
Usefulness of Melatonin and Other Compounds as Antioxidants and Epidrugs in the Treatment of Head and Neck Cancer



Abstract
:1. Introduction
2. Carcinogenesis of Head and Neck Squamous Cell Carcinomas
3. Melatonin, a Pleiotropic Molecule
3.1. Melatonin and Head and Neck Squamous Cell Carcinomas
3.2. Epigenetic Effects of Melatonin on Head and Neck Squamous Cell Carcinoma
4. Other Antioxidants with Potential Epigenetic Effect on Squamous Cell Carcinoma of the Head and Neck
4.1. DNA Methylation Inhibitors (DNMTi)
4.1.1. Epigallocatechin Gallate
4.1.2. Sulforaphane
4.1.3. Folate
4.2. Histone Deacetylase Inhibitors (HDACi)
Sodium Butyrate
4.3. Histone Acetyltransferases Inhibitors (HATi)
Curcumin
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, V.; Lefebvre, J.L.; Licitra, L.; Felip, E. Squamous cell carcinoma of the head and neck: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21, v184–v186. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.P.; le Maître, A.; Maillard, E.; Bourhis, J.; MACH-NC Collaborative group. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An update on 93 randomised trials and 17,346 patients. Radiother. Oncol. 2009, 92, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Lubov, J.; Maschietto, M.; Ibrahim, I.; Mlynarek, A.; Hier, M.; Kowalski, L.; Alaoui-Jamali, M.A.; da Silva, S.D. Meta-analysis of microRNAs expression in head and neck cancer: Uncovering association with outcome and mechanisms. Oncotarget 2017, 8, 55511–55524. [Google Scholar] [CrossRef] [Green Version]
- Bais, M.V. Impact of Epigenetic regulation on head and neck squamous cell carcinoma. J. Dent. Res. 2019, 98, 268–276. [Google Scholar] [CrossRef]
- Qi, Y.; Wang, D.; Wang, D.; Jin, T.; Yang, L.; Wu, H.; Li, Y.; Zhao, J.; Du, F.; Song, M.; et al. HEDD: The human epigenetic drug database. Database 2016, 2016, baw159. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Carlos-Reyes, Á.; López-González, J.S.; Meneses-Flores, M.; Gallardo-Rincón, D.; Ruíz-García, E.; Marchat, L.A.; Astudillo-de la Vega, H.; Hernández de la Cruz, O.N.; López-Camarillo, C. Dietary compounds as epigenetic modulating agents in cancer. Front. Genet. 2019, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, G.; Dempsey, A. The role of HPV in head and neck cancer and review of the HPV vaccine. Prev. Med. 2011, 53, S5–S11. [Google Scholar] [CrossRef] [Green Version]
- Hardeland, R. Neurobiology, pathophysiology, and treatment of melatonin deficiency and dysfunction. Sci. World J. 2012, 2012, 640389. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, A.R.; Larrick, J.W. Paradoxical effects of antioxidants on cancer. Rejuvenation Res. 2014, 17, 306–311. [Google Scholar] [CrossRef]
- Singh, K.; Bhori, M.; Kasu, Y.A.; Bhat, G.; Marar, T. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity—Exploring the armoury of obscurity. Saudi Pharm. J. 2018, 26, 177–190. [Google Scholar] [CrossRef]
- Rudin, C.M. Head and Neck Squamous Cell Carcinogenesis: Molecular and Genetic Alterations. Available online: https://www.uptodate.com/contents/head-and-neck-squamous-cell-carcinogenesis-molecular-and-genetic-alterations (accessed on 3 November 2021).
- Yang, H.; Jin, X.; Dan, H.; Chen, Q. Histone modifications in oral squamous cell carcinoma and oral potentially malignant disorders. Oral Dis. 2020, 26, 719–732. [Google Scholar] [CrossRef]
- Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic modifications and head and neck cancer: Implications for tumor progression and resistance to therapy. Int. J. Mol. Sci. 2017, 18, 1506. [Google Scholar] [CrossRef]
- Gaździcka, J.; Gołąbek, K.; Strzelczyk, J.K.; Ostrowska, Z. Epigenetic modifications in head and neck cancer. Biochem. Genet. 2020, 58, 213–244. [Google Scholar] [CrossRef] [Green Version]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.H.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef]
- Luo, G.; Ono, S.; Beukes, N.J.; Wang, D.T.; Xie, S.; Summons, R.E. Rapid oxygenation of earth’s atmosphere 2.33 billion years ago. Sci. Adv. 2016, 2, e1600134. [Google Scholar] [CrossRef] [Green Version]
- Izon, G.; Zerkle, A.L.; Williford, K.H.; Farquhar, J.; Poulton, S.W.; Claire, M.W. Biological regulation of atmospheric chemistry en route to planetary oxygenation. Proc. Natl. Acad. Sci. USA 2017, 114, E2571–E2579. [Google Scholar] [CrossRef] [Green Version]
- Taverne, Y.J.; Merkus, D.; Bogers, A.J.; Halliwell, B.; Duncker, D.J.; Lyons, T.W. Reactive oxygen species: Radical factors in the evolution of animal life: A molecular timescale from earth’s earliest history to the rise of complex life. BioEssays 2018, 40, 1700158. [Google Scholar] [CrossRef]
- Margulis, L. Symbiotic theory of the origin of eukaryotic organelles; Criteria for proof. Symp. Soc. Exp. Biol. 1975, 29, 21–38. [Google Scholar]
- Tan, D.X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuña-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Poeggeler, B.; Alvares, F.L.; Ogden, G.B.; Reiter, R.J. Melatonin immunoreactivity in the photosynthetic prokaryote Rhodospirillum rubrum: Implications for an ancient antioxidant system. Chem. Mol. Biol. Res. 1995, 41, 391–395. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R. Melatonin as a potent and inducible endogenous antioxidant: Synthesis and metabolism. Molecules 2015, 20, 18886. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin: A versatile protector against oxidative DNA damage. Molecules 2018, 23, 530. [Google Scholar] [CrossRef] [Green Version]
- Hevia, D.; Mayo, J.C.; Tan, D.X.; Rodriguez-Garcia, A.; Sainz, R.M. Melatonin enhances photo-oxidation of 2′,7′-dichlorodihydrofluoresceinby an antioxidant reaction that renders N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK). PLoS ONE 2014, 9, e109257. [Google Scholar] [CrossRef] [Green Version]
- Vielma, J.R.; Bonilla, E.; Chacin-Bonilla, L.; Mora, M.; Medina-Leendertz, S.; Bravo, Y. Effects of melatonin on oxidative stress, and resistance to bacterial, parasitic, and viral infections: A review. Acta Trop. 2014, 137, 31–38. [Google Scholar] [CrossRef]
- Lee, K.; Zawadzka, A.; Czarnocki, Z.; Reiter, R.J.; Back, K. Molecular cloning of melatonin 3-hydroxylase and its production of cyclic 3-hydroxymelatonin in rice (Oryza sativa). J. Pineal Res. 2016, 61, 470–478. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Hardeland, R. Taxon- and site-specific melatonin catabolism. Molecules 2017, 22, 2015. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Hester, R.J. Interrelationships of the pineal gland, the superior ganglia and the photoperiod in the regulation of the endocrine systems of hamsters. Endocrinology 1966, 79, 1168–1170. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, D.H.; Wang, M.L.; Jiao, D.R.; Pang, S.F. Rhythms of serum melatonin in patients with spinal lesions at the cervical, thoracic or lumbar region. Clin. Endocrinol. 1989, 30, 47–56. [Google Scholar] [CrossRef]
- Zeitzer, J.M.; Ayas, N.T.; Shea, S.A.; Brown, R.; Czeisler, C.A. Absence of detectable melatonin and preservation of cortisol and thyrotropin rhythms in tetraplegia. J. Clin. Endocrinol. Metab. 2000, 85, 2189–2196. [Google Scholar] [CrossRef]
- Devesa, J.; Segade, N.L.; Isorna, J.; Devesa, P.; Castellanos, S.; Puell, C.I. Is the use of growth hormone and melatonin justified in spinal cord injuries? MOJ Anat. Physiol. 2017, 4, 00128. [Google Scholar] [CrossRef] [Green Version]
- Hardeland, R. Melatonin and the electron transporter chain. Cell. Mol. Life Sci. 2017, 74, 3883–3986. [Google Scholar] [CrossRef]
- Suofu, V.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [Green Version]
- Mayo, J.C.; Sainz, R.M.; González-Menéndez, P.; Cepas, V.; Tan, D.X.; Reiter, R.J. Melatonin and sirtuins: A “not-so unexpected” relationship. J. Pineal Res. 2017, 62, e12391. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.; Zhou, X.; Tan, D.X. Role of SIRT3/SOD2 signaling in mediating the antioxidant actions of melatonin in mitochondria. Curr. Trends Endocrinol. 2017, 9, 45–49. [Google Scholar]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammosome Inhibition. Oxid. Med. Cell Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Harderland, R. Melatonin, its Metabolites and their Interference with Reactive Nitrogen Compounds. Molecules 2021, 26, 4105. [Google Scholar] [CrossRef]
- Bonmati-Carrion, M.A.; Tomas-Loba, A. Melatonin and cancer: A polyhedral network where the source matters. Antioxidants 2021, 10, 210. [Google Scholar] [CrossRef]
- Karasek, M. Melatonin, human aging, and age-related diseases. Exp. Gerontol. 2004, 39, 1723–1729. [Google Scholar] [CrossRef]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Therapeutic actions of melatonin in cancer: Possible mechanisms. Integr. Cancer Ther. 2008, 7, 189–203. [Google Scholar] [CrossRef]
- Jockers, R.; Maurice, P.; Boutin, J.A.; Delagrange, P. Melatonin receptors heterodimerization, signal transduction and binding sites: What’s new? Br. J. Pharmacol. 2008, 154, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Jockers, R.; Delagrange, P.; Dubocovich, M.L.; Markus, R.P.; Renault, N.; Tosini, G.; Cecon, E.; Zlotos, D.P. Update on melatonin receptors: IUPHAR Review 20. Br. J. Pharmacol. 2016, 173, 2702–2725. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International union of basic and clinical pharmacology. LXXV Nomenclature, classification, and pharmacology of G- protein coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [Green Version]
- Alkozi, H.A.; Sánchez, J.M.; Doadrio, A.L.; Pintor, J. Docking studies for melatonin receptors. Expert Opin. Drug Discov. 2017, 13, 241–248. [Google Scholar] [CrossRef]
- Boutin, J.A.; Ferry, G. Is there sufficient evidence that the melatonin binding Site MT3 is Quinone Reductase 2? J. Pharmacol. Exp. Ther. 2019, 368, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.M.; Frasch, T.; Xiang, S.; Yuan, L.; Duplessis, T.; Mao, L. Molecular mechanisms of melatonin anticancer effects. Integr. Cancer Ther. 2009, 8, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Liu, H.; Xu, L.; Zhang, H.; Zhou, R.X. Involvement of nuclear receptor RZR/ROR gamma in melatonin-induced HIF-1alpha inactivation in SGC-7901 human gastric cancer cells. Oncol. Rep. 2015, 34, 2541–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xu, L.; Ding, S.; Lin, N.; Ji, Q.; Gao, L.; Su, Y.; He, B.; Pu, J. Novel protective role of the circadian nuclear receptor retinoic acid-related orphan receptor-alpha in diabetic cardiomyopathy. J. Pineal Res. 2017, 62, e12378. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.A. Quinone reductase 2 as a promising target of melatonin therapeutic actions. Expert Opin. Ther. Targets 2016, 20, 303–317. [Google Scholar] [CrossRef]
- Benitez-King, G.; Huerto-Delgadillo, L.; Anton-Tay, F. Binding of 3H-melatonin to calmodulin. Life Sci. 1993, 53, 201–207. [Google Scholar] [CrossRef]
- Menendez-Menendez, J.; Martinez-Campa, C. Melatonin: An anti-tumor agent in hormone-dependent cancers. Int. J. Endocrinol. 2018, 2018, 3271948. [Google Scholar] [CrossRef]
- Cutando, A.; Aneiros-Fernández, J.; López-Valverde, A.; Arias-Santiago, S.; Aneiros-Cachaza, J.; Reiter, R.J. A new perspective in oral health: Potential importance and actions of melatonin receptors MT1, MT2, MT3, and RZR/ROR in the oral cavity. Arch. Oral Biol. 2011, 56, 944–950. [Google Scholar] [CrossRef]
- Winczyk, K.; Pawlikowski, M.; Guerrero, J.M.; Karasek, M. Possible involvement of the nuclear RZR/ROR-alpha receptor in the antitumor action of melatonin on murine Colon 38 cancer. Tumour Biol. 2002, 23, 298–302. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Touitou, Y.; Reinberg, A.; Touitou, D. Association between light at night, melatonin secretion, sleep deprivation, and the internal clock: Health impacts and mechanisms of circadian disruption. Life Sci. 2017, 173, 94–106. [Google Scholar] [CrossRef]
- Davis, S.; Mirick, D.K. Circadian disruption, shift work and the risk of cancer: A summary of the evidence and studies in Seattle. Cancer Causes Control 2006, 17, 539–545. [Google Scholar] [CrossRef]
- Davis, S.; Mirick, D.K.; Chen, C.; Stanczyk, F.Z. Night shift work and hormone levels in women. Cancer Epidemiol. Biomark. Prev. 2012, 21, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Maiese, K. Moving to the rhythm with clock (circadian) genes, autophagy, mTOR, and SIRT1 in degenerative disease and cancer. Curr. Neurovasc. Res. 2017, 14, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Reiter-Russel, J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Lilan, Q.; Yang, S.F.; Xu, K. Melatonin, a full service anti-cancer agent: Inhibition of initiation, progression and metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [Green Version]
- Moloudizargari, M.; Moradkhani, F.; Hekmatirad, S.; Fallah, M.; Asghari, M.H.; Reiter, R.J. Therapeutic targets of cancer drugs: Modulation by melatonin. Life Sci. 2021, 267, 118934. [Google Scholar] [CrossRef]
- Rakici, S.Y.; Tumkaya, L.; Edirvanli, O.C.; Yazici, U.; Dursun, E.; Arpa, M.; Mercantepe, T. Radioprotective effect of endogenous melatonin secretion associated with the circadian rhythm in irradiated rats. Int. J. Radiat. Biol. 2019, 95, 1236–1241. [Google Scholar] [CrossRef]
- Onseng, K.; Johns, N.P.; Khuayjarernpanishk, T.; Subongkot, S.; Priprem, A.; Hurst, C.; Johns, J. Beneficial effects of adjuvant melatonin in minimizing oral mucositis complications in head and neck cancer patients receiving concurrent chemoradiation. J. Altern. Complement. Med. 2017, 23, 957–963. [Google Scholar] [CrossRef]
- Elsabagh, H.H.; Moussa, E.; Mahmoud, S.A.; Elsaka, R.O.; Abdelrahman, H. Efficacy of Melatonin in prevention of radiation-induced oral mucositis: A randomized clinical trial. Oral Dis. 2020, 26, 566–572. [Google Scholar] [CrossRef]
- Lozano, A.; Marruecos, J.; Rubió, J.; Farré, N.; Gómez-Millán, J.; Morera, R.; Planas, I.; Lanzuela, M.; Vázquez-Masedo, M.G.; Cascallar, L.; et al. Randomized placebo-controlled phase II trial of high-dose melatonin mucoadhesive oral gel for the prevention and treatment of oral mucositis in patients with head and neck cancer undergoing radiation therapy concurrent with systemic treatment. Clin. Transl. Oncol. 2021, 23, 1801–1810. [Google Scholar] [CrossRef]
- Stanciu, A.E.; Zamfir-Chiru-Anton, A.; Stanciu, M.M.; Pantea-Stoian, A.; Nitipir, C.; Gheorghe, D.C. Serum melatonin is inversely associated with matrix metalloproteinase-9 in oral squamous cell carcinoma. Oncol. Lett. 2020, 19, 3011–3020. [Google Scholar] [CrossRef]
- Shin, Y.Y.; Seo, Y.; Oh, S.J.; Ahn, J.S.; Song, M.H.; Kang, M.J.; Oh, J.M.; Lee, D.; Kim, Y.H.; Sung, E.S.; et al. Melatonin and verteporfin synergistically suppress the growth and stemness of head and neck squamous cell carcinoma through the regulation of mitochondrial dynamics. J. Pineal Res. 2021, 26, e12779. [Google Scholar] [CrossRef]
- Salarić, I.; Karmelić, I.; Lovrić, J.; Baždarić, K.; Rožman, M.; Čvrljević, I.; Zajc, I.; Brajdić, D.; Macan, D. Salivary melatonin in oral squamous cell carcinoma patients. Sci. Rep. 2021, 11, 13201. [Google Scholar] [CrossRef]
- Nuszkiewicz, J.; Czuczejko, J.; Maruszak, M.; Pawłowska, M.; Woźniak, A.; Małkowski, B.; Szewczyk-Golec, K. Parameters of oxidative stress, vitamin d, osteopontin, and melatonin in patients with lip, oral cavity, and pharyngeal cancer. Oxid. Med. Cell. Longev. 2021, 2021, 2364931. [Google Scholar] [CrossRef]
- Liu, R.; Wang, H.L.; Deng, M.J.; Wen, X.J.; Mo, Y.Y.; Chen, F.M.; Zou, C.L.; Duan, W.F.; Li, L.; Nie, X. Melatonin inhibits reactive oxygen species-driven proliferation, epithelial-mesenchymal transition, and vasculogenic mimicry in oral cancer. Oxid. Med. Cell. Longev. 2018, 2018, 3510970. [Google Scholar] [CrossRef] [PubMed]
- Sung, E.S.; Kim, J.Y.; Ahn, Y.T.; Lee, I.W.; Choi, S.W.; Jang, H.B. Melatonin exerts anticancer effects in human tongue squamous cell carcinoma cells by promoting autophagy. Anticancer Res. 2020, 40, 6295–6303. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Pi, H.; Li, M.; Ren, Z.; He, Z.; Zhu, F.; Tian, L.; Tu, M.; Xie, J.; Liu, M.; et al. Inhibiting MT2-TFE3-dependent autophagy enhances melatonin-induced apoptosis in tongue squamous cell carcinoma. J. Pineal Res. 2018, 64, e12457. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Q.; Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; García-López, S.; Martinez-Ruiz, L.; Mendivil-Perez, M.; Soto-Mercado, V.; Acuña-Castroviejo, D.; Ortega-Arellano, H.; et al. Combination of melatonin and rapamycin for head and neck cancer therapy: Suppression of AKT/mTOR pathway activation, and activation of mitophagy and apoptosis via mitochondrial function regulation. J. Pineal Res. 2018, 64, e12461. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gil, B.I.; Guerra-Librero, A.; Shen, Y.Q.; Florido, J.; Martínez-Ruiz, L.; García-López, S.; Adan, C.; Rodríguez-Santana, C.; Acuña-Castroviejo, D.; Quiñones-Hinojosa, A.; et al. Melatonin enhances cisplatin and radiation cytotoxicity in head and neck squamous cell carcinoma by stimulating mitochondrial ros generation, apoptosis, and autophagy. Oxid. Med. Cell. Longev. 2019, 2019, 7187128. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Librero, A.; Fernandez-Gil, B.I.; Florido, J.; Martinez-Ruiz, L.; Rodríguez-Santana, C.; Shen, Y.Q.; García-Verdugo, J.M.; López-Rodríguez, A.; Rusanova, I.; Quiñones-Hinojosa, A.; et al. Melatonin targets metabolism in head and neck cancer cells by regulating mitochondrial structure and function. Antioxidants 2021, 10, 603. [Google Scholar] [CrossRef]
- Nakamura, E.; Kozaki, K.; Tsuda, H.; Suzuki, E.; Pimkhaokham, A.; Yamamoto, G.; Irie, T.; Tachikawa, T.; Amagasa, T.; Inazawa, J.; et al. Frequent silencing of a putative tumor suppressor gene melatonin receptor 1 A (MTNR1A) in oral squamous-cell carcinoma. Cancer Sci. 2008, 99, 1390–1400. [Google Scholar] [CrossRef]
- Yeh, C.; Lin, C.; Yang, J.; Yang, W.; Su, S.; Yang, S. Melatonin inhibits TPA-induced oral cancer cell migration by suppressing matrix metalloproteinase-9 activation through the histone acetylation. Oncotarget 2016, 7, 21952–21967. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.Y.; Lin, C.W.; Chien, M.H.; Reiter, R.J.; Su, S.C.; Hsieh, Y.H.; Yang, S.F. Melatonin suppresses TPA-induced metastasis by downregulating matrix metalloproteinase-9 expression through JNK/SP-1 signaling in nasopharyngeal carcinoma. J. Pineal Res. 2016, 61, 479–492. [Google Scholar] [CrossRef]
- Yang, C.Y.; Lin, C.K.; Tsao, C.H.; Hsieh, C.C.; Lin, G.J.; Ma, K.H.; Shieh, Y.S.; Sytwu, H.K.; Chen, Y.W. Melatonin exerts anti-oral cancer effect via suppressing LSD1 in patient-derived tumor xenograft models. Oncotarget 2017, 8, 33756–33769. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.J.; Lin, C.W.; Su, S.C.; Reiter, R.J.; Chen, A.W.; Chen, M.K.; Yang, S.F. Effects of miRNA-34b/miRNA-892a Upregulation and Inhibition of ABCB1/ABCB4 on melatonin-induced apoptosis in VCR-Resistant oral cancer cells. Mol. Ther. Nucleic Acids 2020, 19, 877–889. [Google Scholar] [CrossRef]
- Hunsaker, M.; Barba, G.; Kingsley, K.; Howard, K.M. Differential MicroRNA Expression of miRNA-21 and miRNA-155 within oral cancer extracellular vesicles in response to melatonin. Dent. J. 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tao, B.; Li, J.; Mao, X.; He, W.; Chen, Q. Melatonin inhibits the progression of oral squamous cell carcinoma via inducing mirna-25-5p expression by directly targeting NEDD9. Front. Oncol. 2020, 10, 2642. [Google Scholar] [CrossRef]
- Su, S.C.; Yeh, C.M.; Lin, C.W.; Hsieh, Y.H.; Chuang, C.Y.; Tang, C.H.; Lee, Y.C.; Yang, S.F. A novel melatonin-regulated lncRNA suppresses TPA-induced oral cancer cell motility through replenishing PRUNE2 expression. J. Pineal Res. 2021, 71, e12760. [Google Scholar] [CrossRef]
- Lang, L.; Xiong, Y.; Prieto-Dominguez, N.; Loveless, R.; Jensen, C.; Shay, C.; Teng, Y. FGF19/FGFR4 signaling axis confines and switches the role of melatonin in head and neck cancer metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 93. [Google Scholar] [CrossRef]
- Kartini, D.; Taher, A.; Panigoro, S.S.; Setiabudy, R.; Jusman, S.W.; Haryana, S.M.; Abdullah, M.; Rustamadji, P.; Purwanto, D.J.; Sutandyo, N.; et al. Effect of melatonin supplementation in combination with neoadjuvant chemotherapy to miRNA-210 and CD44 expression and clinical response improvement in locally advanced oral squamous cell carcinoma: A randomized controlled trial. J. Egypt Natl. Canc. Inst. 2020, 32, 12. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xu, L.; Yang, L.; Wang, X. Epigallocatechin gallate is the most effective catechin against antioxidant stress via hydrogen peroxide and radical scavenging activity. Med. Sci. Monit. 2018, 24, 8198–8206. [Google Scholar] [CrossRef]
- Huang, Z.; Jiang, H.; Liu, X.; Chen, Y.; Wong, J.; Wang, Q.; Huang, W.; Shi, T.; Zhang, J. HEMD: An integrated tool of human epigenetic enzymes and chemical modulators for therapeutics. PLoS ONE 2012, 7, e39917. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Park, J.; Lambert, J. Differential prooxidative effects of the green tea polyphenol, (-)-epigallocatechin-3-gallate, in normal and oral cancer cells are related to differences in sirtuin 3 signaling. Mol. Nutr. Food Res. 2014, 59, 203–211. [Google Scholar] [CrossRef]
- Hu, J.; Webster, D.; Cao, J.; Shao, A. The safety of green tea and green tea extract consumption in adults—Results of a systematic review. Regul. Toxicol. Pharmacol. 2018, 95, 412–433. [Google Scholar] [CrossRef]
- Zhu, W.; Mei, H.; Jia, L.; Zhao, H.; Li, X.; Meng, X.; Zhao, X.; Xing, L.; Yu, J. Epigallocatechin-3-gallate mouthwash protects mucosa from radiation-induced mucositis in head and neck cancer patients: A prospective, non-randomized, phase 1 trial. Investig. New Drugs 2020, 38, 1129–1136. [Google Scholar] [CrossRef]
- Gu, L.T.; Yang, J.; Su, S.Z.; Liu, W.W.; Shi, Z.G.; Wang, Q.R. Green Tea Polyphenols Protects Cochlear Hair Cells from Ototoxicity by Inhibiting Notch Signaling. Neurochem. Res. 2015, 40, 1211–1219. [Google Scholar] [CrossRef]
- Li, N.; Sun, Z.; Han, C.; Chen, J. The chemopreventive effects of tea on human oral precancerous mucosa lesions. Proc. Soc. Exp. Biol. Med. 1999, 220, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Liu, D.; Martin, J.; Tang, X.M.; Lee, J.J.; El-Naggar, A.K.; Wistuba, I.; Culotta, K.S.; Mao, L.; Gillenwater, A.; et al. Phase II randomized, placebo-controlled trial of green tea extract in patients with high-risk oral premalignant lesions. Cancer Prev. Res. 2009, 2, 931–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Long, N.K.; Makita, H.; Toida, M.; Yamashita, T.; Hatakeyama, D.; Hara, A.; Mori, H.; Shibata, T. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br. J. Cancer 2008, 99, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhazzazi, T.Y.; Kamarajan, P.; Verdin, E.; Kapila, Y.L. Sirtuin-3 (SIRT3) and the Hallmarks of Cancer. Genes Cancer 2013, 4, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Chen, P.; Peng, C.; Yu, C.; Chou, M. Suppression of miRNA-204 enables oral squamous cell carcinomas to promote cancer stemness, EMT traits, and lymph node metastasis. Oncotarget 2016, 7, 20180–20192. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.S.; Kang, S.U.; Park, J.K.; Kim, Y.E.; Kim, Y.S.; Baek, S.J.; Lee, S.H.; Kim, C.H. Anti-cancer effect of (-)-epigallocatechin-3-gallate (EGCG) in head and neck cancer through repression of transactivation and enhanced degradation of β-catenin. Phytomedicine 2016, 23, 1344–1355. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Tighiouart, M.; Lee, J.E.; Shin, H.J.; Khuri, F.R.; Yang, C.S.; Chen, Z.; Shin, D.M. Synergistic inhibition of head and neck tumor growth by green tea (-)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer 2008, 123, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.P.; Hung, P.F.; Ku, W.Y.; Chang, C.Y.; Wu, B.H.; Wu, M.H.; Yao, J.Y.; Yang, J.R.; Lee, C.H. The inhibitory activity of gallic acid against DNA methylation: Application of gallic acid on epigenetic therapy of human cancers. Oncotarget 2018, 9, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef]
- Shishu; Singla, A.K.; Kaur, I. Inhibition of Mutagenicity of Food-Derived Heterocyclic Amines by Sulphoraphene—An Isothiocyanate Isolated from Radish. Planta Med. 2003, 69, 184–186. [Google Scholar] [CrossRef]
- Bauman, J.E.; Zang, Y.; Sen, M.; Li, C.; Wang, L.; Egner, P.A.; Fahey, J.W.; Normolle, D.P.; Grandis, J.R.; Kensler, T.W.; et al. Prevention of carcinogen-induced oral cancer by sulforaphane. Cancer Prev. Res. 2016, 9, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Li, H.; Lee, E.; Grandis, J.; Bauman, J.; Johnson, D. Gene targets of sulforaphane in head and neck squamous cell carcinoma. Mol. Med. Rep. 2019, 20, 5335–5344. [Google Scholar] [CrossRef]
- Chen, C.T.; Hsieh, M.J.; Hsieh, Y.H.; Hsin, M.C.; Chuang, Y.T.; Yang, S.F.; Lin, C.W. Sulforaphane suppresses oral cancer cell migration by regulating cathepsin S expression. Oncotarget 2018, 9, 17564–17575. [Google Scholar] [CrossRef]
- Chen, L.; Chan, L.; Lung, H.; Yip, T.T.C.; Ngan, R.K.C.; Wong, J.W.C.; Lo, K.W.; Ng, W.T.; Lee, A.W.M.; Tsao, G.S.W.; et al. Crucifera sulforaphane (SFN) inhibits the growth of nasopharyngeal carcinoma through DNA methyltransferase 1 (DNMT1)/Wnt inhibitory factor 1 (WIF1) axis. Phytomedicine 2019, 63, 153058. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Z.; Li, M.; Liu, M.; Bahena, A.; Zhang, Y.; Zhang, Y.; Nambiar, C.; Liu, G. Sulforaphane promotes apoptosis, and inhibits proliferation and self-renewal of nasopharyngeal cancer cells by targeting STAT signal through miRNA-124-3p. Biomed. Pharmacother. 2018, 103, 473–481. [Google Scholar] [CrossRef]
- Elkashty, O.A.; Ashry, R.; Elghanam, G.A.; Pham, H.M.; Su, X.; Stegen, C.; Tran, S.D. Broccoli extract improves chemotherapeutic drug efficacy against head-neck squamous cell carcinomas. Med. Oncol. 2018, 35, 124. [Google Scholar] [CrossRef]
- Gliszczynskaswiglo, A. Folates as antioxidants. Food Chem. 2007, 101, 1480–1483. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate and cancer: A tale of Dr. Jekyll and Mr. Hyde. Am. J. Clin. Nutr. 2018, 107, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Kawakita, D.; Lee, Y.A.; Gren, L.H.; Buys, S.S.; La Vecchia, C.; Hashibe, M. The impact of folate intake on the risk of head and neck cancer in the prostate, lung, colorectal, and ovarian cancer screening trial (PLCO) cohort. Br. J. Cancer 2018, 118, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Kraunz, K.S.; Hsiung, D.; McClean, M.D.; Liu, M.; Osanyingbemi, J.; Nelson, H.H.; Kelsey, K.T. Dietary folate is associated with p16(INK4A) methylation in head and neck squamous cell carcinoma. Int. J. Cancer 2006, 119, 1553–1557. [Google Scholar] [CrossRef]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Stilling, R.M.; van de Wow, M.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int. 2016, 99, 110–132. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Xu, P.; Xue, T.; Wang, J.; Guo, Y.; Jia, L.; Qiao, X.; Li, L.; Zhai, Y. Melatonin orchestrates lipid homeostasis through the hepatointestinal circadian clock and microbiota during constant light exposure. Cells 2020, 9, 489. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Wang, P.; Yan, J.; Liu, G.; Zeng, B.; Hussain, T.; Peng, C.; Yin, J.; Li, T.; Wei, H.; et al. Melatonin alleviates weanling stress in mice: Involvement of intestinal microbiota. J. Pineal Res. 2018, 64, e12448. [Google Scholar] [CrossRef]
- Ranganna, K.; Mathew, O.P.; Yatsu, F.M.; Yousefipour, Z.; Hayes, B.E.; Milton, S.G. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. 2007, 274, 5962–5978. [Google Scholar] [CrossRef] [PubMed]
- Gillenwater, A.; Xu, X.C.; Estrov, Y.; Sacks, P.G.; Lotan, D.; Lotan, R. Modulation of galectin-1 content in human head and neck squamous carcinoma cells by sodium butyrate. Int. J. Cancer 1998, 75, 217–224. [Google Scholar] [CrossRef]
- Gillenwater, A.; Zou, C.P.; Zhong, M.; Lotan, R. Effects of sodium butyrate on growth, differentiation, and apoptosis in head and neck squamous carcinoma cell lines. Head Neck 2000, 22, 247–256. [Google Scholar] [CrossRef]
- Wang, A.; Zeng, R.; Huang, H. Retinoic acid and sodium butyrate as cell cycle regulators in the treatment of oral squamous carcinoma cells. Oncol. Res. 2008, 17, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Matsumoto, N.; Matsue, Y.; Okudera, M.; Nishikawa, Y.; Abiko, Y.; Komiyama, K. Sodium butyrate, a histone deacetylase inhibitor, regulates lymphangiogenic factors in oral cancer cell line HSC-3. Anticancer Res. 2014, 34, 1701–1708. [Google Scholar] [PubMed]
- Provenzano, M.J.; Fitzgerald, M.P.; Krager, K.; Domann, F.E. Increased iodine uptake in thyroid carcinoma after treatment with sodium butyrate and decitabine (5-Aza-dC). Otolaryngol. Head Neck Surg. 2007, 137, 722–728. [Google Scholar] [CrossRef]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar] [CrossRef]
- Lindsay, C.; Kostiuk, M.; Conrad, D.; O’Connell, D.A.; Harris, J.; Seikaly, H.; Biron, V.I. Antitumor effects of metformin and curcumin in human papillomavirus positive and negative head and neck cancer cells. Mol. Carcinog. 2019, 58, 1946–1959. [Google Scholar] [CrossRef]
- Dei Cas, M.; Ghidoni, R. Dietary Curcumin: Correlation between bioavailability and health potential. Nutrients 2019, 11, 2147. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tang, G.; Wei, Z. Prophylactic and Therapeutic Effects of Curcumin on Treatment-Induced Oral Mucositis in Patients with Head and Neck Cancer: A Meta-Analysis of Randomized Controlled Trials. Nutr. Cancer 2021, 73, 740–749. [Google Scholar] [CrossRef]
- Boven, L.; Holmes, S.P.; Latimer, B.; McMartin, K.; Ma, X.; Moore-Medlin, T.; Khandelwal, A.R.; McLarty, J.; Nathan, C.O. Curcumin gum formulation for prevention of oral cavity head and neck squamous cell carcinoma. Laryngoscope 2019, 129, 1597–1603. [Google Scholar] [CrossRef]
- Hu, A.; Huang, J.; Li, R.L.; Lu, Z.Y.; Duan, J.L.; Xu, W.H.; Chen, X.P.; Fan, J.P. Curcumin as therapeutics for the treatment of head and neck squamous cell carcinoma by activating SIRT1. Sci. Rep. 2015, 5, 13429. [Google Scholar] [CrossRef] [Green Version]
- Borges, G.A.; Elias, S.T.; Amorim, B.; de Lima, C.L.; Coletta, R.D.; Castilho, R.M.; Squarize, C.H.; Guerra, E.N.S. Curcumin downregulates the PI3K-AKT-mTOR pathway and inhibits growth and progression in head and neck cancer cells. Phytother. Res. 2020, 34, 3311–3324. [Google Scholar] [CrossRef]
- Arif, M.; Vedamurthy, B.; Choudhari, R.; Ostwal, Y.B.; Mantelingu, K.; Kodaganur, G.D.; Kundu, T.K. Nitric oxide-mediated histone hyperacetylation in oral cancer: Target for a water-soluble HAT inhibitor, CTK7A. Chem. Biol. 2010, 17, 903–913. [Google Scholar] [CrossRef] [Green Version]

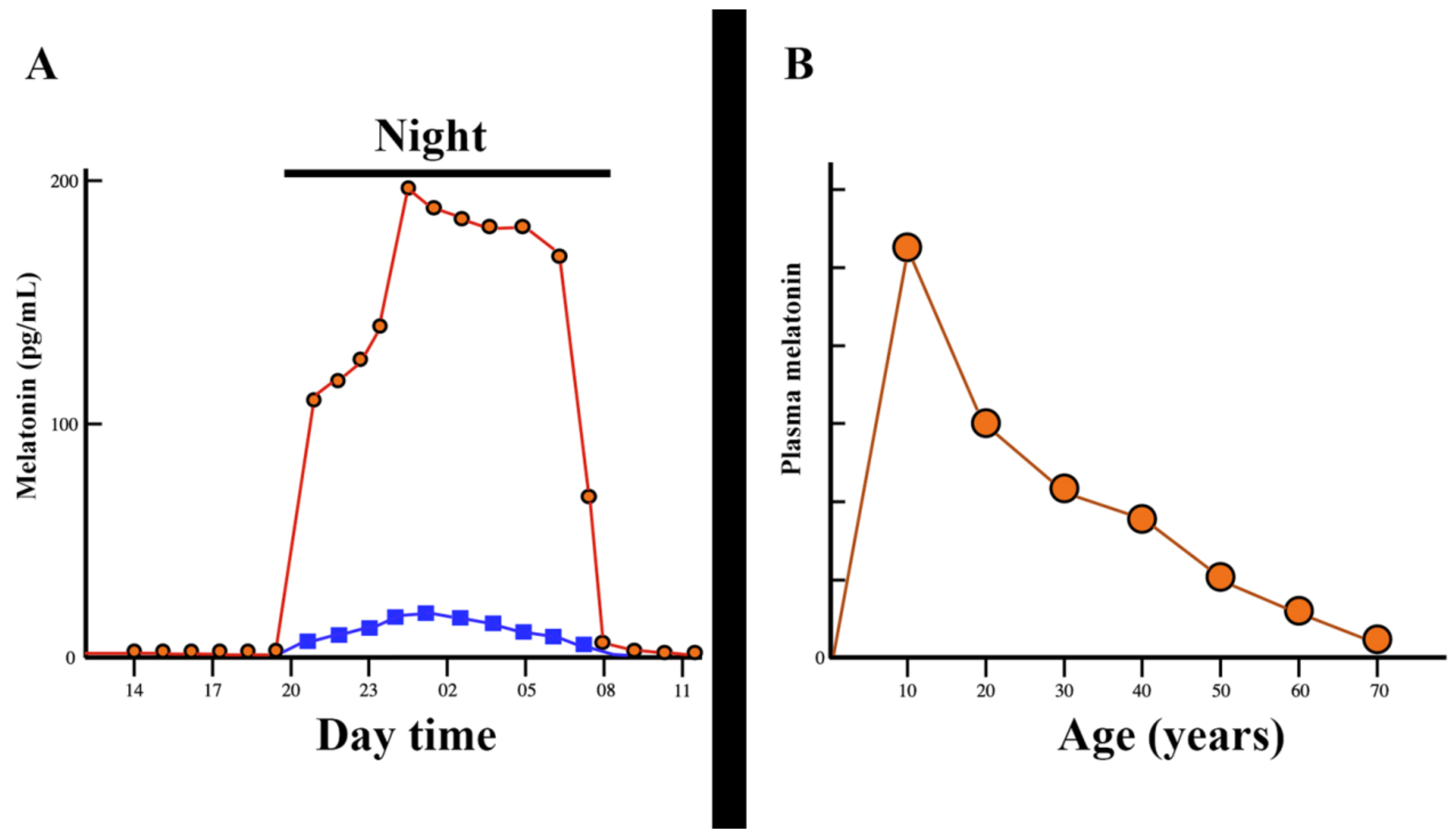

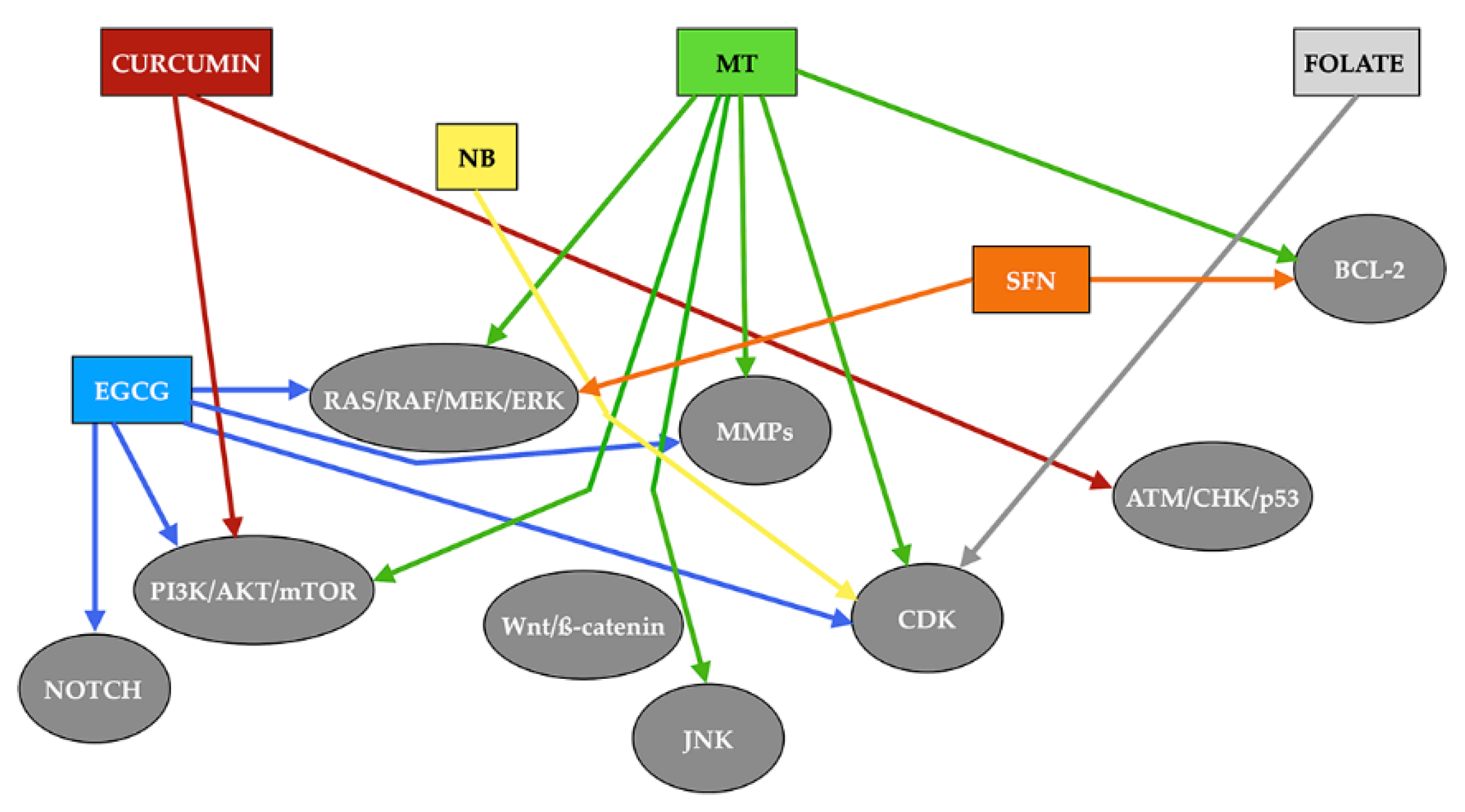

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Effect | Target | |
|---|---|---|---|
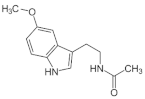 | Melatonin | HDACi | SIRT1 SIRT3 |
| HDMi | LSD1 | ||
| miRNAs | miRNA-892a miRNA-34-5p miRNA-155 miRNA-21 miRNA-25-5p miRNA-210 | ||
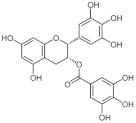 | Epigallocatechin-3-gallate | DNMTi | DNMT1 DNMT3A/3B |
| HDACi | SIRT3 | ||
| miRNAs | miRNA-204 | ||
 | Sulforaphane | DNMTi | DNMT1 DNMT3A/3B Trn |
| miRNAs | miRNA-124-3p | ||
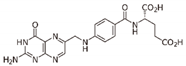 | Folate | DNMTi | DNMT1 DNMT3A/3B |
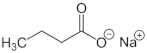 | Sodium butyrate | HDACi | HDACI/II |
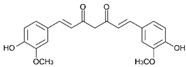 | Curcumin | HATi | CTK7A CREBBP p300 |
| DNMTi | DNMT1 | ||
| HDACi | SIRT1 | ||
| Other (Histone phosporylation inhibitor) | GSK3β | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, J.; Devesa, J. Usefulness of Melatonin and Other Compounds as Antioxidants and Epidrugs in the Treatment of Head and Neck Cancer. Antioxidants 2022, 11, 35. https://doi.org/10.3390/antiox11010035
Guerra J, Devesa J. Usefulness of Melatonin and Other Compounds as Antioxidants and Epidrugs in the Treatment of Head and Neck Cancer. Antioxidants. 2022; 11(1):35. https://doi.org/10.3390/antiox11010035
Chicago/Turabian StyleGuerra, Joaquín, and Jesús Devesa. 2022. "Usefulness of Melatonin and Other Compounds as Antioxidants and Epidrugs in the Treatment of Head and Neck Cancer" Antioxidants 11, no. 1: 35. https://doi.org/10.3390/antiox11010035
APA StyleGuerra, J., & Devesa, J. (2022). Usefulness of Melatonin and Other Compounds as Antioxidants and Epidrugs in the Treatment of Head and Neck Cancer. Antioxidants, 11(1), 35. https://doi.org/10.3390/antiox11010035