
Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg?


{kind=link}
{kind=link}
Abstract
:1. Introduction
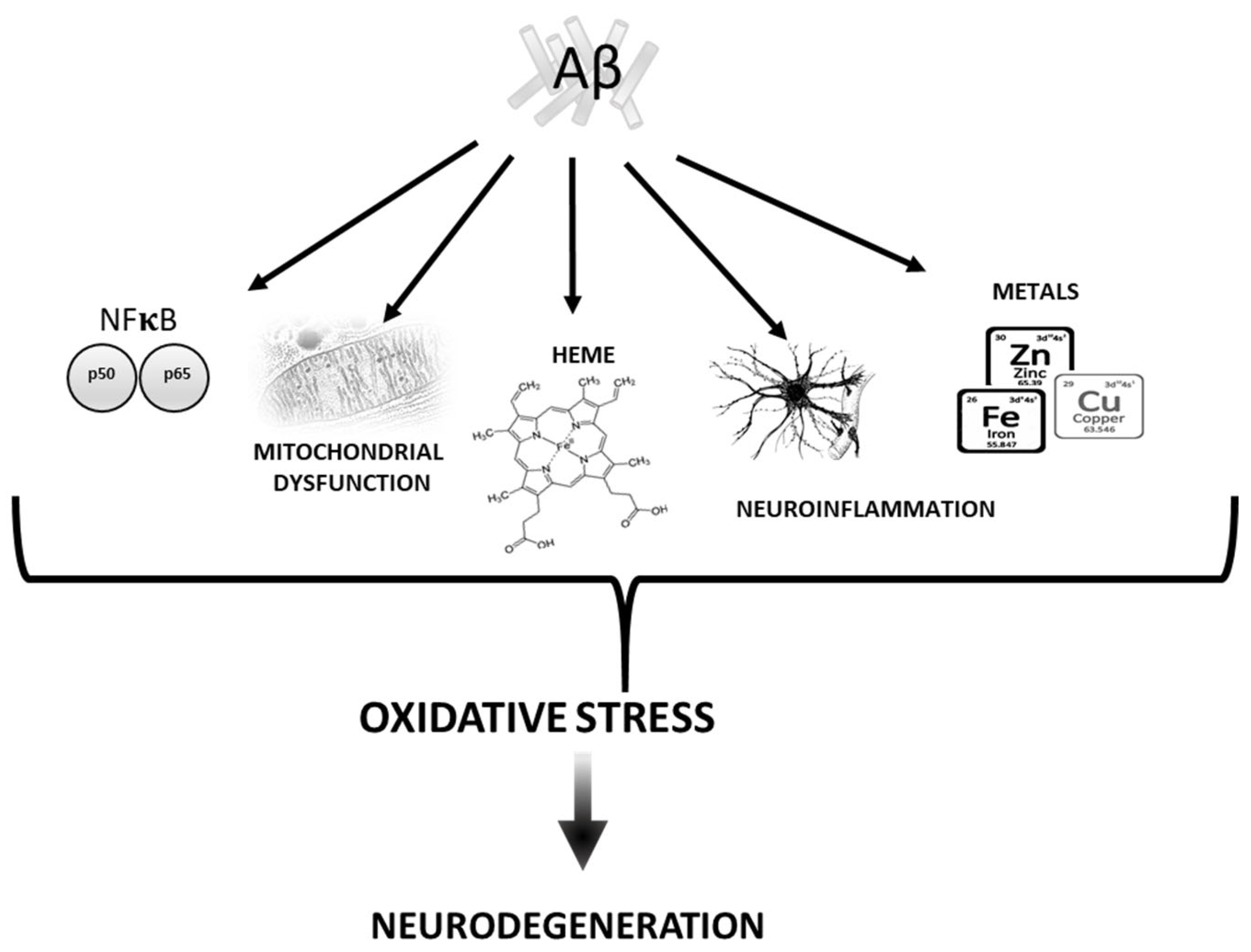
2. Aβ vs. Oxidative stress
2.1. Mitochondria
2.2. Transition Metals
2.3. Heme
2.4. Neuroinflammation
2.5. NF-kB Pathway
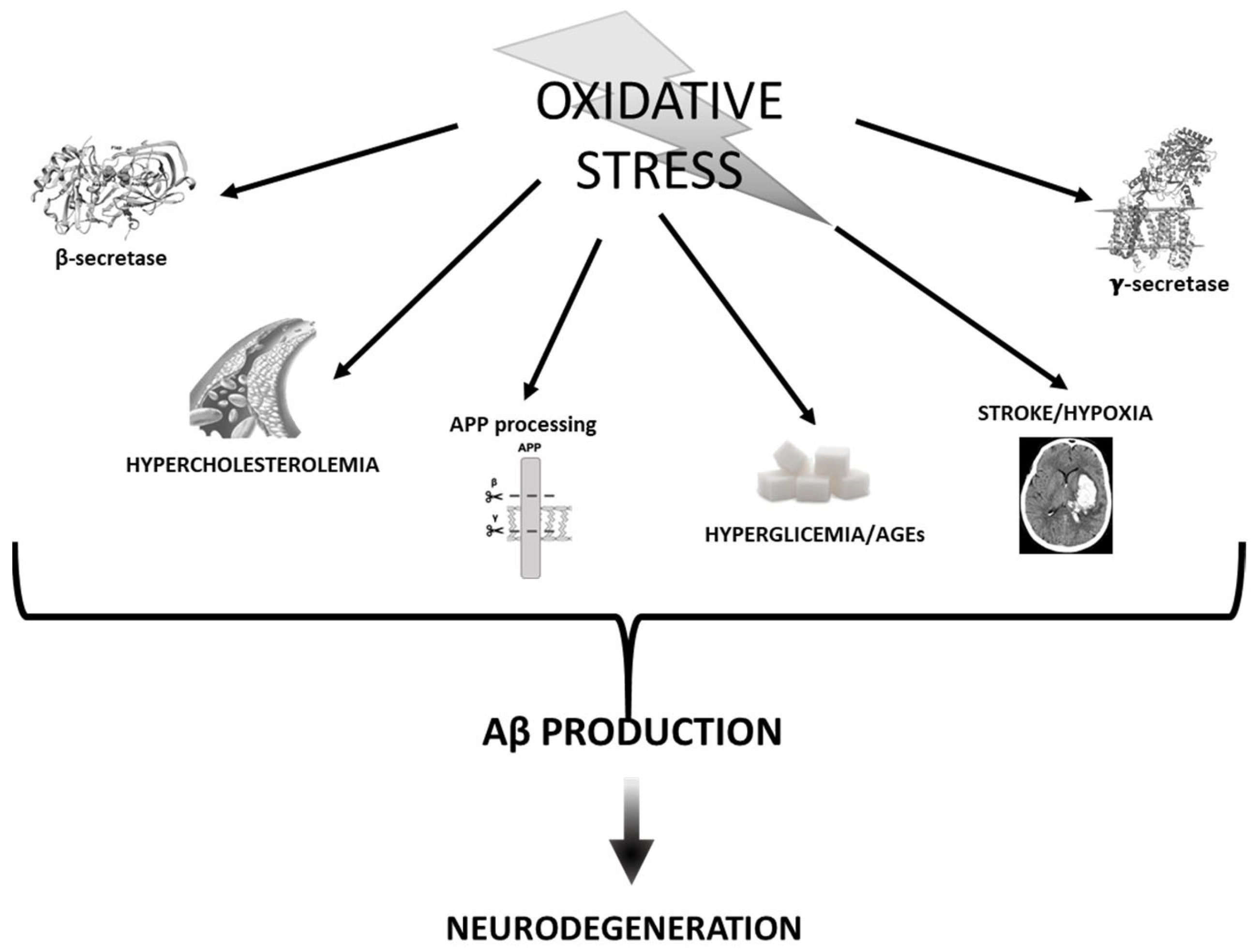
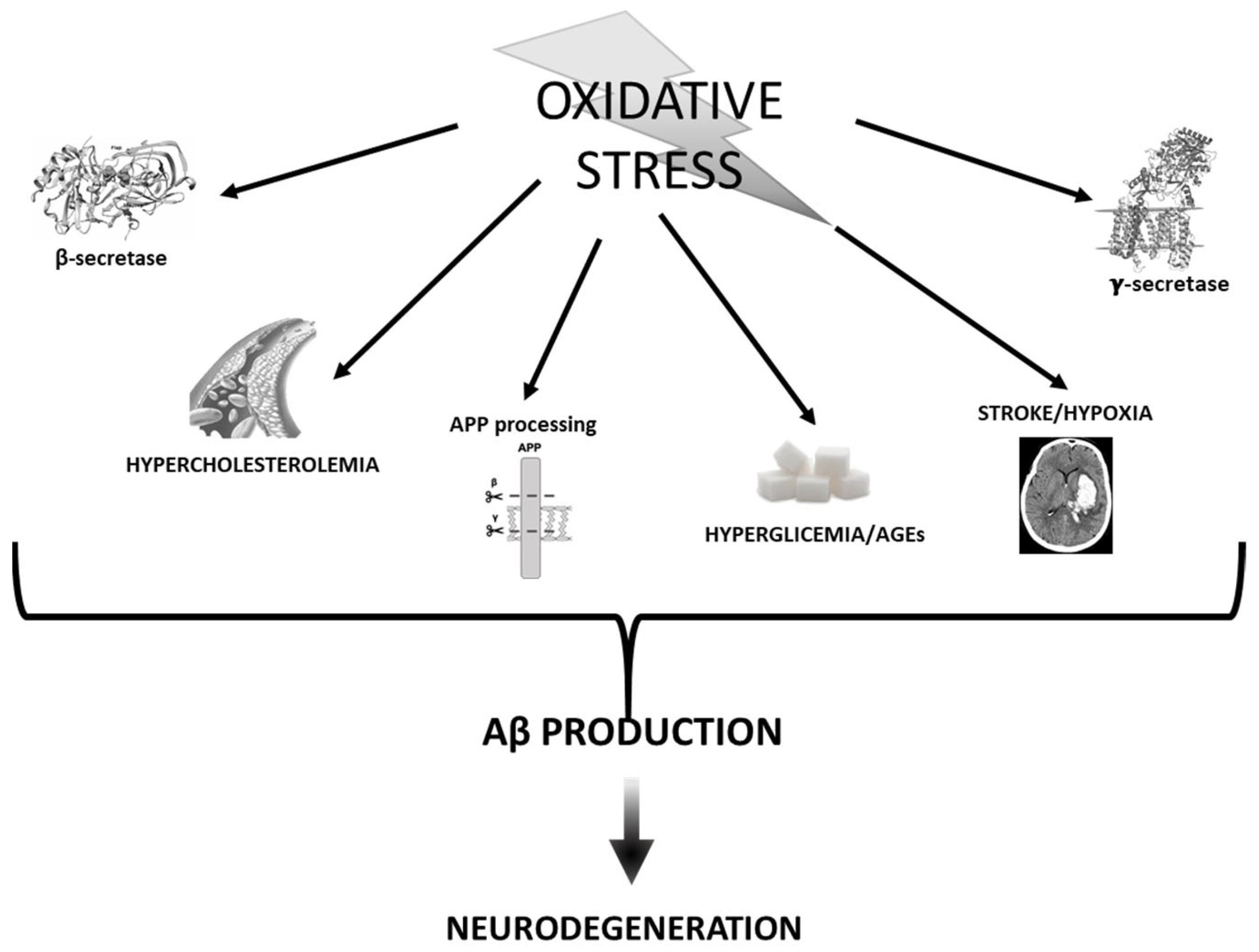
3. Oxidative Stress vs. Aβ
3.1. APP Processing and Secretases
3.2. Stroke and Hypoxia
3.3. Hyperglycemia and AGEs
3.4. Cholesterol
4. Concluding Remarks
Funding
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wu, J. Amyloid-β: A double agent in Alzheimer’s disease? Biomed. Pharm. 2021, 139, 111575. [Google Scholar] [CrossRef]
- Ji, C.; Sigurdsson, E.M. Current Status of Clinical Trials on Tau Immunotherapies. Drugs 2021, 8, 1135–1152. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haaksma, M.L.; Vilela, L.R.; Marengoni, A.; Calderón-Larrañaga, A.; Leoutsakos, J.S.; Olde Rikkert, M.G.M.; Melis, R.J.F. Comorbidity and progression of late onset Alzheimer’s disease: A systematic review. PLoS ONE 2017, 12, e0177044. [Google Scholar]
- Lemche, E. Early Life Stress and Epigenetics in Late-onset Alzheimer’s Dementia: A Systematic Review. Curr. Genom. 2018, 19, 522–602. [Google Scholar] [CrossRef]
- Zhou, X.; Fu, A.K.; Ip, N.Y. APOE signaling in neurodegenerative diseases: An integrative approach targeting APOE coding and noncoding variants for disease intervention. Curr. Opin. Neurobiol. 2021, 69, 58–67. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Repetto, I.E.; Monteleone, D.; Vasciaveo, V.; Franchino, C.; Rinaldi, S.; Tabaton, M.; Tamagno, E. Stroke and Amyloid-β Downregulate TREM-2 and Uch-L1 Expression that Synergistically Promote the Inflammatory Response. J. Alzheimers Dis. 2019, 71, 907–920. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Extracellular signal-regulated kinase regulates microglial immune responses in Alzheimer’s disease. J. Neurosci. Res. 2021, 99, 1704–1721. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1-42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Mitsumori, R.; Sakaguchi, K.; Shigemizu, D.; Mori, T.; Akiyama, S.; Ozaki, K.; Niida, S.; Shimoda, N. Lower DNA methylation levels in CpG island shores of CR1, CLU, and PICALM in the blood of Japanese Alzheimer’s disease patients. PLoS ONE 2020, 15, e0239196. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tan, C.C.; Cao, X.P.; Tan, L. Alzheimer’s Disease Neuroimaging Initiative. Association of Alzheimer’s disease risk variants on the PICALM gene with PICALM expression, core biomarkers, and feature neurodegeneration. Aging 2020, 12, 21202–21219. [Google Scholar] [CrossRef] [PubMed]
- Franzmeier, N.; Ossenkoppele, R.; Brendel, M.; Rubinski, A.; Smith, R.; Kumar, A.; Mattsson-Carlgren, N.; Strandberg, O.; Duerin, M.; Buerger, K.; et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI)* and the Swedish BioFINDER study. The BIN1 rs744373 Alzheimer’s disease risk SNP is associated with faster Aβ-associated tau accumulation and cognitive decline. Alzheimers Dement. 2021. in print. [Google Scholar] [CrossRef] [PubMed]
- Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603. [Google Scholar] [CrossRef]
- An, N.; Fu, Y.; Shi, J.; Guo, H.N.; Yang, Z.W.; Li, Y.C.; Li, S.; Wang, Y.; Yao, Z.J.; Hu, B. Alzheimer’s Disease Neuroimaging Initiative. Synergistic Effects of APOE and CLU May Increase the Risk of Alzheimer’s Disease: Acceleration of Atrophy in the Volumes and Shapes of the Hippocampus and Amygdala. J. Alzheimers Dis. 2021, 80, 1311–1327. [Google Scholar] [CrossRef]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.V. The human β-amyloid precursor protein: Biomolecular and epigenetic aspects. Biomol. Concepts. 2015, 6, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Huse, J.T.; Doms, R.W. Closing in on the amyloid cascade: Recent insights into the cell biology of Alzheimer’s disease. Mol. Neurobiol. 2000, 22, 81–98. [Google Scholar] [PubMed]
- Sun, Z.T.; Ma, C.; Li, G.J.; Zheng, X.Y.; Hao, Y.T.; Yang, Y.; Wang, X. Application of Antibody Fragments Against Aβ With Emphasis on Combined Application with Nanoparticles in Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 654611. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, F.; Lanni, C.; Racchi, M.; Govoni, S. (Dys)regulation of Synaptic Activity and Neurotransmitter Release by β-Amyloid: A Look Beyond Alzheimer’s Disease Pathogenesis. Front. Mol. Neurosci. 2021, 14, 635880. [Google Scholar] [CrossRef]
- Wang, K.; Na, L.; Duan, M. The Pathogenesis Mechanism, Structure Properties, Potential Drugs and Therapeutic Nanoparticles against the Small Oligomers of Amyloid-β. Curr. Top Med. Chem. 2021, 21, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Boumelhem, F.; Pope, E.D., 3rd; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical Appraisal of Amyloid Lowering Agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Tabaton, M.; Gambetti, P. Soluble amyloid-beta in the brain: The scarlet pimpernel. J. Alzheimers Dis. 2006, 9, 127–132. [Google Scholar] [CrossRef]
- Johannesson, M.; Sahlin, C.; Söderberg, L.; Basun, H.; Fälting, J.; Möller, C.; Zachrisson, O.; Sunnemark, D.; Svensson, A.; Odergren, T.; et al. Elevated soluble amyloid beta protofibrils in Down syndrome and Alzheimer’s disease. Mol. Cell Neurosci. 2021, 114, 103641. [Google Scholar] [CrossRef] [PubMed]
- Klyubin, I.; Walsh, D.M.; Lemere, C.A.; Cullen, W.K.; Shankar, G.M.; Betts, V.; Spooner, E.T.; Jiang, L.; Anwyl, R.; Selkoe, D.J.; et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat. Med. 2005, 11, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Meng, X.; El Fatimy, R.; Sun, B.; Mai, D.; Zhang, J.; Arora, R.; Zeng, A.; Xu, P.; Qu, S.; et al. Environmental enrichment prevents Aβ oligomer-induced synaptic dysfunction through mirna-132 and hdac3 signaling pathways. Neurobiol. Dis. 2020, 134, 104617. [Google Scholar] [CrossRef]
- Koike, H.; Iguchi, Y.; Sahashi, K.; Katsuno, M. Significance of Oligomeric and Fibrillar Species in Amyloidosis: Insights into Pathophysiology and Treatment. Molecules 2021, 26, 5091. [Google Scholar] [CrossRef]
- Morgan, D.; Diamond, D.M.; Gottschall, P.E.; Ugen, K.E.; Dickey, C.; Hardy, J.; Duff, K.; Jantzen, P.; DiCarlo, G.; Wilcock, D.; et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000, 408, 982–985. [Google Scholar] [CrossRef]
- Piccini, A.; Russo, C.; Gliozzi, A.; Relini, A.; Vitali, A.; Borghi, R.; Giliberto, L.; Armirotti, A.; D’Arrigo, C.; Bachi, A.; et al. beta-amyloid is different in normal aging and in Alzheimer disease. J. Biol. Chem. 2005, 280, 34186–34192. [Google Scholar] [CrossRef] [Green Version]
- Lana, E.; Gellerbring, A.; Jung, S.; Nordberg, A.; Unger Lithner, C.; Darreh-Shori, T. Homomeric and Heteromeric Aβ Species Exist in Human Brain and CSF Regardless of Alzheimer’s Disease Status and Risk Genotype. Front. Mol. Neurosci. 2019, 12, 176. [Google Scholar] [CrossRef] [Green Version]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Campagnani, I.; et al. Molecular subtypes of Alzheimer’s disease. Sci. Rep. 2018, 8, 3269. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Liu, L.; Dang, Y.; Ostaszewski, B.L.; Selkoe, D.J. Decoding the synaptic dysfunction of bioactive human AD brain soluble Aβ to inspire novel therapeutic avenues for Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Chang, Y.T.; Chang, W.N.; Tsai, N.W.; Huang, C.C.; Kung, C.T.; Su, Y.J.; Lin, W.C.; Cheng, B.C.; Su, C.M.; Chiang, Y.F.; et al. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer’s disease: A systematic review. Biomed. Res. Int. 2014, 2014, 182303. [Google Scholar] [CrossRef]
- Tofighi, N.; Asle-Rousta, M.; Rahnema, M.; Amini, R. Protective effect of alpha-linoleic acid on Aβ-induced oxidative stress, neuroinflammation, and memory impairment by alteration of α7 nAChR and NMDAR gene expression in the hippocampus of rats. Neurotoxicology 2021, 85, 245–253. [Google Scholar] [CrossRef]
- Yang, X.; Zhi, J.; Leng, H.; Chen, Y.; Gao, H.; Ma, J.; Ji, J.; Hu, Q. The piperine derivative HJ105 inhibits Aβ1-42-induced neuroinflammation and oxidative damage via the Keap1-Nrf2-TXNIP axis. Phytomedicine 2021, 20, 153571. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.M.; Tsuji, M.; Yasumoto, T.; Mori, Y.; Oguchi, T.; Tsuji, Y.; Umino, M.; Umino, A.; Nishikawa, T.; Nakamura, S.; et al. Myricetin prevents high molecular weight Aβ1-42 oligomer-induced neurotoxicity through antioxidant effects in cell membranes and mitochondria. Free Radic. Biol. Med. 2021, 171, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Zhang, L.; Ai, D.; Wu, H.; Peng, W. β-Asarone Ameliorates β-Amyloid-Induced Neurotoxicity in PC12 Cells by Activating P13K/Akt/Nrf2 Signaling Pathway. Front. Pharmacol. 2021, 12, 659955. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Robino, G.; Obbili, A.; Bardini, P.; Aragno, M.; Parola, M.; Danni, O. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp. Neurol. 2003, 180, 144–155. [Google Scholar] [CrossRef]
- Zuo, L.; Hemmelgarn, B.T.; Chuang, C.C.; Best, T.M. The Role of Oxidative Stress-Induced Epigenetic Alterations in Amyloid-β Production in Alzheimer’s Disease. Oxid. Med. Cell Longev. 2015, 2015, 604658. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [Green Version]
- Desler, C.; Lillenes, M.S.; Tønjum, T.; Rasmussen, L.J. The Role of Mitochondrial Dysfunction in the Progression of Alzheimer’s Disease. Curr. Med. Chem. 2018, 25, 5578–5587. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, M.; Tangpong, J.; Keller, J.N.; Murphy, M.P.; Markesbery, W.R.; Kiningham, K.K.; St Clair, D.K. Beta-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol. 2006, 168, 1608–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, A.; Nelson, T.J.; Alkon, D.L.; Hongpaisan, J. Loss in PKC Epsilon Causes Downregulation of MnSOD and BDNF Expression in Neurons of Alzheimer’s Disease Hippocampus. J. Alzheimers Dis. 2018, 63, 1173–1189. [Google Scholar] [CrossRef]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Detaille, D.; Pasdois, P.; Sémont, A.; Dos Santos, P.; Diolez, P. An old medicine as a new drug to prevent mitochondrial complex I from producing oxygen radicals. PLoS ONE 2019, 14, e0216385. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.P. Medical hypothesis: Neurodegenerative diseases arise from oxidative damage to electron tunneling proteins in mitochondria. Med. Hypotheses 2019, 127, 1–4. [Google Scholar] [CrossRef]
- Qi, Z.; Liu, K.J. The interaction of zinc and the blood-brain barrier under physiological and ischemic conditions. Toxicol. Appl. Pharmacol. 2019, 364, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Duck, K.A.; Connor, J.R. Iron uptake and transport across physiological barriers. Biometals 2016, 29, 573–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Sahoo, A.; Anand, S.; Ganguly, A.; Righi, G.; Bovicelli., P.; Saso, L.; Chakrabarti, S. Multiple mechanisms of iron-induced amyloid beta-peptide accumulation in SHSY5Y cells: Protective action of negletein. Neuromolecular Med. 2014, 16, 787–798. [Google Scholar] [CrossRef]
- Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000, 852, 274–278. [Google Scholar] [CrossRef]
- Zhang, L.H.; Wang, X.; Zheng, Z.H.; Ren, H.; Stoltenberg, M.; Danscher, G.; Huang, L.; Rong, M.; Wang, Z.Y. Altered expression and distribution of zinc transporters in APP/PS1 transgenic mouse brain. Neurobiol. Aging 2010, 31, 74–87. [Google Scholar] [CrossRef]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef] [PubMed]
- Hooda, J.; Shah, A.; Zhang, L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients 2014, 6, 1080–1102. [Google Scholar] [CrossRef] [Green Version]
- Faux, N.G.; Rembach, A.; Wiley, J.; Ellis, K.A.; Ames, D.; Fowler, C.J.; Martins, R.N.; Pertile, K.K.; Rumble, R.L.; Trounson, B.; et al. An anemia of Alzheimer’s disease. Mol. Psychiatry 2014, 19, 1227–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. [Google Scholar] [CrossRef]
- Vanacore, R.; Eskew, J.D.; Sung, L.; Davis, T.; Smith, A. Safe coordinated trafficking of heme and iron with copper maintain cell homeostasis: Modules from the hemopexin system. Biometals 2019, 2, 355–367. [Google Scholar] [CrossRef]
- Sankar, S.B.; Donegan, R.K.; Shah, K.J.; Reddi, A.R.; Wood, L.B. Heme and hemoglobin suppress amyloid β-mediated inflammatory activation of mouse astrocytes. J. Biol. Chem. 2018, 293, 11358–11373. [Google Scholar] [CrossRef] [Green Version]
- Atamna, H. Heme binding to Amyloid-beta peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 255–266. [Google Scholar] [CrossRef]
- Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Revelance to aging. J. Biol. Chem. 2001, 276, 48410–48416. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, C.; Seal, M.; Mukherjee, S.; Ghosh Dey, S. Alzheimer’s Disease: A Heme-Aβ Perspective. Acc. Chem. Res. 2015, 48, 2556–2564. [Google Scholar] [CrossRef]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.R.; Jonas, L.A.; Li, Y.M. Friend, Foe or Both? Immune Activity in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90-231 or lipopolysaccharide. Pharmacol. Res. 2016, 113, 500–514. [Google Scholar] [CrossRef]
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflamm. 2021, 18, 124. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Haroutunian, V.; Ho, L.; Purohit, D.; Pasinetti, G.M. Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Dis. Markers 2006, 22, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez Leo, E.E.; Segura Campos, M.R. Systemic Oxidative Stress: A key Point in Neurodegeneration—A Review. J. Nutr. Health Aging 2019, 23, 694–699. [Google Scholar] [CrossRef]
- Ashour, N.H.; El-Tanbouly, D.M.; El Sayed, N.S.; Khattab, M.M. Roflumilast ameliorates cognitive deficits in a mouse model of amyloidogenesis and tauopathy: Involvement of nitric oxide status, Aβ extrusion transporter ABCB1, and reversal by PKA inhibitor H89. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 111, 110366. [Google Scholar] [CrossRef]
- Xu, J.J.; Guo, S.; Xue, R.; Xiao, L.; Kou, J.N.; Liu, Y.Q.; Han, J.Y.; Fu, J.J.; Wei, N. Adalimumab ameliorates memory impairments and neuroinflammation in chronic cerebral hypoperfusion rats. Aging 2021, 13, 14001–14014. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener 2016, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Chiu, Y.J.; Lin, C.H.; Lee, M.C.; Hsieh-Li, H.M.; Chen, C.M.; Wu, Y.R.; Chang, K.H.; Lee-Chen, G.J. Formulated Chinese medicine Shaoyao Gancao Tang reduces NLRP1 and NLRP3 in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. Aging 2021, 9, 13. [Google Scholar]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa β as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Han, J.; Lee, N.; Yoon, J.H.; Youn, K.; Ha, H.J.; Yoon, E.; Kim, D.H.; Jun, M. Neuroprotective Effects of Baicalein, Wogonin, and Oroxylin A on Amyloid Beta-Induced Toxicity via NF-κB/MAPK Pathway Modulation. Molecules 2020, 25, 5087. [Google Scholar] [CrossRef]
- Youn, K.; Jun, M. Geraniin Protects PC12 Cells Against Aβ25-35-Mediated Neuronal Damage: Involvement of NF-κB and MAPK Signaling Pathways. J. Med. Food 2020, 23, 928–937. [Google Scholar] [CrossRef]
- Ma, L.Y.; Liu, S.F.; Du, J.H.; Niu, Y.; Hou, P.F.; Shu, Q.; Ma, R.R.; Wu, S.D.; Qu, Q.M.; Lv, Y.L. Chronic ghrelin administration suppresses IKK/NF-κB/BACE1 mediated Aβ production in primary neurons and improves cognitive function via upregulation of PP1 in STZ-diabetic rats. Neurobiol. Learn Mem. 2020, 169, 107155. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Arimon, M.; Takeda, S.; Post, K.; Svirsky, S.; Hyman, B.T.; Berezovska, O. Oxidative stress and lipid peroxidation are upstream of amyloid pathology. Neurobiol. Dis. 2015, 84, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, G.; Nunomura, A.; Hirai, K.; Takeda, A.; Aliev, G.; Smith, M.A. Oxidative damage in Alzheimer’s disease: The metabolic dimension. Int. J. Dev. Neurosci. 2000, 18, 417–421. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A. Proteomic analysis of oxidatively modified proteins in Alzheimer’s disease brain: Insights into neurodegeneration. Cell Mol. Biol. 2003, 49, 747–751. [Google Scholar]
- Butterfield, D.A.; Gnjec, A.; Poon, H.F.; Castegna, A.; Pierce, W.M.; Klein, J.B.; Martins, R.N. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: An initial assessment. J. Alzheimers Dis. 2006, 10, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhao, X.; Li, B.; Cai, Y.; Zhang, S.; Yu, F.; Wan, Q. Biomarkers for evaluating the effects of exercise interventions in patients with MCI or dementia: A systematic review and meta-analysis. Exp. Gerontol. 2021, 151, 111424. [Google Scholar] [CrossRef]
- Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β Peptide Impact on Synaptic Function and Neuroepigenetic Gene Control Reveal New Therapeutic Strategies for Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 577622. [Google Scholar] [CrossRef]
- Saeedi, M.; Rashidy-Pour, A. Association between chronic stress and Alzheimer’s disease: Therapeutic effects of Saffron. Biomed. Pharmacother. 2021, 133, 110995. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; Lerner, A. A Fundamental Role for Oxidants and Intracellular Calcium Signals in Alzheimer’s Pathogenesis-And How a Comprehensive Antioxidant Strategy May Aid Prevention of This Disorder. Int. J. Mol. Sci. 2021, 22, 2140. [Google Scholar] [CrossRef]
- Patil, S.; Sheng, L.; Masserang, A.; Chan, C. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci. Lett. 2006, 406, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, M.; Arciniega-Martínez, I.M.; García-Marín, I.D.; Correa-Basurto, J.; Rosales-Hernández, M.C. Chronic Administration of Scopolamine Increased GSK3βP9, Beta Secretase, Amyloid Beta, and Oxidative Stress in the Hippocampus of Wistar Rats. Mol. Neurobiol. 2020, 57, 3979–3988. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J. Neural. Transm. 2005, 112, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Zhou, S.F.; Wang, Q.; Guo, J.N.; Liang, H.M.; Deng, J.B.; He, W.Y. Gastrodin suppresses BACE1 expression under oxidative stress condition via inhibition of the PKR/eIF2α pathway in Alzheimer’s disease. Neuroscience 2016, 25, 1–9. [Google Scholar] [CrossRef]
- Syeda, T.; Cannon, J.R. Environmental exposures and the etiopathogenesis of Alzheimer’s disease: The potential role of BACE1 as a critical neurotoxic target. J. Biochem. Mol. Toxicol. 2021, 35, e22694. [Google Scholar] [CrossRef] [PubMed]
- Borghi, R.; Patriarca, S.; Traverso, N.; Piccini, A.; Storace, D.; Garuti, A.; Cirmena, G.; Odetti, P.; Tabaton, M. The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Bromley-Brits, K.; Song, W. Regulation of β-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s disease. J. Neurochem. 2012, 120, 62–70. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Bardini, P.; Santoro, G.; Davit, A.; Di Simone, D.; Danni, O.; Tabaton, M. Dehydroepiandrosterone reduces expression and activity of BACE in NT2 neurons exposed to oxidative stress. Neurobiol. Dis. 2003, 14, 291–301. [Google Scholar] [CrossRef]
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A.; et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar]
- Jo, D.G.; Arumugam, T.V.; Woo, H.N.; Park, J.S.; Tang, S.C.; Mughal, M.; Hyun, D.H.; Park, J.-H.; Choi, Y.H.; Gwon, A.R.; et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Giliberto, L.; Borghi, R.; Piccini, A.; Mangerini, R.; Sorbi, S.; Cirmena, G.; Garuti, A.; Ghetti, B.; Tagliavini, F.; Mughal, M.R.; et al. Mutant presenilin 1 increases the expression and activity of BACE1. J. Biol. Chem. 2009, 284, 9027–9038. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Cho, H.J.; Jung, E.S.; Shim, M.Y.; Mook-Jung, I. DNA damage-inducing agents elicit gamma-secretase activation mediated by oxidative stress. Cell Death Differ. 2008, 15, 1375–1384. [Google Scholar] [CrossRef]
- Sheng, B.; Gong, K.; Niu, Y.; Liu, L.; Yan, Y.; Lu, G.; Zhang, L.; Hu, M.; Zhao, N.; Zhang, X.; et al. Inhibition of gamma-secretase activity reduces Abeta production, reduces oxidative stress, increases mitochondrial activity and leads to reduced vulnerability to apoptosis: Implications for the treatment of Alzheimer’s disease. Free Radic. Biol. Med. 2009, 46, 1362–1375. [Google Scholar] [CrossRef]
- Masand, N.; Gupta, S.P.; Khosa, R.L. Designing of Selective γ-Secretase Inhibitory Benzenesulfonamides through Comparative In Vitro and In Silico Analysis. Curr. Drug Discov. Technol. 2018, 15, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, J.T.; Wang, H.F.; Meng, X.F.; Tan, C.C.; Wang, J.; Wang, C.; Tan, L. Association between stroke and Alzheimer’s disease: Systematic review and meta-analysis. J. Alzheimers Dis. 2015, 43, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Shabir, O.; Berwick, J.; Francis, S.E. Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neurosci. 2018, 19, 62. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, X.; Yang, D.; Luo, G.; Chen, S.; Le, W. Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol. Aging 2009, 30, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Peers, C.; Pearson, H.A.; Boyle, J.P. Hypoxia and Alzheimer’s disease. Essays Biochem. 2007, 43, 153–164. [Google Scholar] [CrossRef]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef]
- Guzy, R.D.; Mack, M.M.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and gene transcription in yeast. Antioxid. Redox Signal. 2007, 9, 1317–1328. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, J.; Chang, Y.; ShiDu Yan, S.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 2011, 18, 4335–4343. [Google Scholar] [CrossRef] [Green Version]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Diniz Pereira, J.; Gomes Fraga, V.; Morais Santos, A.L.; Carvalho, M.D.G.; Caramelli, P.; Braga Gomes, K. Alzheimer’s disease and type 2 diabetes mellitus: A systematic review of proteomic studies. J. Neurochem. 2021, 156, 753–776. [Google Scholar] [CrossRef]
- Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Tan, Y.Q.; Yu, X.Y.; Zhao, Y.Y. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic. Biol. Med. 2021, 171, 260–271. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. Differential effects of glycation on protein aggregation and amyloid formation. Front. Mol. Biosci. 2014, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawver, J.N.; Schall, H.E.; Petrofes Chapa, R.D.; Zhu, X.; Murray, I.V. Amyloid-β metabolite sensing: Biochemical linking of glycation modification and misfolding. J. Alzheimers Dis. 2012, 30, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Muscat, S.; Pelka, J.; Hegele, J.; Weigle, B.; Münch, G.; Pischetsrieder, M. Coffee and Maillard products activate NF-kappaB in macrophages via H2O2 production. Mol. Nutr. Food Res. 2007, 51, 525–535. [Google Scholar] [CrossRef]
- Hu, Z.; Fang, W.; Liu, Y.; Liang, H.; Chen, W.; Wang, H. Acute glucose fluctuation promotes RAGE expression via reactive oxygen species-mediated NF-κB activation in rat podocytes. Mol. Med. Rep. 2021, 23, 330. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. AGE-RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.H.; Humpert, P.M.; Nawroth, P.P.; Bierhaus, A. Reactive metabolites and AGE/RAGE-mediated cellular dysfunction affect the aging process: A mini-review. Gerontology 2011, 57, 435–443. [Google Scholar] [CrossRef]
- Prasad, K. AGE-RAGE stress play a role in aortic aneurysm: A comprehensive review and novel potential therapeutic target. Rev. Cardiovasc. Med. 2019, 20, 201–208. [Google Scholar]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campora, M.; Francesconi, V.; Schenone, S.; Tasso, B.; Tonelli, M. Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Samant, N.P.; Gupta, G.L. Novel therapeutic strategies for Alzheimer’s disease targeting brain cholesterol homeostasis. Eur. J. Neurosci. 2021, 53, 673–686. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Ribe, E.M.; Lovestone, S. Insulin signalling in Alzheimer’s disease and diabetes: From epidemiology to molecular links. J. Intern Med. 2016, 280, 430–442. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–13353. [Google Scholar] [CrossRef]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Rebelos, E.; Rinne, J.O.; Nuutila, P.; Ekblad, L.L. Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline-Does Insulin Have Anything to Do with It? A Narrative Review. J. Clin. Med. 2021, 10, 1532. [Google Scholar] [CrossRef]
- Wang, D.; Pascual, J.M.; Yang, H.; Engelstad, K.; Mao, X.; Cheng, J.; Yoo, J.; Noebels, J.L.; De Vivo, D.C. A mouse model for Glut-1 haploinsufficiency. Hum. Mol. Genet. 2006, 15, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Ullner, P.M.; Di Nardo, A.; Goldman, J.E.; Schobel, S.; Yang, H.; Engelstad, K.; Wang, D.; Sahin, M.; De Vivo, D.C. Murine Glut-1 transporter haploinsufficiency: Postnatal deceleration of brain weight and reactive astrocytosis. Neurobiol. Dis. 2009, 36, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007, 1181, 93–103. [Google Scholar] [CrossRef]
- Pang, R.; Wang, X.; Pei, F.; Zhang, W.; Shen, J.; Gao, X.; Chang, C. Regular Exercise Enhances Cognitive Function and Intracephalic GLUT Expression in Alzheimer’s Disease Model Mice. J. Alzheimers Dis. 2019, 72, 83–96. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzier-Sore, A.K.; Pellerin, L. Unraveling the complex metabolic nature of astrocytes. Front. Cell Neurosci. 2013, 7, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Vaishnavi, S.N.; Couture, L.; Sacco, D.; Shannon, B.J.; Mach, R.H.; Morris, J.C.; Raichle, M.E.; Mintun, M.A. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl. Acad. Sci. USA 2010, 107, 17763–17767. [Google Scholar] [CrossRef] [Green Version]
- Paul, R.; Borah, A. Global loss of acetylcholinesterase activity with mitochondrial complexes inhibition and inflammation in brain of hypercholesterolemic mice. Sci. Rep. 2017, 7, 17922. [Google Scholar] [CrossRef]
- Duong, M.T.; Nasrallah, I.M.; Wolk, D.A.; Chang, C.C.Y.; Chang, T.Y. Cholesterol, Atherosclerosis, and APOE in Vascular Contributions to Cognitive Impairment and Dementia (VCID): Potential Mechanisms and Therapy. Front. Aging Neurosci. 2021, 13, 647990. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Staurenghi, E.; Giannelli, S.; Gargiulo, S.; Guglielmotto, M.; Tabaton, M.; Tamagno, E.; Gamba, P.; Leonarduzzi, G. A silver lining for 24-hydroxycholesterol in Alzheimer’s disease: The involvement of the neuroprotective enzyme sirtuin 1. Redox Biol. 2018, 17, 423–431. [Google Scholar] [CrossRef]
- Gamba, P.; Guglielmotto, M.; Testa, G.; Monteleone, D.; Zerbinati, C.; Gargiulo, S.; Biasi, F.; Iuliano, L.; Giaccone, G.; Mauro, A.; et al. Up-regulation of β-amyloidogenesis in neuron-like human cells by both 24- and 27-hydroxycholesterol: Protective effect of N-acetyl-cysteine. Aging Cell 2014, 13, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lee, H.G.; Perry, G.; Smith, M.A. Alzheimer disease, the two-hit hypothesis: An update. Biochim. Biophys. Acta 2007, 1772, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.; Puli, L.; Patil, C.R. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug Discov. Today 2021, 26, 794–803. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. https://doi.org/10.3390/antiox10091479
Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants. 2021; 10(9):1479. https://doi.org/10.3390/antiox10091479
Chicago/Turabian StyleTamagno, Elena, Michela Guglielmotto, Valeria Vasciaveo, and Massimo Tabaton. 2021. "Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg?" Antioxidants 10, no. 9: 1479. https://doi.org/10.3390/antiox10091479
APA StyleTamagno, E., Guglielmotto, M., Vasciaveo, V., & Tabaton, M. (2021). Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants, 10(9), 1479. https://doi.org/10.3390/antiox10091479