
Structural Studies of Aliphatic Glucosinolate Chain-Elongation Enzymes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
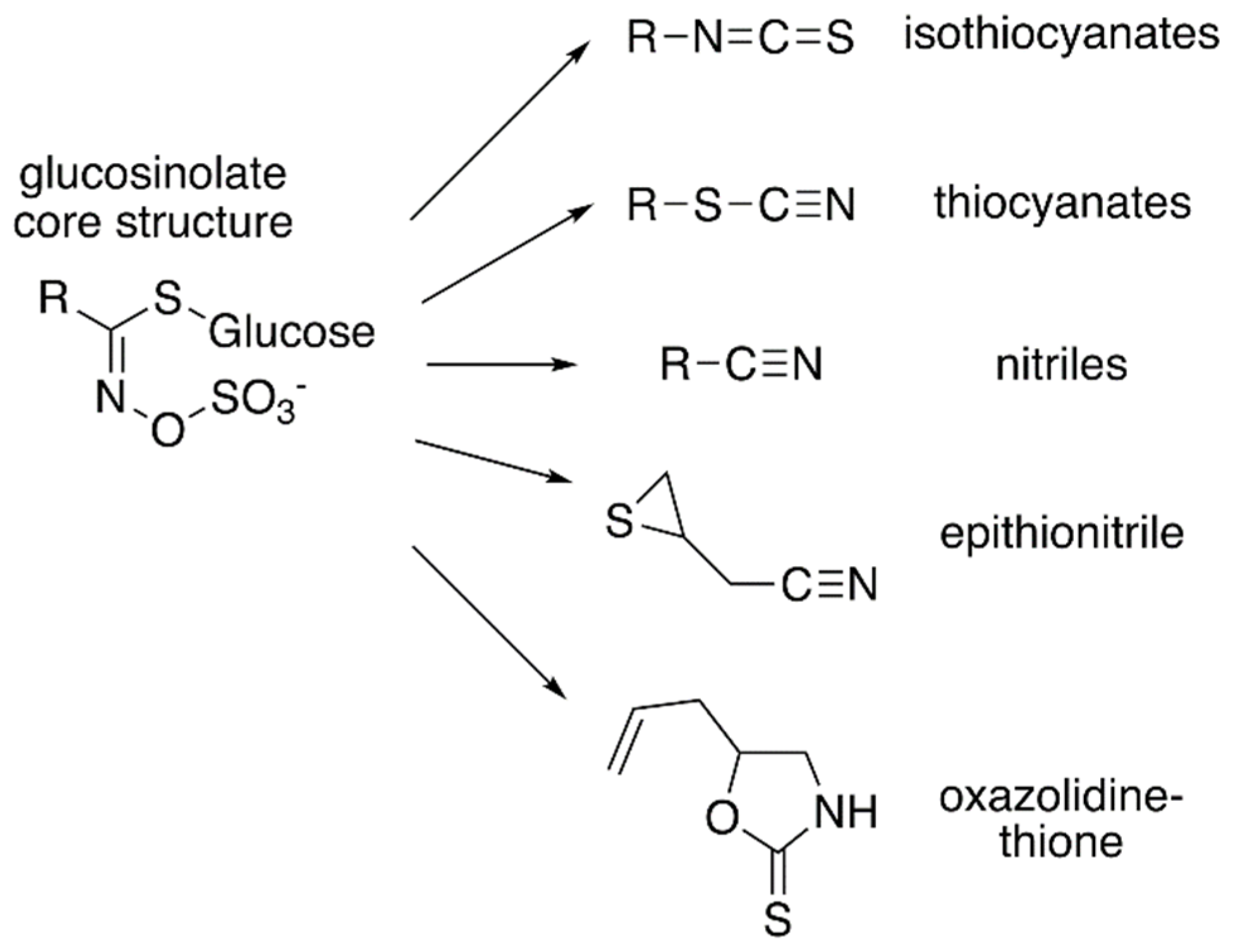
:1. Introduction to the Glucosinolates
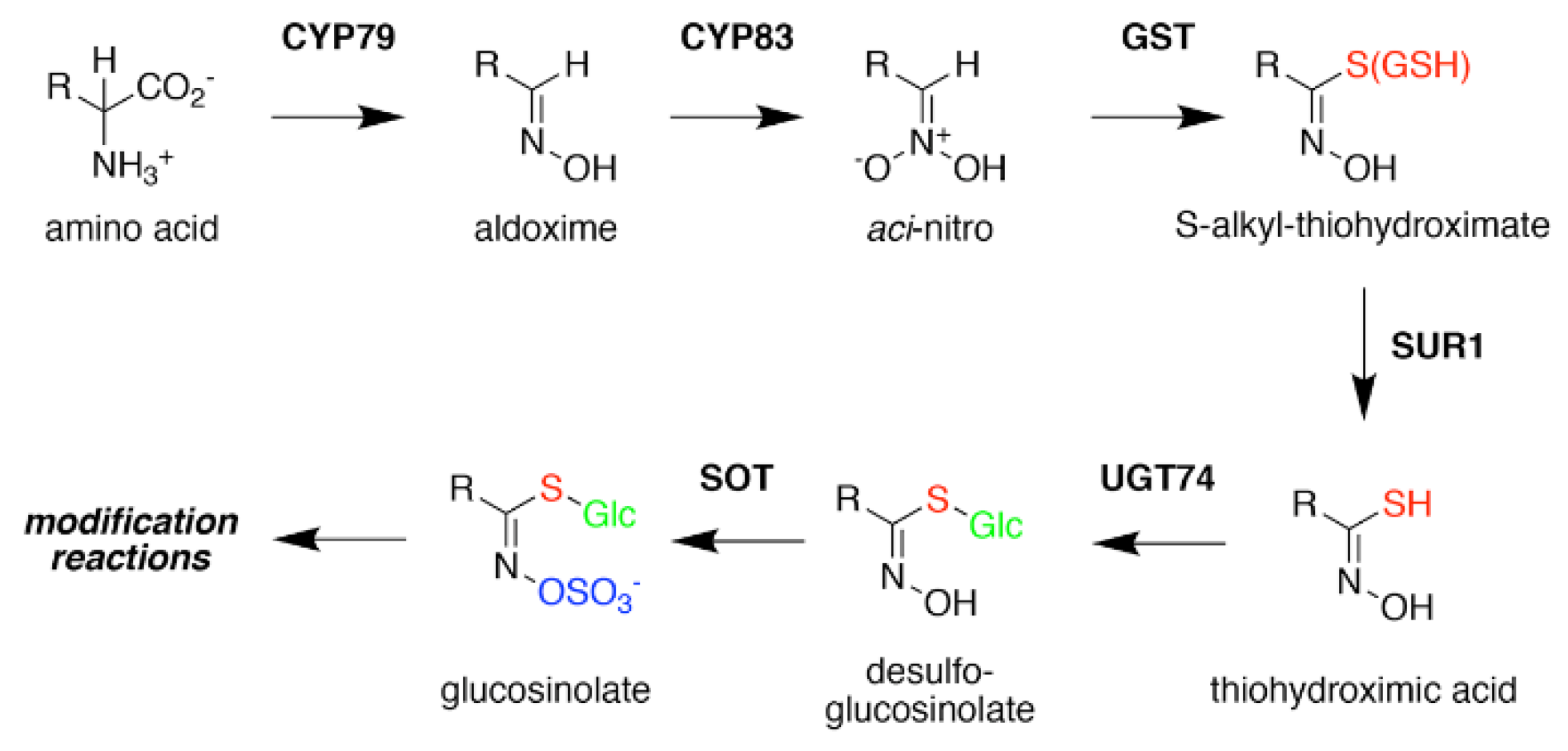
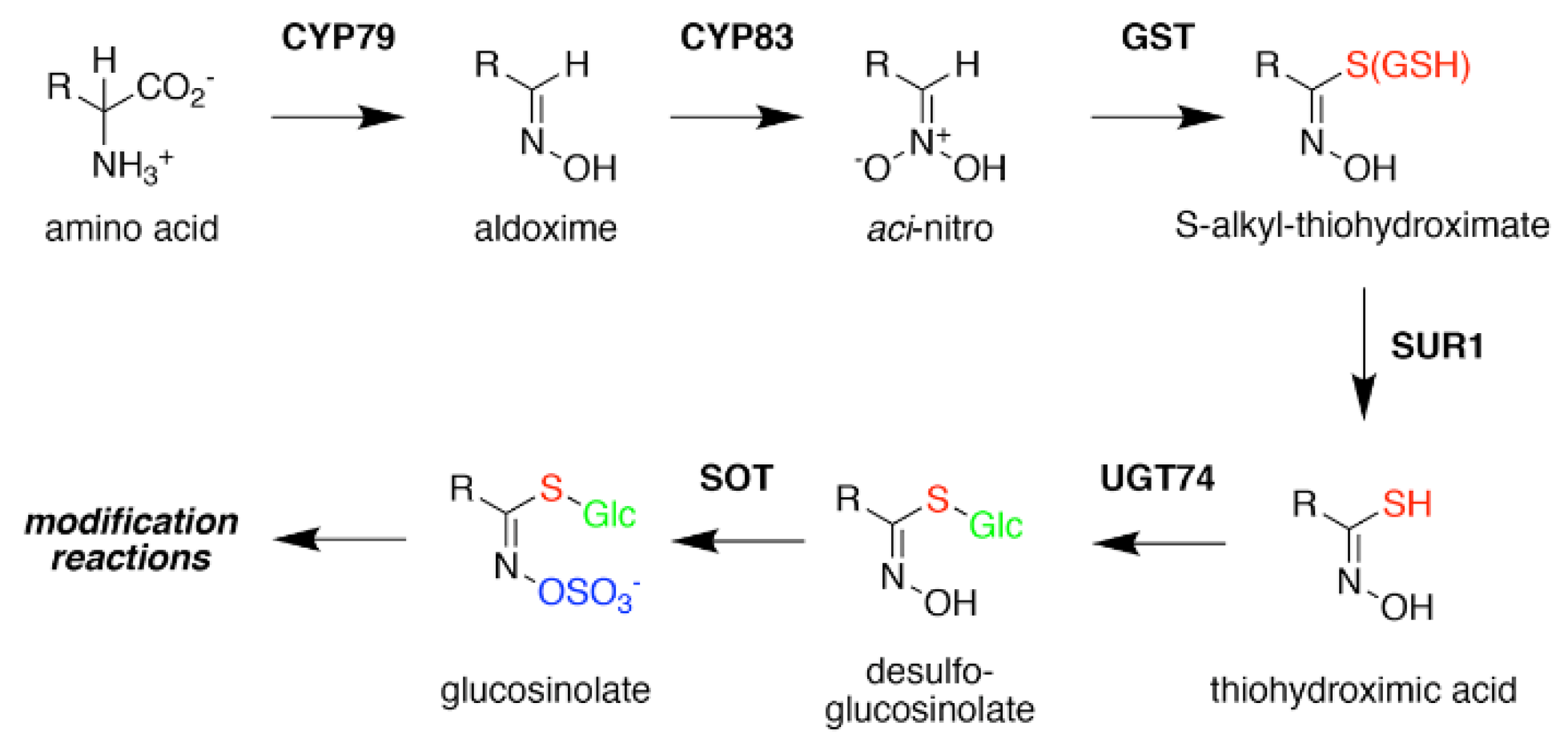
2. Glucosinolate Biosynthesis
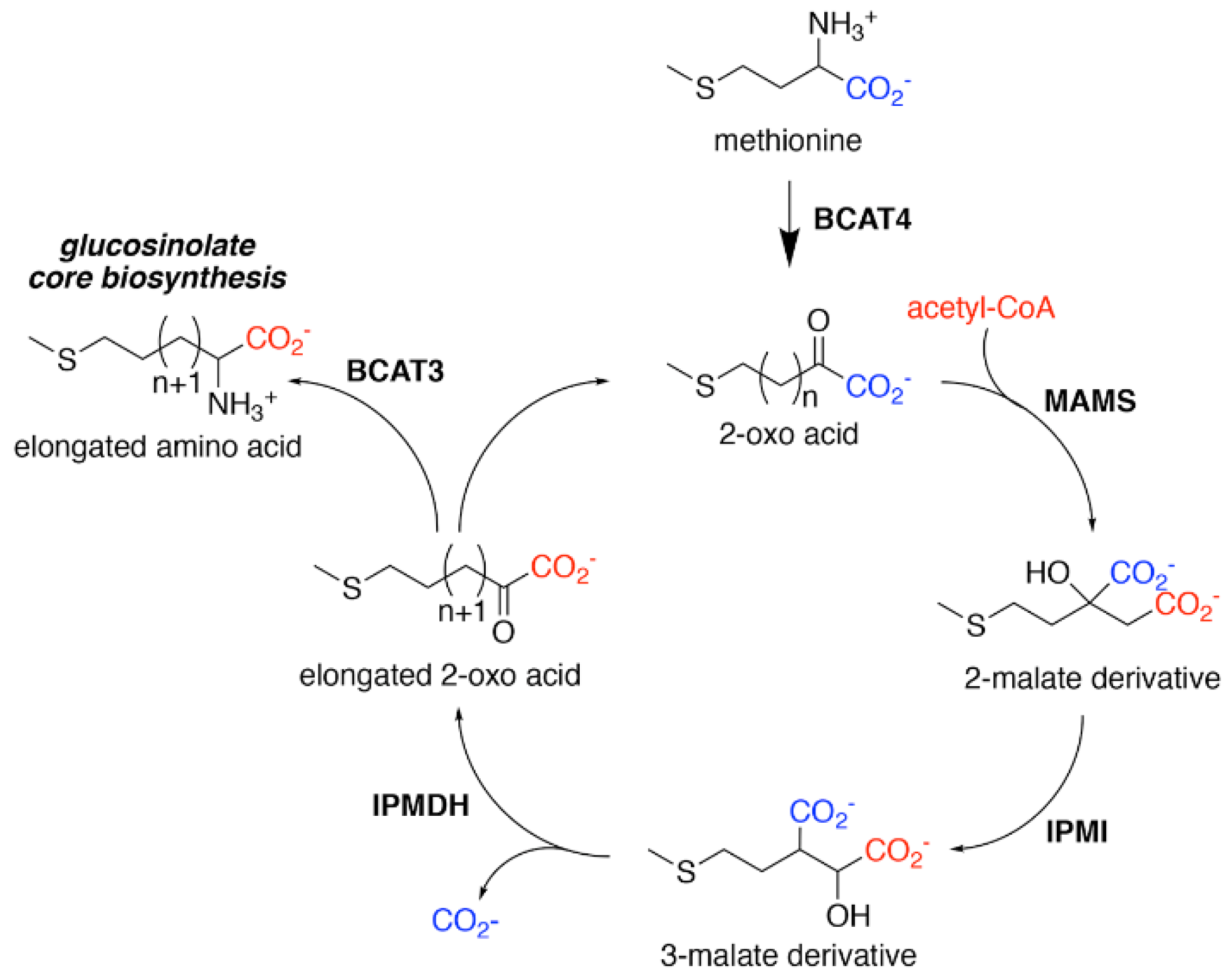
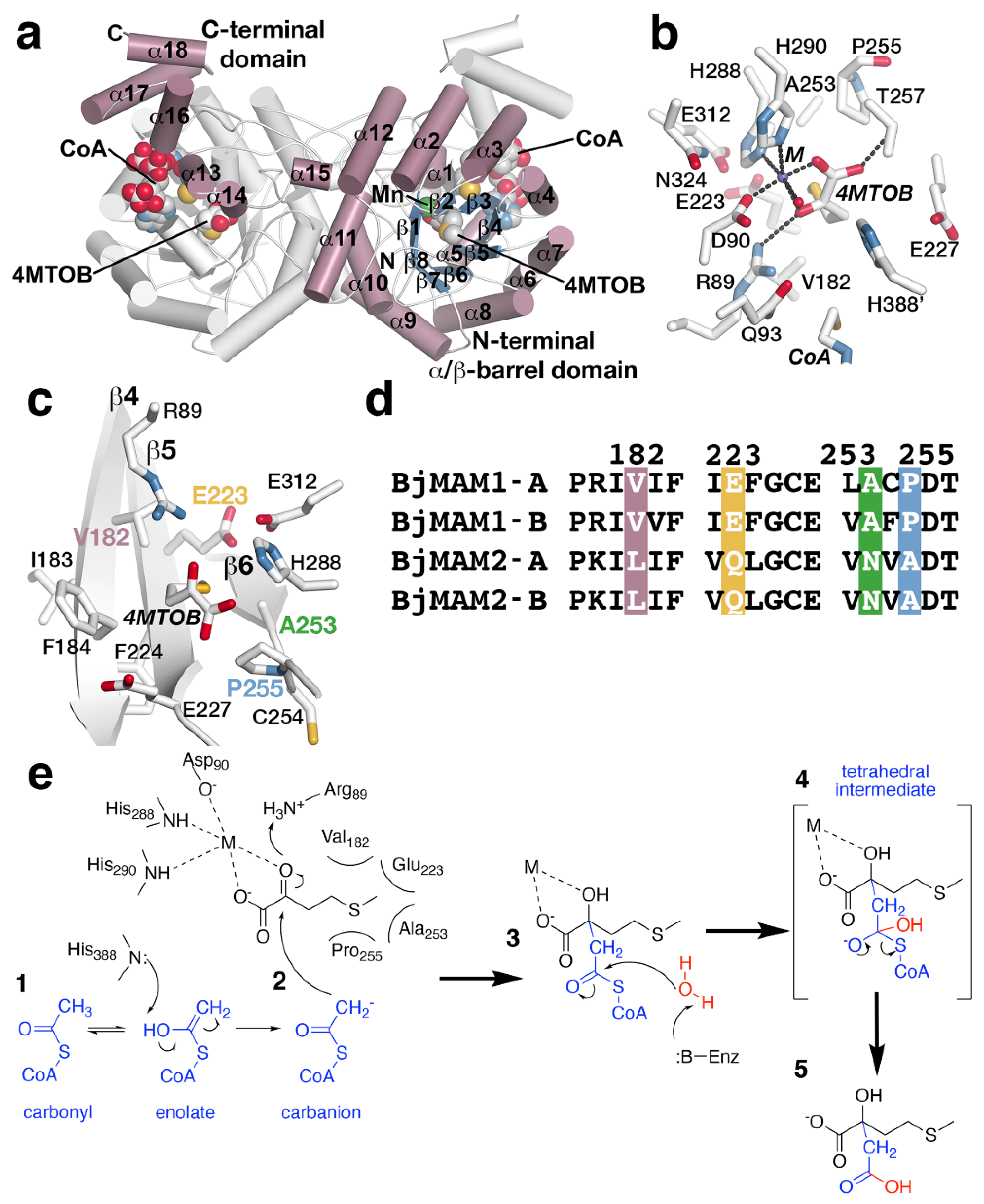
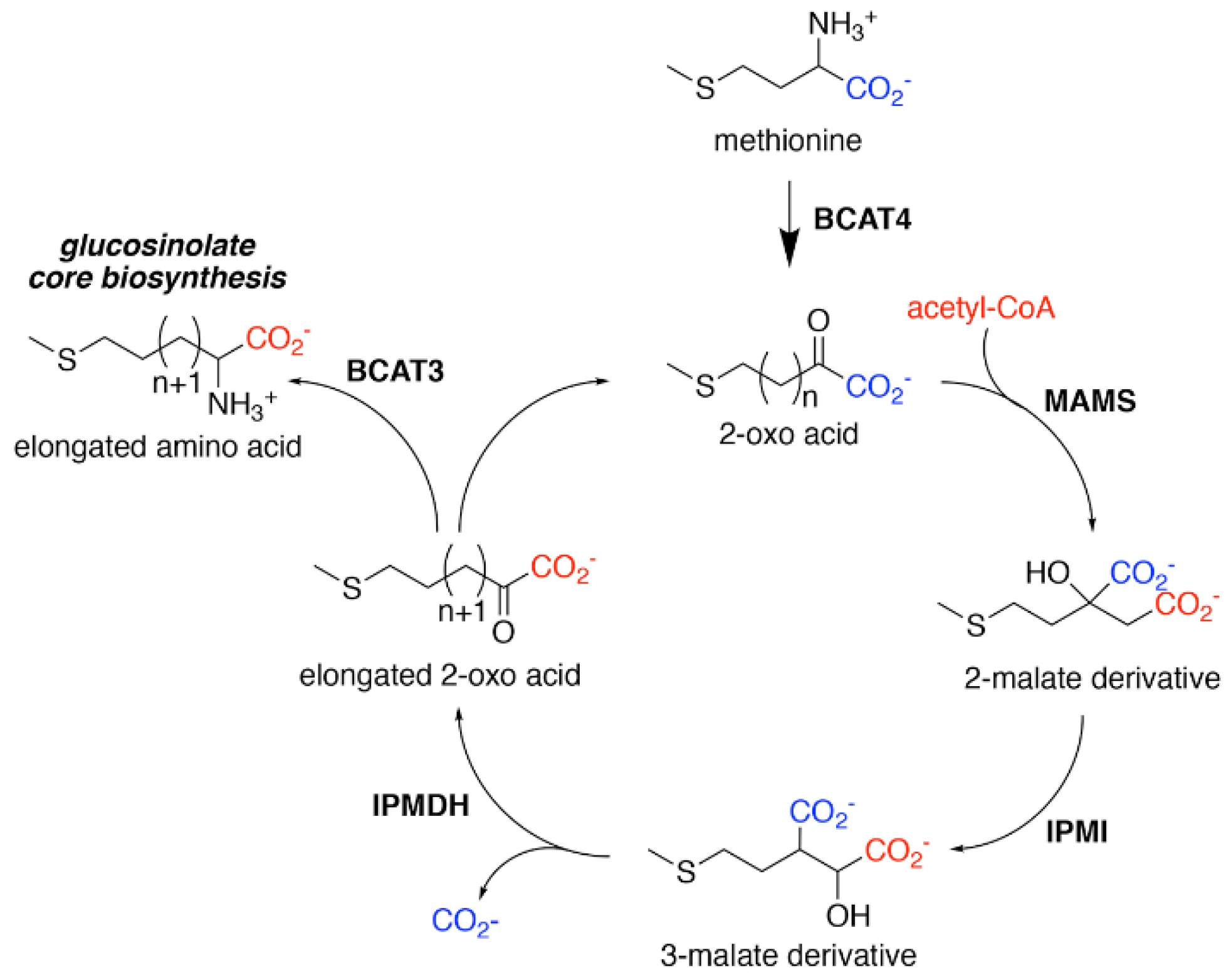
3. Aliphatic Chain-Elongation: Methylthioalkylmalate Synthase (MAMS)
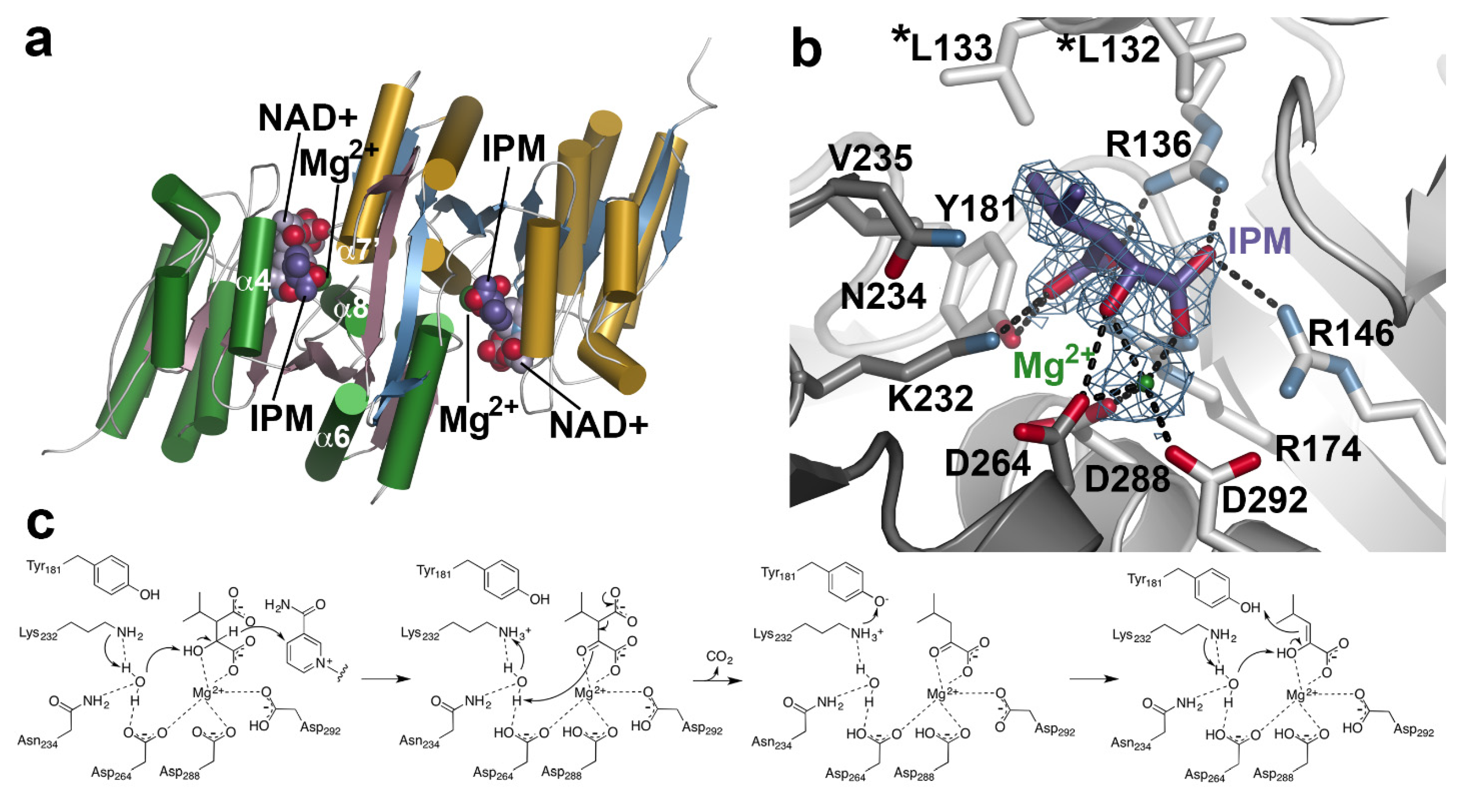
4. Aliphatic Chain-Elongation: Isopropylmalate Dehydrogenase (IPMDH)
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodman, J.; Karol, K.; Price, R.; Sytsma, K. Molecules, morphology, and Dahlgren's expanded order Capparales. Syst. Bot. 1996, 21, 289–307. [Google Scholar] [CrossRef]
- Sønderby, I.E.; Geu-Flores, F.; Halkier, B.A. Biosynthesis of glucosinolates- gene discovery and beyond. Trends Plant Sci. 2010, 15, 283–290. [Google Scholar] [CrossRef]
- Singh, A. Glucosinolates and plant defense. In Glucosinolates- Reference Series in Phytochemistry; Mérillon, J.M., Ramawat, K., Eds.; Springer: Cham, Switzerland, 2017; pp. 237–246. [Google Scholar]
- Nguyen, V.P.T.; Stewart, J.; Lopez, M.; Ioannou, I.; Allais, F. Glucosinolates: Natural occurrence, biosynthesis, accessibility, isolation, structures, and biological activities. Molecules 2020, 25, 4537. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Cottaz, S.; Driguez, H.; Iori, R.; Palmieri, S.; Henrissat, B. The crystal structures of Sinapis alba myrosinase and a covalent glycosyl-enzyme intermediate provide insights into the substrate recognition and active-site machinery of an S-glycosidase. Structure 1997, 5, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Ravilious, G.E.; Jez, J.M. Structural biology of plant sulfur metabolism: From assimilation to biosynthesis. Nat. Prod. Rep. 2012, 29, 1138–1152. [Google Scholar] [CrossRef]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Engel, E.; Baty, C.; Le Corre, D.; Souchon, I.; Martin, N. Flavor-active compounds potentially implicated in cooked cauliflower acceptance. J. Agric. Food Chem. 2002, 50, 6459–6467. [Google Scholar] [CrossRef]
- Nour-Eldin, H.H.; Madsen, S.R.; Engelen, S.; Jørgensen, M.E.; Olsen, C.E.; Andersen, J.S.; Seynnaeve, D.; Verhoye, T.; Fulawka, R.; Denolf, P.; et al. Reduction of antinutritional glucosinolates in Brassica oilseeds by mutation of genes encoding transporters. Nat. Biotechnol. 2017, 35, 377–382. [Google Scholar] [CrossRef]
- Griffiths, D.W.; Birch, A.N.E.; Hillman, J.R. Antinutritional compounds in the Brasi analysis, biosynthesis, chemistry and dietary effects. J. Hort. Sci. Biotech. 1998, 73, 1–18. [Google Scholar] [CrossRef]
- Brown, P.D.; Morra, M.J. Glucosinolate-containing plant tissues as bioherbicides. J. Agric. Food Chem. 1995, 43, 3070–3074. [Google Scholar] [CrossRef]
- Vaugh, S.F.; Palmquist, D.E.; Duval, S.M.; Berhow, M.A. Herbicidal activity of glucosinolate-containing seedmeals. Weed Sci. 2006, 54, 743–748. [Google Scholar] [CrossRef]
- Zasada, I.A.; Ferris, H. Nematode suppression with brassicaceous amendments: Application based upon glucosinolate profiles. Soil Biol. Biochem. 2004, 36, 1017–1024. [Google Scholar] [CrossRef]
- Hopkins, R.J.; van Dam, N.M.; van Loon, J.J. Role of glucosinolates in insect-plant relationships and multitrophic interactions. Annu. Rev. Entomol. 2009, 54, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Mithen, R. Leaf glucosinolate profiles and their relationship to pest and disease resistance in oilseed rape. Euphytica 1992, 64, 71–83. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J. Isothiocyanates: Mechanism of cancer chemopreventive action. Anticancer Drugs 2002, 13, 331–338. [Google Scholar] [CrossRef]
- Keum, Y.S.; Jeong, W.S.; Kong, A.N. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat. Res. 2004, 555, 191–202. [Google Scholar] [CrossRef]
- Hayes, J.D.; Kelleher, M.O.; Eggleston, I.M. The cancer chemopreventive actions of phytochemicals derived from glucosinolates. Eur. J. Nutr. 2008, 47, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibdah, M.; Chen, Y.T.; Wilkerson, C.G.; Pichersky, E. An aldehyde oxidase in developing seeds of Arabidopsis converts benzaldehyde to benzoic acid. Plant Physiol. 2009, 150, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hansen, B.G.; Ober, J.A.; Kliebenstein, D.J.; Halkier, B.A. Subclade of flavin-monooxygenases involved in aliphatic glucosinolate biosynthesis. Plant Physiol. 2008, 148, 1721–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, B.G.; Kliebenstein, D.J.; Halkier, B.A. Identification of a flavin-monooxygenase as the S-oxygenating enzyme in aliphatic glucosinolate biosynthesis in Arabidopsis. Plant J. 2007, 50, 902–910. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; D’Auria, J.C.; Behere, A.S.; Kim, J.H.; Gunderson, K.L.; Breen, J.N.; Lee, G.; Gershenzon, J.; Last, R.L.; Jander, G. Characterization of seed-specific benzoyloxyglucosinolate mutations in Arabidopsis thaliana. Plant J. 2007, 51, 1062–1076. [Google Scholar] [CrossRef]
- Brown, P.D.; Tokuhisa, J.G.; Reichelt, M.; Gershenzon, J. Variation of glucosinolate accumulation among different organs and developmental stages of Arabidopsis thaliana. Phytochemistry 2003, 62, 471–481. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; Lambrix, V.M.; Reichelt, M.; Gershenzon, J.; Mitchell-Olds, T. Gene duplication in the diversification of secondary metabolism: Tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 2001, 13, 681–693. [Google Scholar]
- Diebold, R.; Schuster, J.; Däschner, K.; Binder, S. The branched-chain amino acid transaminase gene family in Arabidopsis encodes plastid and mitochondrial proteins. Plant Physiol. 2002, 129, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, J.; Knill, T.; Reichelt, M.; Gershenzon, J.; Binder, S. Branched-chain aminotransferase4 is part of the chain elongation pathway in the biosynthesis of methionine-derived glucosinolates in Arabidopsis. Plant Cell 2006, 18, 2664–2679. [Google Scholar] [CrossRef] [Green Version]
- Falk, K.L.; Vogel, C.; Textor, S.; Bartram, S.; Hick, A.; Pickett, J.A.; Gershenzon, J. Glucosinolate biosynthesis: Demonstration and characterization of the condensing enzyme of the chain elongation cycle in Eruca sativa. Phytochemistry 2004, 65, 1073–1084. [Google Scholar] [CrossRef]
- Textor, S.; de Kraker, J.W.; Hause, B.; Gershenzon, J.; Tokuhisa, J.G. MAM3 catalyzes the formation of all aliphatic glucosinolate chain lengths in Arabidopsis. Plant Physiol. 2007, 144, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, Y.; Kuwahara, A.; Nagano, M.; Narisawa, T.; Sakata, A.; Saito, K.; Hirai, M.Y. Omics-based approaches to methionine side chain elongation in Arabidopsis: Characterization of the genes encoding methylthioalkylmalate isomerase and methylthioalkylmalate dehydrogenase. Plant Cell. Physiol. 2009, 50, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Knill, T.; Reichelt, M.; Paetz, C.; Gershenzon, J.; Binder, S. Arabidopsis thaliana encodes a bacterial-type heterodimeric isopropylmalate isomerase involved in both Leu biosynthesis and the Met chain elongation pathway of glucosinolate formation. Plant Mol. Biol. 2009, 71, 227–239. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Mawhinney, T.P.; Preuss, M.L.; Schroeder, A.C.; Chen, B.; Abraham, L.; Jez, J.M.; Chen, S. A redox-active isopropylmalate dehydrogenase functions in the biosynthesis of glucosinolates and leucine in Arabidopsis. Plant J. 2009, 60, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Gigolashvili, T.; Yatusevich, R.; Rollwitz, I.; Humphry, M.; Gershenzon, J.; Flügge, U.I. The plastidic bile acid transporter 5 is required for the biosynthesis of methionine-derived glucosinolates in Arabidopsis thaliana. Plant Cell 2009, 21, 1813–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Textor, S.; Bartram, S.; Kroymann, J.; Falk, K.L.; Hick, A.; Pickett, J.A.; Gershenzon, J. Biosynthesis of methionine-derived glucosinolates in Arabidopsis thaliana: Recombinant expression and characterization of methylthioalkylmalate synthase, the condensing enzyme of the chain-elongation cycle. Planta 2004, 218, 1026–1035. [Google Scholar] [CrossRef]
- de Kraker, J.W.; Gershenzon, J. From amino acid to glucosinolate biosynthesis: Protein sequence changes in the evolution of methylthioalkylmalate synthase in Arabidopsis. Plant Cell 2011, 23, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Lee, S.G.; Augustine, R.; Reichelt, M.; Vassão, D.G.; Palavalli, M.H.; Allen, A.; Gershenzon, J.; Jez, J.M.; Bisht, N.C. Molecular basis of the evolution of methylthioalkylmalate synthase and the diversity of methionine-derived glucosinolates. Plant Cell 2019, 31, 1633–1647. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, B.; Pang, Q.; Strul, J.M.; Chen, S. Functional specification of Arabidopsis isopropylmalate isomerases in glucosinolate and leucine biosynthesis. Plant Cell. Physiol. 2010, 51, 1480–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Galant, A.; Pang, Q.; Strul, J.M.; Balogun, S.F.; Jez, J.M.; Chen, S. Structural and functional evolution of isopropylmalate dehydrogenases in the leucine and glucosinolate pathways of Arabidopsis thaliana. J. Biol. Chem. 2011, 286, 28794–28801. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Chen, L.; Zhou, Y.; Mawhinney, T.P.; Chen, B.; Kang, B.H.; Hauser, B.A.; Chen, S. Functional characterization of Arabidopsis thaliana isopropylmalate dehydrogenases reveals their important roles in gametophyte development. New Phytol. 2011, 189, 160–175. [Google Scholar] [CrossRef]
- Knill, T.; Schuster, J.; Reichelt, M.; Gershenzon, J.; Binder, S. Arabidopsis branched-chain aminotransferase 3 functions in both amino acid and glucosinolate biosynthesis. Plant Physiol. 2008, 146, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- Lächler, K.; Imhof, J.; Reichelt, M.; Gershenzon, J.; Binder, S. The cytosolic branched-chain aminotransferases of Arabidopsis thaliana influence methionine supply, salvage and glucosinolate metabolism. Plant Mol. Biol. 2015, 88, 119–131. [Google Scholar] [CrossRef]
- Mikkelsen, M.D.; Hansen, C.H.; Wittstock, U.; Halkier, B.A. Cytochrome P450 CYP79B2 from Arabidopsis catalyzes the conversion of tryptophan to indole-3-acetaldoxime, a precursor of indole glucosinolates and indole-3-acetic acid. J. Biol. Chem. 2000, 275, 33712–33717. [Google Scholar] [CrossRef] [Green Version]
- Wittstock, U.; Halkier, B.A. Cytochrome P450 CYP79A2 from Arabidopsis thaliana L. catalyzes the conversion of L-phenylalanine to phenylacetaldoxime in the biosynthesis of benzylglucosinolate. J. Biol. Chem. 2000, 275, 14659–14666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, A.K.; Vij, R.; Celenza, J.L. Arabidopsis cytochrome P450s that catalyze the first step of tryptophan-dependent indole-3-acetic acid biosynthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2379–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.H.; Wittstock, U.; Olsen, C.E.; Hick, A.J.; Pickett, J.A.; Halkier, B.A. Cytochrome P450 CYP79F1 from Arabidopsis catalyzes the conversion of dihomomethionine and trihomomethionine to the corresponding aldoximes in the biosynthesis of aliphatic glucosinolates. J. Biol. Chem. 2001, 276, 11078–11085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Glawischnig, E.; Jørgensen, K.; Naur, P.; Jørgensen, B.; Olsen, C.E.; Hansen, C.H.; Rasmussen, H.; Pickett, J.A.; Halkier, B.A. CYP79F1 and CYP79F2 have distinct functions in the biosynthesis of aliphatic glucosinolates in Arabidopsis. Plant J. 2003, 33, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Glawischnig, E. The role of cytochrome P450 enzymes in the biosynthesis of camalexin. Biochem. Soc. Trans. 2006, 34, 1206–1208. [Google Scholar] [CrossRef] [Green Version]
- Bak, S.; Tax, F.E.; Feldmann, K.A.; Galbraith, D.W.; Feyereisen, R. CYP83B1, a cytochrome P450 at the metabolic branch point in auxin and indole glucosinolate biosynthesis in Arabidopsis. Plant Cell 2001, 13, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Bak, S.; Feyereisen, R. The involvement of two P450 enzymes, CYP83B1 and CYP83A1, in auxin homeostasis and glucosinolate biosynthesis. Plant Physiol. 2001, 127, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Hansen, C.H.; Du, L.; Naur, P.; Olsen, C.E.; Axelsen, K.B.; Hick, A.J.; Pickett, J.A.; Halkier, B.A. CYP83B1 is the oxime-metabolizing enzyme in the glucosinolate pathway in Arabidopsis. J. Biol. Chem. 2001, 276, 24790–24796. [Google Scholar] [CrossRef] [Green Version]
- Hemm, M.R.; Ruegger, M.O.; Chapple, C. The Arabidopsis ref2 mutant is defective in the gene encoding CYP83A1 and shows both phenylpropanoid and glucosinolate phenotypes. Plant Cell 2003, 15, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Naur, P.; Petersen, B.L.; Mikkelsen, M.D.; Bak, S.; Rasmussen, H.; Olsen, C.E.; Halkier, B.A. CYP83A1 and CYP83B1, two nonredundant cytochrome P450 enzymes metabolizing oximes in the biosynthesis of glucosinolates in Arabidopsis. Plant Physiol. 2003, 133, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, M.D.; Naur, P.; Halkier, B.A. Arabidopsis mutants in the C-S lyase of glucosinolate biosynthesis establish a critical role for indole-3-acetaldoxime in auxin homeostasis. Plant J. 2004, 37, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Jez, J.M. Structural biology of plant sulfur metabolism: From sulfate to glutathione. J. Exp. Bot. 2019, 70, 4089–4103. [Google Scholar] [CrossRef]
- Grubb, C.D.; Zipp, B.J.; Ludwig-Müller, J.; Masuno, M.N.; Molinski, T.F.; Abel, S. Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J. 2004, 40, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Gachon, C.M.; Langlois-Meurinne, M.; Henry, Y.; Saindrenan, P. Transcriptional co-regulation of secondary metabolism enzymes in Arabidopsis: Functional and evolutionary implications. Plant Mol. Biol. 2005, 58, 229–245. [Google Scholar] [CrossRef]
- Klein, M.; Papenbrock, J. The multi-protein family of Arabidopsis sulphotransferases and their relatives in other plant species. J. Exp. Bot. 2004, 55, 1809–1820. [Google Scholar] [CrossRef]
- Klein, M.; Reichelt, M.; Gershenzon, J.; Papenbrock, J. The three desulfoglucosinolate sulfotransferase proteins in Arabidopsis have different substrate specificities and are differentially expressed. FEBS J. 2006, 273, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, M.; Schemenewitz, A.; Lopukhina, A.; Müller, A.; Janowitz, T.; Weiler, E.W.; Oecking, C. Desulfoglucosinolate sulfotransferases from Arabidopsis thaliana catalyze the final step in the biosynthesis of the glucosinolate core structure. J. Biol. Chem. 2004, 279, 50717–50725. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Papenbrock, J. Kinetics and substrate specificities of desulfo-glucosinolate sulfotransferases in Arabidopsis thaliana. Physiol. Plant. 2009, 135, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann, F.; Papenbrock, J. The fusion of genomes leads to more options: A comparative investigation on the desulfo-glucosinolate sulfotransferases of Brassica napus and homologous proteins of Arabidopsis thaliana. Plant Physiol. Biochem. 2015, 91, 10–19. [Google Scholar] [CrossRef]
- Mugford, S.G.; Yoshimoto, N.; Reichelt, M.; Wirtz, M.; Hill, L.; Mugford, S.T.; Nakazato, Y.; Noji, M.; Takahashi, H.; Kramell, R.; et al. Disruption of adenosine-5'-phosphosulfate kinase in Arabidopsis reduces levels of sulfated secondary metabolites. Plant Cell 2009, 21, 910–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugford, S.G.; Matthewman, C.A.; Hill, L.; Kopriva, S. Adenosine-5′-phosphosulfate kinase is essential for Arabidopsis viability. FEBS Lett. 2010, 584, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Yatusevich, R.; Mugford, S.G.; Matthewman, C.; Gigolashvili, T.; Frerigmann, H.; Delaney, S.; Koprivova, A.; Flügge, U.I.; Kopriva, S. Genes of primary sulfate assimilation are part of the glucosinolate biosynthetic network in Arabidopsis thaliana. Plant J. 2010, 62, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mugford, S.G.; Lee, B.R.; Koprivova, A.; Matthewman, C.; Kopriva, S. Control of sulfur partitioning between primary and secondary metabolism. Plant J. 2011, 65, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Ravilious, G.E.; Nguyen, A.; Francois, J.A.; Jez, J.M. Structural basis and evolution of redox regulation in plant adenosine-5’-phosphosulfate kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Ravilious, G.E.; Jez, J.M. Nucleotide binding site communication in Arabidopsis thaliana adenosine 5′-phosphosulfate kinase. J. Biol. Chem. 2012, 287, 30385–30389. [Google Scholar] [CrossRef] [Green Version]
- Ravilious, G.E.; Westfall, C.S.; Jez, J.M. Redox-linked gating of nucleotide binding by the N-terminal domain of adenosine 5'-phosphosulfate kinase. J. Biol. Chem. 2013, 288, 6107–6115. [Google Scholar] [CrossRef] [Green Version]
- Jez, J.M.; Ravilious, G.E.; Herrmann, J. Structural insights on the regulation of the plant sulfation pathway. Chem.-Bio. Interact. 2016, 259, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Hirschmann, F.; Krause, F.; Baruch, P.; Chizhov, I.; Mueller, J.W.; Manstein, D.J.; Papenbrock, J.; Fedorov, R. Structural and biochemical studies of sulphotransferase 18 from Arabidopsis thaliana explain its substrate specificity and reaction mechanism. Sci. Rep. 2017, 7, 4160. [Google Scholar] [CrossRef] [PubMed]
- Kliebenstein, D.J.; Kroymann, J.; Brown, P.; Figuth, A.; Pedersen, D.; Gershenzon, J.; Mitchell-Olds, T. Genetic control of natural variation in Arabidopsis glucosinolate accumulation. Plant Physiol. 2001, 126, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Kroymann, J.; Donnerhacke, S.; Schnabelrauch, D.; Mitchell-Olds, T. Evolutionary dynamics of an Arabidopsis insect resistance quantitative trait locus. Proc. Natl. Acad. Sci. USA 2003, 100, 14587–14592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, B.; Cardon, G.; Traka, M.; Botterman, J.; Vancanneyt, G.; Mithen, R. Glucosinolate and amino acid biosynthesis in Arabidopsis. Plant Physiol. 2004, 135, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Benderoth, M.; Textor, S.; Windsor, A.J.; Mitchell-Olds, T.; Gershenzon, J.; Kroymann, J. Positive selection driving diversification in plant secondary metabolism. Proc. Natl. Acad. Sci. USA 2006, 103, 9118–9123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kraker, J.W.; Luck, K.; Textor, S.; Tokuhisa, J.G.; Gershenzon, J. Two Arabidopsis genes (IPMS1 and IPMS2) encode isopropylmalate synthase, the branchpoint step in the biosynthesis of leucine. Plant Physiol. 2007, 143, 970–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agerbirk, N.; Olsen, C.E. Glucosinolate structures in evolution. Phytochemistry 2012, 77, 16–45. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; Gershenzon, J.; Mitchell-Olds, T. Comparative quantitative trait loci mapping of aliphatic, indolic and benzylic glucosinolate production in Arabidopsis thaliana leaves and seeds. Genetics 2001, 159, 359–370. [Google Scholar] [CrossRef]
- Kumar, G.; Johnson, J.L.; Frantom, P.A. Improving functional annotation in the DRE-TIM metallolyase superfamily through identification of active site fingerprints. Biochemistry 2016, 55, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Koon, N.; Squire, C.J.; Baker, E.N. Crystal structure of LeuA from Mycobacterium tuberculosis, a key enzyme in leucine biosynthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 8295–8300. [Google Scholar] [CrossRef] [Green Version]
- Huisman, F.H.; Koon, N.; Bulloch, E.M.; Baker, H.M.; Baker, E.N.; Squire, C.J.; Parker, E.J. Removal of the C-terminal regulatory domain of α-isopropylmalate synthase disrupts functional substrate binding. Biochemistry 2012, 51, 2289–2297. [Google Scholar] [CrossRef] [PubMed]
- Ellerström, M.; Josefsson, L.G.; Rask, L.; Ronne, H. Cloning of a cDNA for rape chloroplast 3-isopropylmalate dehydrogenase by genetic complementation in yeast. Plant Mol. Biol. 1992, 18, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.D.; Sonnewald, U.; Willmitzer, L. Cloning and expression analysis of β-isopropylmalate dehydrogenase from potato. Mol. Gen. Genet. 1993, 236, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, A.; Takano, J.; Miwa, K.; Nakagawa, Y.; Fujiwara, T. Cloning of cDNAs encoding isopropylmalate dehydrogenase from Arabidopsis thaliana and accumulation patterns of their transcripts. Biosci. Biotech. Biochem. 2005, 69, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Aktas, D.F.; Cook, P.F. A lysine-tyrosine pair carries out acid-base chemistry in the metal ion-dependent pyridine dinucleotide-linked beta-hydroxyacid oxidative decarboxylases. Biochemistry 2009, 48, 3565–3577. [Google Scholar] [CrossRef]
- Lee, S.G.; Nwumeh, R.; Jez, J.M. Structure and mechanism of isopropylmalate dehydrogenase from Arabidopsis thaliana: Insights on leucine and aliphatic glucosinolate biosynthesis. J. Biol. Chem. 2016, 291, 13421–13430. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitainda, V.; Jez, J.M. Structural Studies of Aliphatic Glucosinolate Chain-Elongation Enzymes. Antioxidants 2021, 10, 1500. https://doi.org/10.3390/antiox10091500
Kitainda V, Jez JM. Structural Studies of Aliphatic Glucosinolate Chain-Elongation Enzymes. Antioxidants. 2021; 10(9):1500. https://doi.org/10.3390/antiox10091500
Chicago/Turabian StyleKitainda, Vivian, and Joseph M. Jez. 2021. "Structural Studies of Aliphatic Glucosinolate Chain-Elongation Enzymes" Antioxidants 10, no. 9: 1500. https://doi.org/10.3390/antiox10091500
APA StyleKitainda, V., & Jez, J. M. (2021). Structural Studies of Aliphatic Glucosinolate Chain-Elongation Enzymes. Antioxidants, 10(9), 1500. https://doi.org/10.3390/antiox10091500