
Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress

 ,
,  and
and
Abstract
:
1. Introduction
2. Atherosclerosis
2.1. Atherosclerosis Initiation
2.2. Early Stages of Atherosclerosis
2.3. Atheroma Plaque Progression
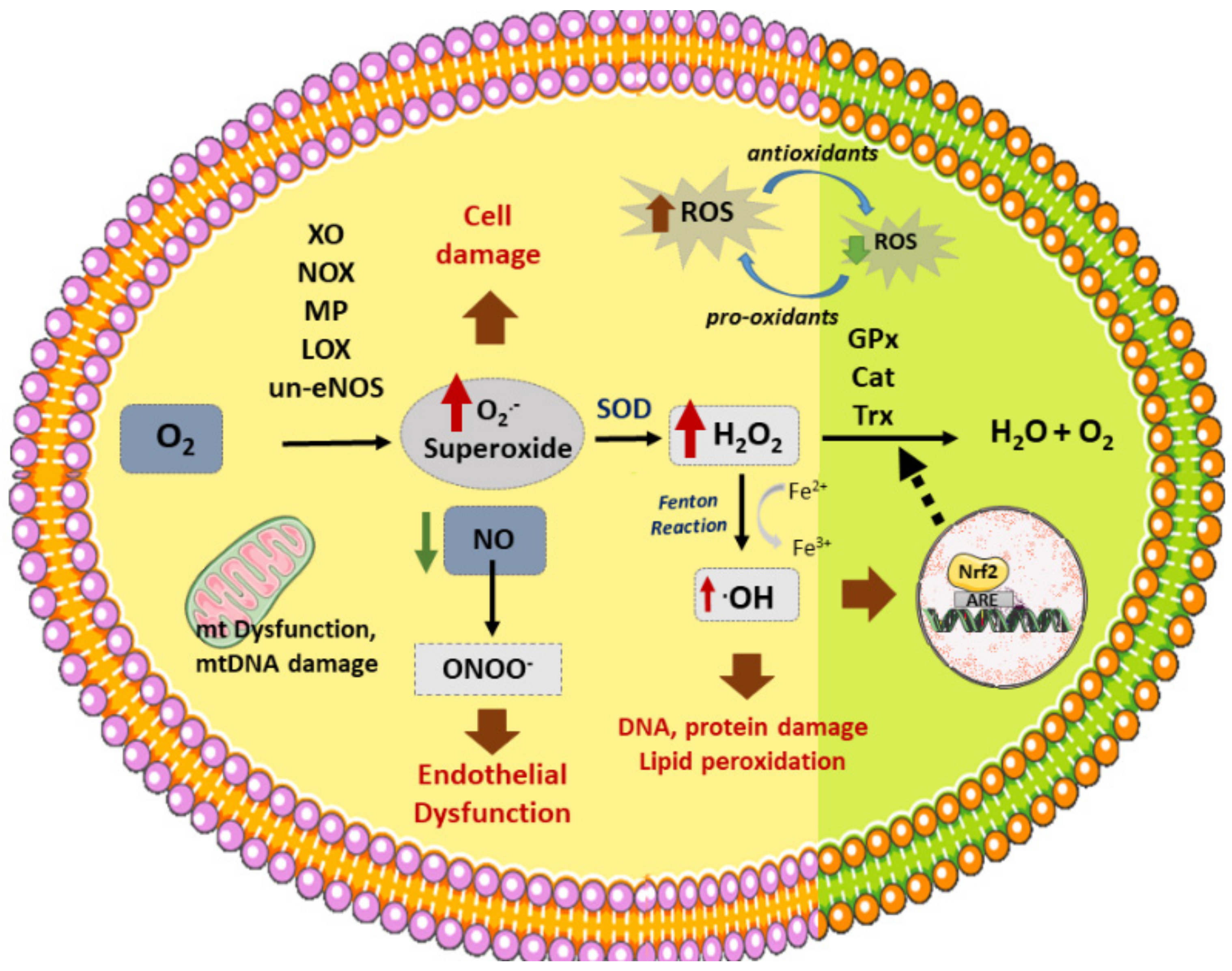
3. Atherosclerosis and Oxidative Stress
3.1. Vascular Sources of ROS
3.2. Vascular Antioxidant Enzymes
4. NRF2 Antioxidant Roles in Atherosclerosis
Modulation of Nrf2 Activity
5. HO-1 Antioxidant Role in Atherosclerosis
6. Therapeutic Approaches
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Libby, P.; Aikawa, M. Stabilization of atherosclerotic plaques: New mechanisms and clinical targets. Nat. Med. 2002, 8, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis-from experimental insights to the clinic. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Yazdani, S.K.; Vorpahl, M.; Ladich, E.; Virmani, R. Pathology and vulnerability of atherosclerotic plaque: Identification, treatment options, and individual patient differences for prevention of stroke. Curr. Treat. Options Cardiovasc. Med. 2010, 12, 297–314. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.K.; Orekhov, A.N. Oxidative Stress and Antioxidants in Atherosclerosis Development and Treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Hue, M.; Rayego-Mateos, S.; Vazquez-Carballo, C.; Palomino-Antolin, A.; Garcia-Caballero, C.; Opazo-Rios, L.; Morgado-Pascual, J.L.; Herencia, C.; Mas, S.; Ortiz, A.; et al. Protective Role of Nrf2 in Renal Disease. Antioxidants 2020, 10, 39. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a Potential Mediator of Cardiovascular Risk in Metabolic Diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howden, R. Nrf2 and cardiovascular defense. Oxid. Med. Cell. Longev. 2013, 2013, 104308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Li, W.; Su, Z.Y.; Kong, A.N. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Lee, S.H.; Kwak, M.K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxid. Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [Green Version]
- Bento-Pereira, C.; Dinkova-Kostova, A.T. Activation of transcription factor Nrf2 to counteract mitochondrial dysfunction in Parkinson’s disease. Med. Res. Rev. 2021, 41, 785–802. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Schirinzi, T.; Di Lazzaro, G.; D’Amico, J.; Colona, V.L.; Bertini, E.; Pierantozzi, M.; Mari, L.; Mercuri, N.B.; Piemonte, F.; et al. Systemic activation of Nrf2 pathway in Parkinson’s disease. Mov. Disord. 2020, 35, 180–184. [Google Scholar] [CrossRef]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Vinchi, F.; Muckenthaler, M.U.; Da Silva, M.C.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis and iron: From epidemiology to cellular level. Front. Pharmacol. 2014, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Chien, S. Mechanotransduction and endothelial cell homeostasis: The wisdom of the cell. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1209–H1224. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, S.M.; Topper, J.N. Adaptation of the endothelium to fluid flow: In vitro analyses of gene expression and in vivo implications. Vasc. Med. 2004, 9, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Conklin, B.S.; Zhong, D.S.; Zhao, W.; Lin, P.H.; Chen, C. Shear stress regulates occludin and VEGF expression in porcine arterial endothelial cells. J. Surg. Res. 2002, 102, 13–21. [Google Scholar] [CrossRef]
- Chiu, J.J.; Wung, B.S.; Shyy, J.Y.; Hsieh, H.J.; Wang, D.L. Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arterioscler Thromb. Vasc. Biol. 1997, 17, 3570–3577. [Google Scholar] [CrossRef]
- Kolovou, G.; Kolovou, V.; Mavrogeni, S. Lipidomics in vascular health: Current perspectives. Vasc. Health Risk Manag. 2015, 11, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sata, M.; Walsh, K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J. Clin. Investig. 1998, 102, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Tao, Y.K.; Bai, Y.P.; Yan, S.T.; Zhao, S.P. Inhibitory Effects of Simvastatin on Oxidized Low-Density Lipoprotein-Induced Endoplasmic Reticulum Stress and Apoptosis in Vascular Endothelial Cells. Chin. Med. J. 2018, 131, 950–955. [Google Scholar] [CrossRef]
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, M.L.; Ho, F.M.; Yang, R.S.; Chao, K.F.; Lin, W.W.; Lin-Shiau, S.Y.; Liu, S.H. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 539–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Lei, M. alpha-Mangostin protects against high-glucose induced apoptosis of human umbilical vein endothelial cells. Biosci. Rep. 2017, 37, 779. [Google Scholar] [CrossRef] [Green Version]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2010, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Eslava-Alcon, S.; Extremera-Garcia, M.J.; Gonzalez-Rovira, A.; Rosal-Vela, A.; Rojas-Torres, M.; Beltran-Camacho, L.; Sanchez-Gomar, I.; Jimenez-Palomares, M.; Alonso-Pinero, J.A.; Conejero, R.; et al. Molecular signatures of atherosclerotic plaques: An up-dated panel of protein related markers. J. Proteomics. 2020, 221, 103757. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Tousoulis, D.; Psaltopoulou, T.; Androulakis, E.; Papageorgiou, N.; Papaioannou, S.; Oikonomou, E.; Synetos, A.; Stefanadis, C. Oxidative stress and early atherosclerosis: Novel antioxidant treatment. Cardiovasc. Drugs Ther. 2015, 29, 75–88. [Google Scholar] [CrossRef]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Nicholson, A.C.; Han, J.; Febbraio, M.; Silversterin, R.L.; Hajjar, D.P. Role of CD36, the macrophage class B scavenger receptor, in atherosclerosis. Ann. N. Y. Acad. Sci. 2001, 947, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Wassmann, K.; Nickenig, G. Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 2004, 44, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Guzik, T.J.; Harrison, D.G. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug. Discov. Today 2006, 11, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Weseler, A.R.; Bast, A. Oxidative stress and vascular function: Implications for pharmacologic treatments. Curr. Hypertens. Rep. 2010, 12, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, D.J.; Barman, S.A. Clarity on the Isoform-Specific Roles of NADPH Oxidases and NADPH Oxidase-4 in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Lozhkin, A.; Vendrov, A.E.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J. Mol. Cell. Cardiol. 2017, 102, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative Stress in Aging-Associated Cardiovascular Disease. Antioxid. Redox. Signal. 2015, 23, 1389–1409. [Google Scholar] [CrossRef]
- Di Marco, E.; Gray, S.P.; Kennedy, K.; Szyndralewiez, C.; Lyle, A.N.; Lassegue, B.; Griendling, K.K.; Cooper, M.E.; Schmidt, H.; Jandeleit-Dahm, K.A.M. NOX4-derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free Radic. Biol. Med. 2016, 97, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef]
- Battelli, M.G.; Bolognesi, A.; Polito, L. Pathophysiology of circulating xanthine oxidoreductase: New emerging roles for a multi-tasking enzyme. Biochim. Biophys. Acta 2014, 1842, 1502–1517. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Patetsios, P.; Song, M.; Shutze, W.P.; Pappas, C.; Rodino, W.; Ramirez, J.A.; Panetta, T.F. Identification of uric acid and xanthine oxidase in atherosclerotic plaque. Am. J. Cardiol. 2001, 88, 188–191, A186. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddu, P.; Puddu, G.M.; Galletti, L.; Cravero, E.; Muscari, A. Mitochondrial dysfunction as an initiating event in atherogenesis: A plausible hypothesis. Cardiology 2005, 103, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Shoffner, J.M.; Lott, M.T.; Wallace, D.C. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat. Res. 1992, 275, 169–180. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Guerby, P.; Gayral, S.; Laffargue, M.; Salvayre, R. Role of reactive oxygen species in atherosclerosis: Lessons from murine genetic models. Free Radic. Biol. Med. 2020, 149, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef]
- Ruotsalainen, A.K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Makinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkila, J.; Vatanen, T.; et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Mimura, J.; Itoh, K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 2015, 88, 221–232. [Google Scholar] [CrossRef]
- Hosoya, T.; Maruyama, A.; Kang, M.I.; Kawatani, Y.; Shibata, T.; Uchida, K.; Warabi, E.; Noguchi, N.; Itoh, K.; Yamamoto, M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J. Biol. Chem. 2005, 280, 27244–27250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, A.C.; Murray, T.V.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A.M. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaiahgari, S.; Kleeberger, S.R.; Cho, H.Y.; Kalvakolanu, D.V.; Reddy, S.P. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J. Biol. Chem. 2004, 279, 42302–42312. [Google Scholar] [CrossRef] [Green Version]
- Smyrnias, I.; Zhang, X.; Zhang, M.; Murray, T.V.; Brandes, R.P.; Schroder, K.; Brewer, A.C.; Shah, A.M. Nicotinamide adenine dinucleotide phosphate oxidase-4-dependent upregulation of nuclear factor erythroid-derived 2-like 2 protects the heart during chronic pressure overload. Hypertension 2015, 65, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef] [Green Version]
- Pae, H.O.; Oh, G.S.; Lee, B.S.; Rim, J.S.; Kim, Y.M.; Chung, H.T. 3-Hydroxyanthranilic acid, one of L-tryptophan metabolites, inhibits monocyte chemoattractant protein-1 secretion and vascular cell adhesion molecule-1 expression via heme oxygenase-1 induction in human umbilical vein endothelial cells. Atherosclerosis 2006, 187, 274–284. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Kunsch, C. Sulforaphane inhibits TNF-alpha-induced activation of p38 MAP kinase and VCAM-1 and MCP-1 expression in endothelial cells. Inflamm. Res. 2009, 58, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Vaughn, S.; Zhang, Y.; Wang, E.T.; Garcia-Cardena, G.; Gimbrone, M.A., Jr. Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ. Res. 2007, 101, 723–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Jeoung, N.H.; Oh, C.J.; Choi, Y.K.; Lee, H.J.; Kim, H.J.; Kim, J.Y.; Hwang, J.H.; Tadi, S.; Yim, Y.H.; et al. Activation of NAD(P)H:quinone oxidoreductase 1 prevents arterial restenosis by suppressing vascular smooth muscle cell proliferation. Circ. Res. 2009, 104, 842–850. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Haasdijk, R.A.; Tempel, D.; den Dekker, W.K.; Chrifi, I.; Blonden, L.A.; van de Kamp, E.H.; de Boer, M.; Burgisser, P.E.; Noorderloos, A.; et al. PDGF-induced migration of vascular smooth muscle cells is inhibited by heme oxygenase-1 via VEGFR2 upregulation and subsequent assembly of inactive VEGFR2/PDGFRbeta heterodimers. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1289–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [Green Version]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Yuji Fuse, M.K. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.; Hawkins, A.E.; Eggler, A.L.; Mesecar, A.D.; Fabris, D.; Fishbein, J.C. Prospective type 1 and type 2 disulfides of Keap1 protein. Chem. Res. Toxicol. 2008, 21, 2051–2060. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ma, Q. Critical cysteine residues of Kelch-like ECH-associated protein 1 in arsenic sensing and suppression of nuclear factor erythroid 2-related factor 2. J. Pharmacol. Exp. Ther. 2010, 332, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell. Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Gao, L.; Zucker, I.H. Regulation of Nrf2 signaling pathway in heart failure: Role of extracellular vesicles and non-coding RNAs. Free Radic. Biol. Med. 2021, 167, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.K.; Jaiswal, A.K. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem. 2006, 281, 12132–12142. [Google Scholar] [CrossRef] [Green Version]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.; Hebbar, V.; Nair, S.; Xu, C.; Li, W.; Lin, W.; Keum, Y.S.; Han, J.; Gallo, M.A.; Kong, A.N. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J. Biol. Chem. 2004, 279, 23052–23060. [Google Scholar] [CrossRef] [Green Version]
- Elbirt, K.K.; Whitmarsh, A.J.; Davis, R.J.; Bonkovsky, H.L. Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J. Biol. Chem. 1998, 273, 8922–8931. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef]
- Huang, X.; Gao, Y.; Qin, J.; Lu, S. The role of miR-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS ONE 2014, 9, e113305. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yuan, B.; Cheng, B.; Liu, Y.; Zhang, B.; Wang, X.; Lin, X.; Yang, B.; Gong, G. Crocin Alleviates Myocardial Ischemia/Reperfusion-Induced Endoplasmic Reticulum Stress via Regulation of miR-34a/Sirt1/Nrf2 Pathway. Shock 2019, 51, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Stachurska, A.; Ciesla, M.; Kozakowska, M.; Wolffram, S.; Boesch-Saadatmandi, C.; Rimbach, G.; Jozkowicz, A.; Dulak, J.; Loboda, A. Cross-talk between microRNAs, nuclear factor E2-related factor 2, and heme oxygenase-1 in ochratoxin A-induced toxic effects in renal proximal tubular epithelial cells. Mol. Nutr. Food Res. 2013, 57, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhao, L.; Zheng, J.; Wang, K.; Deng, H.; Liu, P.; Chen, L.; Mu, H. MicroRNA-144 modulates oxidative stress tolerance in SH-SY5Y cells by regulating nuclear factor erythroid 2-related factor 2-glutathione axis. Neurosci. Lett. 2017, 655, 21–27. [Google Scholar] [CrossRef]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Mao, L.; Wang, S.G.; Chen, F.L.; Ji, F.; Fei, H.D. MicroRNA-200a activates Nrf2 signaling to protect osteoblasts from dexamethasone. Oncotarget 2017, 8, 104867–104876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Fu, Z.; Zou, Y.; Wen, D.; Ma, H.; Zhou, F.; Chen, Y.; Zhang, M.; Zhang, W. MicroRNA-140-5p attenuated oxidative stress in Cisplatin induced acute kidney injury by activating Nrf2/ARE pathway through a Keap1-independent mechanism. Exp. Cell. Res. 2017, 360, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ishfaq, M.; Xu, L.; Xia, C.; Chen, C.; Li, J. METTL3/m(6)A/miRNA-873-5p Attenuated Oxidative Stress and Apoptosis in Colistin-Induced Kidney Injury by Modulating Keap1/Nrf2 Pathway. Front. Pharmacol. 2019, 10, 517. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ku, C.H.; Siow, R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef]
- Yin, Y.; Zhao, X.; Yang, Z.; Min, X. Downregulation of miR-93 elevates Nrf2 expression and alleviates reactive oxygen species and cell apoptosis in diabetic retinopathy. Int. J. Clin. Exp. Med. 2019, 12, 10235–10243. [Google Scholar]
- Zhu, J.; Wang, S.; Qi, W.; Xu, X.; Liang, Y. Overexpression of miR-153 promotes oxidative stress in MPP(+)-induced PD model by negatively regulating the Nrf2/HO-1 signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 4179–4187. [Google Scholar]
- Yamamoto, S.; Inoue, J.; Kawano, T.; Kozaki, K.; Omura, K.; Inazawa, J. The impact of miRNA-based molecular diagnostics and treatment of NRF2-stabilized tumors. Mol. Cancer Res. 2014, 12, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Navab, M.; Lusis, A.J. Vasculitis, Atherosclerosis, and Altered HDL Composition in Heme-Oxygenase-1-Knockout Mice. Int. J. Hypertens. 2012, 2012, 948203. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Ferrandiz, M.L.; Devesa, I. Inducers of heme oxygenase-1. Curr Pharm Des. 2008, 14, 473–486. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Fernandez-Fierro, A.; Covian, C.; Bueno, S.M.; Riedel, C.A.; Mackern-Oberti, J.P.; Kalergis, A.M. Naturally Derived Heme-Oxygenase 1 Inducers and Their Therapeutic Application to Immune-Mediated Diseases. Front. Immunol. 2020, 11, 1467. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Kondo, K.; Momiyama, Y. The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. Int. J. Mol. Sci. 2019, 20, 3628. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Sugawara, D.; Wang, X.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme oxygenase-1 inhibits atherosclerotic lesion formation in ldl-receptor knockout mice. Circ. Res. 2001, 88, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Orozco, L.D.; Kapturczak, M.H.; Barajas, B.; Wang, X.; Weinstein, M.M.; Wong, J.; Deshane, J.; Bolisetty, S.; Shaposhnik, Z.; Shih, D.M.; et al. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ. Res. 2007, 100, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; He, Z.; Wu, L.; Fang, Y. Effects of Induction/Inhibition of Endogenous Heme Oxygenase-1 on Lipid Metabolism, Endothelial Function, and Atherosclerosis in Rabbits on a High Fat Diet. J. Pharmacol. Sci. 2012, 118, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Song, F.; Li, X.; Rong, S.; Yang, W.; Zhang, M.; Yao, P.; Hao, L.; Yang, N.; Hu, F.B.; et al. Plasma heme oxygenase-1 concentration is elevated in individuals with type 2 diabetes mellitus. PLoS ONE 2010, 5, e12371. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [Green Version]
- Fiorelli, S.; Porro, B.; Cosentino, N.; Di Minno, A.; Manega, C.M.; Fabbiocchi, F.; Niccoli, G.; Fracassi, F.; Barbieri, S.; Marenzi, G.; et al. Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability:an In Vitro and In Vivo Study. Cells 2019, 8, 356. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, Y.; Sasaki, K.; Saita, E.; Niki, H.; Ohmori, R.; Kondo, K.; Momiyama, Y. Plasma Heme Oxygenase-1 Levels and Carotid Atherosclerosis. Stroke 2018, 49, 2230–2232. [Google Scholar] [CrossRef]
- Cheng, C.; Noordeloos, A.M.; Jeney, V.; Soares, M.P.; Moll, F.; Pasterkamp, G.; Serruys, P.W.; Duckers, H.J. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 2009, 119, 3017–3027. [Google Scholar] [CrossRef] [Green Version]
- Schwertner, H.A.; Jackson, W.G.; Tolan, G. Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem. 1994, 40, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, D.; Gullu, H.; Yildirim, E.; Tok, D.; Kirbas, I.; Ciftci, O.; Baycan, S.T.; Muderrisoglu, H. Low serum bilirubin levels are independently and inversely related to impaired flow-mediated vasodilation and increased carotid intima-media thickness in both men and women. Atherosclerosis 2006, 184, 431–437. [Google Scholar] [CrossRef]
- Novotny, L.; Vitek, L. Inverse relationship between serum bilirubin and atherosclerosis in men: A meta-analysis of published studies. Exp. Biol. Med. 2003, 228, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug. Targets 2010, 11, 1504–1516. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.W.; Smith, D.L.; Zucker, S.D. Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 2004, 40, 424–433. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Srisook, K.; Han, S.S.; Choi, H.S.; Li, M.H.; Ueda, H.; Kim, C.; Cha, Y.N. CO from enhanced HO activity or from CORM-2 inhibits both O2- and NO production and downregulates HO-1 expression in LPS-stimulated macrophages. Biochem. Pharmacol. 2006, 71, 307–318. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Foresti, R.; Motterlini, R. Heme Oxygenase-1 and Carbon Monoxide in the Heart: The Balancing Act Between Danger Signaling and Pro-Survival. Circ. Res. 2016, 118, 1940–1959. [Google Scholar] [CrossRef] [Green Version]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, J.; Vercellotti, G.M.; Jeney, V.; Yachie, A.; Varga, Z.; Jacob, H.S.; Eaton, J.W.; Balla, G. Heme, heme oxygenase, and ferritin: How the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox. Signal. 2007, 9, 2119–2137. [Google Scholar] [CrossRef] [PubMed]
- Brunet, S.; Thibault, L.; Delvin, E.; Yotov, W.; Bendayan, M.; Levy, E. Dietary iron overload and induced lipid peroxidation are associated with impaired plasma lipid transport and hepatic sterol metabolism in rats. Hepatology 1999, 29, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Rooyakkers, T.M.; Stroes, E.S.; Kooistra, M.P.; van Faassen, E.E.; Hider, R.C.; Rabelink, T.J.; Marx, J.J. Ferric saccharate induces oxygen radical stress and endothelial dysfunction in vivo. Eur. J. Clin. Investig. 2002, 32 (Suppl. 1), 9–16. [Google Scholar] [CrossRef]
- Sullivan, J.L. Iron in arterial plaque: Modifiable risk factor for atherosclerosis. Biochim. Biophys. Acta 2009, 1790, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792. [Google Scholar] [CrossRef]
- Durante, W. Targeting Heme Oxygenase-1 in the Arterial Response to Injury and Disease. Antioxidants 2020, 9, 829. [Google Scholar] [CrossRef]
- Sabaawy, H.E.; Zhang, F.; Nguyen, X.; ElHosseiny, A.; Nasjletti, A.; Schwartzman, M.; Dennery, P.; Kappas, A.; Abraham, N.G. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension 2001, 38, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Hamedi-Asl, P.; Halabian, R.; Bahmani, P.; Mohammadipour, M.; Mohammadzadeh, M.; Roushandeh, A.M.; Jahanian-Najafabadi, A.; Kuwahara, Y.; Roudkenar, M.H. Adenovirus-mediated expression of the HO-1 protein within MSCs decreased cytotoxicity and inhibited apoptosis induced by oxidative stresses. Cell. Stress Chaperones. 2012, 17, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulis, D.A.; Durante, W.; Liu, X.; Evans, A.J.; Peyton, K.J.; Schafer, A.I. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation 2001, 104, 2710–2715. [Google Scholar] [CrossRef] [Green Version]
- Duckers, H.J.; Boehm, M.; True, A.L.; Yet, S.F.; San, H.; Park, J.L.; Clinton Webb, R.; Lee, M.E.; Nabel, G.J.; Nabel, E.G. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 2001, 7, 693–698. [Google Scholar] [CrossRef]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Bharucha, A.E.; Kulkarni, A.; Choi, K.M.; Camilleri, M.; Lempke, M.; Brunn, G.J.; Gibbons, S.J.; Zinsmeister, A.R.; Farrugia, G. First-in-human study demonstrating pharmacological activation of heme oxygenase-1 in humans. Clin. Pharmacol. Ther. 2010, 87, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Andreas, M.; Oeser, C.; Kainz, F.M.; Shabanian, S.; Aref, T.; Bilban, M.; Messner, B.; Heidtmann, J.; Laufer, G.; Kocher, A.; et al. Intravenous Heme Arginate Induces HO-1 (Heme Oxygenase-1) in the Human Heart. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Jiang, H.; Shi, Y. Upregulation of heme oxygenase-1 expression by curcumin conferring protection from hydrogen peroxide-induced apoptosis in H9c2 cardiomyoblasts. Cell. Biosci. 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, Y.; Chengyong, T.; Cheng, L.; Haixia, H.; Yuanda, Z.; Weihua, Y. Resveratrol Attenuates the Cytotoxicity Induced by Amyloid-beta1-42 in PC12 Cells by Upregulating Heme Oxygenase-1 via the PI3K/Akt/Nrf2 Pathway. Neurochem. Res. 2018, 43, 297–305. [Google Scholar] [CrossRef]
- Chen, C.Y.; Jang, J.H.; Li, M.H.; Surh, Y.J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Cheng, L.; Jin, Z.; Zhao, R.; Ren, K.; Deng, C.; Yu, S. Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia-reperfusion injury: Role of Nrf2/ARE pathway. Int. J. Clin. Exp. Med. 2015, 8, 10420–10428. [Google Scholar]
- Walle, T. Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15. [Google Scholar] [CrossRef]
- Wung, B.S.; Hsu, M.C.; Wu, C.C.; Hsieh, C.W. Piceatannol upregulates endothelial heme oxygenase-1 expression via novel protein kinase C and tyrosine kinase pathways. Pharmacol. Res. 2006, 53, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Li, J.; Li, S.; Almutairi, M.M. Resveratrol Derivative, Trans-3, 5, 4′-Trimethoxystilbene, Prevents the Developing of Atherosclerotic Lesions and Attenuates Cholesterol Accumulation in Macrophage Foam Cells. Mol. Nutr. Food Res. 2020, 64, e1901115. [Google Scholar] [CrossRef]
- Yamagata, K. Protective Effect of Epigallocatechin Gallate on Endothelial Disorders in Atherosclerosis. J. Cardiovasc. Pharmacol. 2020, 75, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Uchi, H.; Morino-Koga, S.; Shi, W.; Furue, M. Z-ligustilide ameliorated ultraviolet B-induced oxidative stress and inflammatory cytokine production in human keratinocytes through upregulation of Nrf2/HO-1 and suppression of NF-kappaB pathway. Exp. Dermatol. 2015, 24, 703–708. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Ma, H.; Yang, N.; Li, L.; Zheng, D.D.; Wu, M.X.; Zhao, Z.L.; Qi, H.Y. Intranasal Pretreatment with Z-Ligustilide, the Main Volatile Component of Rhizoma Chuanxiong, Confers Prophylaxis against Cerebral Ischemia via Nrf2 and HSP70 Signaling Pathways. J. Agric. Food Chem. 2017, 65, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, Y.; Huang, X.; Xie, Y.; Qu, Y.; Long, H.; Gu, N.; Jiang, W. Z-Ligustilide protects vascular endothelial cells from oxidative stress and rescues high fat diet-induced atherosclerosis by activating multiple NRF2 downstream genes. Atherosclerosis 2019, 284, 110–120. [Google Scholar] [CrossRef]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Targeting heme oxygenase-1 in the treatment of atherosclerosis. Drug. Discov. Today 2005, 2, 201–206. [Google Scholar] [CrossRef]
- Fredenburgh, L.E.; Perrella, M.A.; Barragan-Bradford, D.; Hess, D.R.; Peters, E.; Welty-Wolf, K.E.; Kraft, B.D.; Harris, R.S.; Maurer, R.; Nakahira, K.; et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced ARDS. JCI Insight. 2018, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Mumby, S.; Upton, R.L.; Chen, Y.; Stanford, S.J.; Quinlan, G.J.; Nicholson, A.G.; Gutteridge, J.M.; Lamb, N.J.; Evans, T.W. Lung heme oxygenase-1 is elevated in acute respiratory distress syndrome. Crit. Care Med. 2004, 32, 1130–1135. [Google Scholar] [CrossRef]
- Wollborn, J.; Hermann, C.; Goebel, U.; Merget, B.; Wunder, C.; Maier, S.; Schafer, T.; Heuler, D.; Muller-Buschbaum, K.; Buerkle, H.; et al. Overcoming safety challenges in CO therapy-Extracorporeal CO delivery under precise feedback control of systemic carboxyhemoglobin levels. J. Control. Release 2018, 279, 336–344. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnammanesh, G.; Durante, G.L.; Khanna, Y.P.; Peyton, K.J.; Durante, W. Canagliflozin inhibits vascular smooth muscle cell proliferation and migration: Role of heme oxygenase-1. Redox. Biol. 2020, 32, 101527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, C.; Li, T.; Wang, W.; Ye, W.; Zeng, R.; Ni, L.; Lai, Z.; Wang, X.; Liu, C. The protective effect of melatonin on brain ischemia and reperfusion in rats and humans: In vivo assessment and a randomized controlled trial. J. Pineal. Res. 2018, 65, e12521. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Gonzalez-Rodriguez, A.; Gomez-Guerrero, C.; Moreno, J.A. Editorial: Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2019, 10, 1149. [Google Scholar] [CrossRef]
- Lv, D.; Zhou, Q.; Xia, Y.; You, X.; Zhao, Z.; Li, Y.; Zou, H. The Association Between Oxidative Stress Alleviation via Sulforaphane-Induced Nrf2-HO-1/NQO-1 Signaling Pathway Activation and Chronic Renal Allograft Dysfunction Improvement. Kidney Blood Press Res. 2018, 43, 191–205. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Kinases | Identified in | Effect | References |
|---|---|---|---|
| GSK-3β | HEK293T | Inhibits NRF2 | [93] Rada et al., 2011 [97] Salazar et al., 2006 |
| HepG2 | Inhibits NRF2 | [98] Jain et al., 2006 | |
| FYN | HepG2 | Inhibits NRF2, via GSK-3B | [92] Jain et al., 2007 |
| P38 MAP kinase | Thymocytes and 293T; HepG2 | Inhibits HO-1/Nrf2 Activates HO-1/Nrf2 | [99] Thornton et al., 2008; [100] Shen et al., 2004 [101] Elbirt et al. 1998 |
| PI3k /Akt | 293T | Activates NRF2, by Gsk-3β inhibition. | [97] Salazar M et al. 2006 |
| PKC | HepG2 | Activates NRF2 | [95] Huang et al., 2002 |
| JNK | HepG2 | Activates NRF2 | [100] Shen et al., 2004 |
| ERK | HepG2 | Activates NRF2, through GSK-3β inhibition. | [100] Shen et al., 2004 [101] Elbirt et al., 1998 |
| PERK | Mouse embryonic fibroblasts | Activates NRF2 | [102] Cullinan et al. 2003 |
| AMPK | HepG2, HEK293 | Activates Nrf2 | [96] Joo et al. 2016 |
| miRNA | Identified in | Effect | References |
| miR-28 | Breast cancer cell lines | Inhibits | [103] Yang et al., 2011 |
| miR-34a | Hepatocytes | Inhibits | [104] Huang et al., 2014 |
| Cardiomyocytes | [105] Wang et al., 2019 | ||
| miR-132, miR-200c | Renal proximal tubular cell line | Inhibits | [106] Stachurska et al., 2012 |
| miR-144 | Neuronal cell lines; Lymphoblast cell lines (k562 cell line) | Inhibits | [107] Zhou et al., 2017 |
| miR-153, miR27a, miR-142-5p | Neuronal cell lines | Inhibits | [108] Narashimhan et al., 2012 |
| miR-200a | OB-6 osteoblastic cells | Increases | [109] Zhao et al., 2017 |
| miR-140-5p | HK2 cells | Increases NRF2 expression | [110] Liao W et al., 2017 |
| miR873-5p | Mouse renal tubular epithelial cells (mRTECs) | Increases NRF2 and HO-1 expression | [111] Wang J et al., 2019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Piñeiro, J.A.; Gonzalez-Rovira, A.; Sánchez-Gomar, I.; Moreno, J.A.; Durán-Ruiz, M.C. Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress. Antioxidants 2021, 10, 1463. https://doi.org/10.3390/antiox10091463
Alonso-Piñeiro JA, Gonzalez-Rovira A, Sánchez-Gomar I, Moreno JA, Durán-Ruiz MC. Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress. Antioxidants. 2021; 10(9):1463. https://doi.org/10.3390/antiox10091463
Chicago/Turabian StyleAlonso-Piñeiro, Jose Angel, Almudena Gonzalez-Rovira, Ismael Sánchez-Gomar, Juan Antonio Moreno, and Ma Carmen Durán-Ruiz. 2021. "Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress" Antioxidants 10, no. 9: 1463. https://doi.org/10.3390/antiox10091463
APA StyleAlonso-Piñeiro, J. A., Gonzalez-Rovira, A., Sánchez-Gomar, I., Moreno, J. A., & Durán-Ruiz, M. C. (2021). Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress. Antioxidants, 10(9), 1463. https://doi.org/10.3390/antiox10091463