [Google Scholar] [CrossRef]
- Pérez-Boada, M.; Ruiz-Dueñas, F.J.; Pogni, R.; Basosi, R.; Choinowski, T.; Martínez, M.J.; Piontek, K.; Martínez, A.T. Versatile peroxidase oxidation of high redox potential aromatic compounds: Site-directed mutagenesis, spectroscopic and crystallographic investigations of three long-range electron transfer pathways. J. Mol. Biol. 2005, 354, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Ayuso-Fernández, I.; De Lacey, A.L.; Cañada, F.J.; Ruiz-Dueñas, F.J.; Martínez, A.T. Increase of redox potential during the evolution of enzymes degrading recalcitrant lignin. Chem. Eur. J. 2019, 25, 2708–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørlie, M.; Seefeldt, L.C.; Parker, V.D. Use of stopped-flow spectrophotometry to establish midpoint potentials for redox proteins. Anal. Biochem. 2000, 287, 118–125. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Arnhold, J.; Jantschko, W.; Pichler, H.; Obinger, C. Redox properties of the couples compound I/compound II and compound II/native enzyme of human myeloperoxidase. Biochem. Biophys. Res. Commun. 2003, 301, 551–557. [Google Scholar] [CrossRef]
- Prasetyo, E.N.; Kudanga, T.; Ostergaard, L.; Rencoret, J.; Gutiérrez, A.; del Río, J.C.; Santos, J.I.; Nieto, L.; Jimenez-Barbero, J.; Martínez, A.T.; et al. Polymerization of lignosulfonates by the laccase-HBT (1-hydroxybenzotriazole) system improves dispersibility. Bioresour. Technol. 2010, 101, 5054–5062. [Google Scholar] [CrossRef] [Green Version]
- Lebo, S.E.; Braaten, S.M.; Fredheim, G.E.; Lutnaes, B.F.; Lauten, R.A.; Myrvold, B.O.; McNally, T.J. Recent advances in the characterization of lignosulfonates. In Characterization of Lignocellulosic Materials; Hu, T., Ed.; Blackwell Pub.: New York, NY, USA, 2008; pp. 189–205. [Google Scholar]
- Lutnaes, B.F.; Myrvold, B.O.; Lauten, R.A.; Endeshaw, M.M. 1H and 13C NMR data of benzylsulfonic acids—Model compounds for lignosulfonate. Magn. Reson. Chem. 2008, 46, 299–305. [Google Scholar] [CrossRef]
- Magina, S.; Marques, A.P.; Evtuguin, D.V. Study on the residual lignin in Eucalyptus globulus sulphite pulp. Holzforschung 2015, 69, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Martínez, A.T.; Rencoret, J.; Marques, G.; Gutiérrez, A.; Ibarra, D.; Jiménez-Barbero, J.; del Río, J.C. Monolignol acylation and lignin structure in some nonwoody plants: A 2D NMR study. Phytochemistry 2008, 69, 2831–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralph, S.; Ralph, J.; Landucci, L. NMR Database of Lignin and Cell Wall Model Compounds; 2009; Available online: www.glbrc.org/databases_and_software/nmrdatabase (accessed on 16 August 2021).
- Tuisel, H.; Sinclair, R.; Bumpus, J.A.; Ashbaugh, W.; Brock, B.J.; Aust, S.D. Lignin peroxidase H2 from Phanerochaete chrysosporium: Purification, characterization and stability to temperature and pH. Arch. Biochem. Biophys. 1990, 279, 158–166. [Google Scholar] [CrossRef]
- Hiner, A.N.P.; Raven, E.L.; Thorneley, R.N.F.; García-Canovas, F.; Rodríguez-López, J.N. Mechanisms of compound I formation in heme peroxidases. J. Inorg. Biochem. 2002, 91, 27–34. [Google Scholar] [CrossRef]
- Smith, A.T.; Veitch, N.C. Substrate binding and catalysis in heme peroxidases. Curr. Opin. Chem. Biol. 1998, 2, 269–278. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pK(a) values. Proteins 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fueyo, E.; Castanera, R.; Ruiz-Dueñas, F.J.; López-Lucendo, M.F.; Ramírez, L.; Pisabarro, A.G.; Martínez, A.T. Ligninolytic peroxidase gene expression by Pleurotus ostreatus: Differential regulation in lignocellulose medium and effect of temperature and pH. Fungal Genet. Biol. 2014, 72, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Ayuso-Fernández, I.; Rencoret, J.; Gutiérrez, A.; Ruiz-Dueñas, F.J.; Martínez, A.T. Peroxidase evolution in white-rot fungi follows wood lignin evolution in plants. Proc. Natl. Acad. Sci. USA 2019, 116, 17900–17905. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.; Justo, A.; Riley, R.; Salamov, A.; Lopez-Giraldez, F.; Sjokvist, E.; Copeland, A.; Foster, B.; Sun, H.; Larsson, E.; et al. Phylogenetic and phylogenomic overview of the Polyporales. Mycologia 2013, 105, 1350–1373. [Google Scholar] [CrossRef] [Green Version]
- Kirk, P.M.; Cannon, P.F.; Minter, D.W.; Stalpers, J.A. Ainsworth & Bisby’s Dictionary of Fungi, 10th ed.; CABI Europe-UK, Cromwell Press: Trowbridge, UK, 2008. [Google Scholar]
- Martínez, A.T. Molecular biology and structure-function of lignin-degrading heme peroxidases. Enzym. Microb. Technol. 2002, 30, 425–444. [Google Scholar] [CrossRef]
- Miki, Y.; Calviño, F.R.; Pogni, R.; Giansanti, S.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Basosi, R.; Romero, A.; Martínez, A.T. Crystallographic, kinetic, and spectroscopic study of the first ligninolytic peroxidase presenting a catalytic tyrosine. J. Biol. Chem. 2011, 286, 15525–15534. [Google Scholar] [CrossRef] [Green Version]
- Andrawis, A.; Johnson, K.A.; Tien, M. Studies on compound I formation of the lignin peroxidase from Phanerochaete chrysosporium. J. Biol. Chem. 1988, 263, 1195–1198. [Google Scholar] [CrossRef]
- Sáez-Jiménez, V.; Acebes, S.; Guallar, V.; Martínez, A.T.; Ruiz-Dueñas, F.J. Improving the oxidative stability of a high redox potential fungal peroxidase by rational design. PLoS ONE 2015, 10, e0124750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acebes, S.; Ruiz-Dueñas, F.J.; Toubes, M.; Sáez-Jiménez, V.; Pérez-Boada, M.; Lucas, F.; Martínez, A.T.; Guallar, V. Mapping the long-range electron transfer route in ligninolytic peroxidases. J. Phys. Chem. B 2017, 121, 3946–3954. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Verdín, J.; Vázquez-Duhalt, R. Role of oxidizing mediators and tryptophan 172 in the decoloration of industrial dyes by the versatile peroxidase from Bjerkandera adusta. J. Mol. Catal. B Enzym. 2007, 46, 1–7. [Google Scholar] [CrossRef]
- Ruiz-Dueñas, F.J.; Morales, M.; Mate, M.J.; Romero, A.; Martínez, M.J.; Smith, A.T.; Martínez, A.T. Site-directed mutagenesis of the catalytic tryptophan environment in Pleurotus eryngii versatile peroxidase. Biochemistry 2008, 47, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.J.; Schoemaker, H.E.; Palmer, J.M. Veratryl alcohol as a mediator and the role of radical cations in lignin biodegradation by Phanerochaete chrysosporium. FEBS Lett. 1986, 195, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Harvey, P.J.; Candeias, L.P.; King, P.J.; Palmer, J.M.; Wever, R. Lignin peroxidase catalysis: Reaction with veratryl alcohol and a polymeric dye, Poly R. Biochem. Soc. Trans. 1995, 23, S340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinfling, A.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Bergbauer, M.; Szewzyk, U.; Martínez, A.T. A study on reducing substrates of manganese-oxidizing peroxidases from Pleurotus eryngii and Bjerkandera adusta. FEBS Lett. 1998, 428, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Khindaria, A.; Yamazaki, I.; Aust, S.D. Stabilization of the veratryl alcohol cation radical by lignin peroxidase. Biochemistry 1996, 35, 6418–6424. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.T.; Doyle, W.A.; Dorlet, P.; Ivancich, A. Spectroscopic evidence for an engineered, catalytically active Trp radical that creates the unique reactivity of lignin peroxidase. Proc. Natl. Acad. Sci. USA 2009, 106, 16084–16089. [Google Scholar] [CrossRef] [Green Version]
- Verdín, J.; Pogni, R.; Baeza, A.; Baratto, M.C.; Basosi, R.; Vázquez-Duhalt, R. Mechanism of versatile peroxidase inactivation by Ca2+ depletion. Biophys. Chem. 2006, 121, 163–170. [Google Scholar] [CrossRef]
- George, S.J.; Kvaratskhelia, M.; Dilworth, M.J.; Thorneley, R.N.F. Reversible alkaline inactivation of lignin peroxidase involves the release of both the distal and proximal site calcium ions and bishistidine co-ordination of the haem. Biochem. J. 1999, 344, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R.J.; Zapanta, L.S.; Tien, M.; Aust, S.D. Role of calcium in maintaining the heme environment of manganese peroxidase. Biochemistry 1997, 36, 3654–3662. [Google Scholar] [CrossRef]
- Sáez-Jiménez, V.; Fernández-Fueyo, E.; Medrano, F.J.; Romero, A.; Martínez, A.T.; Ruiz-Dueñas, F.J. Improving the pH-stability of versatile peroxidase by comparative structural analysis with a naturally-stable manganese peroxidase. PLoS ONE 2015, 10, e0140984. [Google Scholar]
- González-Pérez, D.; Mateljak, I.; García-Ruiz, E.; Ruiz-Dueñas, F.J.; Martínez, A.T.; Alcalde, M. Alkaline versatile peroxidase by directed evolution. Catal. Sci. Technol. 2016, 6, 6625–6636. [Google Scholar] [CrossRef]
- Sáez-Jiménez, V.; Acebes, S.; García-Ruiz, E.; Romero, A.; Guallar, V.; Alcalde, M.; Medrano, F.J.; Martínez, A.T.; Ruiz-Dueñas, F.J. Unveiling the basis of alkaline stability of an evolved versatile peroxidase. Biochem. J. 2016, 473, 1917–1928. [Google Scholar] [CrossRef] [PubMed]
- Kishi, K.; Wariishi, H.; Marquez, L.; Dunford, H.B.; Gold, M.H. Mechanism of manganese peroxidase compound II reduction. Effect of organic acid chelators and pH. Biochemistry 1994, 33, 8694–8701. [Google Scholar] [CrossRef]
- Wariishi, H.; Márquez, L.; Dunford, H.B.; Gold, M.H. Lignin peroxidase compounds II and III. Spectral and kinetic characterization of reactions with peroxides. J. Biol. Chem. 1990, 265, 11137–11142. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | Refinement | ||
|---|---|---|---|
| Resolution range (Å) | 74.95–1.85 (1.95–1.85) | Resolution (Å) | 55.56–1.85 |
| Space group | P 21 21 21 | Working reflections | 26,256 (2733) |
| Unit cell (Å) | a = 48.13, b = 74.95, c = 82.77 | Reflections test set | 1270 (140) |
| Total reflections | 164,812 (23,933) | R-work (%) | 22.6 (40.8) |
| Unique reflections | 26,316 (3760) | R-free (%) | 26.8 (42.1) |
| Multiplicity | 6.3 (6.4) | Number of non-H atoms | |
| Completeness (%) | 99.8 (100.0) | - protein | 2413 |
| Mean I/sigma (I) | 15.5 (3.0) | - heme group | 43 |
| Wilson B-factor (Å2) | 35.8 | - ions | 4 |
| Rmerge | 0.051 (0.483) | - solvent | 83 |
| Rmeas | 0.061 (0.575) | Average B-factor (Å2) | |
| Rpim | 0.033 (0.309) | - protein | 48.02 |
| CC1/2 | 0.99 (0.93) | - heme group | 33.78 |
| - ions | 43.51 | ||
| - solvent | 45.56 | ||
| rmsd bond lengths (Å) | 0.012 | ||
| rmsd angles (°) | 1.67 | ||
| Ramachandran statistics | |||
| - Preferred (%) | 92.71 | ||
| - Allowed (%) | 5.78 | ||
| - Outliers (%) | 1.52 | ||
| PDB code | 7OO5 |
| ABTS a | DMP | |||||||
|---|---|---|---|---|---|---|---|---|
| H2O2 | Low Efficiency | High Efficiency | pH 3 | pH8 | VA | RB5 | ||
| ApeLiP | KM | 33 ± 4.4 | 693 ± 63 | 8.1 ± 0.6 b | 37 ± 5.6 | 1430 ± 440 | (29.6 ± 4.6) × 103 | 14 ± 1.4 b |
| kcat | 100 ± 4.8 | 179 ± 5 | 42 ± 1.8 | 68.6 ± 2.8 | 5.7 ± 0.7 | 4.3 ± 0.3 | 21 ± 1.5 | |
| kcat/KM | 3070 ± 440 | 258 ± 25 | 5130 ± 460 | 1910 ± 310 | 4.0 ± 1.3 | 0.15 ± 0.03 | 1540 ± 115 | |
| ki | (2.1 ± 0.5) × 103 | - | - | (11 ± 2.2) × 103 | nd d | (406 ± 92) × 103 | - | |
| W166A | KM | 136 ± 12 | 1150 ± 190 c | - | - | 49 ± 8.2 | - | - |
| kcat | 126 ± 4.7 | 117 ± 10 c | - | - | 6.1 ± 0.27 | - | - | |
| kcat/KM | 929 ± 87 | 102 ± 19 c | - | - | 125 ± 22 | - | - | |
| ki | (3.3 ± 0.3) × 103 | nd c | - | - | nd d | - | - | |
| Softwood Lignin | Hardwood Lignin | ||||
|---|---|---|---|---|---|
| Native | Acetylated | Native | Acetylated | ||
| ApeLiP | KD3 | ns b | 31 ± 6.9 | 156 ± 25 | 89 ± 14 |
| k3 | ns | 26 ± 1.7 | 163 ± 38 | 56 ± 4.6 | |
| k3app | 926 ± 47 | 830 ± 195 | 957 ± 270 | 630 ± 111 | |
| W166A | KD3 | - | - | - | - |
| k3 | - | - | - | - | |
| k3app | 0 | 0 | 0 | 0 | |
| P. eryngii VPL d | KD3 | 143 ± 19 | 24 ± 1.9 | 14 ± 1 | 21 ± 2.5 |
| k3 | 48 ± 2 | 14 ± 0.4 | 14 ± 2 | 12 ± 0.5 | |
| k3app | 340 ± 30 | 599 ± 31 | 990 ± 80 | 592 ± 52 | |
| P. chrysosporium LiPA e | KD3 | 95 ± 26 | na c | 19 ± 2 | na |
| k3 | 25 ± 4 | na | 14 ± 0 | na | |
| k3app | 263 ± 83 | na | 764 ± 86 | na | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Ruiz, M.I.; Ayuso-Fernández, I.; Rencoret, J.; González-Ramírez, A.M.; Linde, D.; Davó-Siguero, I.; Romero, A.; Gutiérrez, A.; Martínez, A.T.; Ruiz-Dueñas, F.J. Agaricales Mushroom Lignin Peroxidase: From Structure–Function to Degradative Capabilities. Antioxidants 2021, 10, 1446. https://doi.org/10.3390/antiox10091446
Sánchez-Ruiz MI, Ayuso-Fernández I, Rencoret J, González-Ramírez AM, Linde D, Davó-Siguero I, Romero A, Gutiérrez A, Martínez AT, Ruiz-Dueñas FJ. Agaricales Mushroom Lignin Peroxidase: From Structure–Function to Degradative Capabilities. Antioxidants. 2021; 10(9):1446. https://doi.org/10.3390/antiox10091446
Chicago/Turabian StyleSánchez-Ruiz, María Isabel, Iván Ayuso-Fernández, Jorge Rencoret, Andrés Manuel González-Ramírez, Dolores Linde, Irene Davó-Siguero, Antonio Romero, Ana Gutiérrez, Angel T. Martínez, and Francisco Javier Ruiz-Dueñas. 2021. "Agaricales Mushroom Lignin Peroxidase: From Structure–Function to Degradative Capabilities" Antioxidants 10, no. 9: 1446. https://doi.org/10.3390/antiox10091446
APA StyleSánchez-Ruiz, M. I., Ayuso-Fernández, I., Rencoret, J., González-Ramírez, A. M., Linde, D., Davó-Siguero, I., Romero, A., Gutiérrez, A., Martínez, A. T., & Ruiz-Dueñas, F. J. (2021). Agaricales Mushroom Lignin Peroxidase: From Structure–Function to Degradative Capabilities. Antioxidants, 10(9), 1446. https://doi.org/10.3390/antiox10091446