
Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons

 , ,
, ,  ,
,  , and
, and 
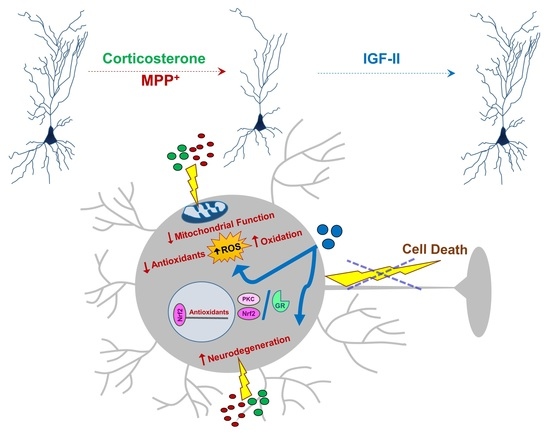
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Visualisation of Cell Morphology
2.4. Determination of Oxidative Stress Parameters
2.4.1. Mitochondrial Levels of ROS
2.4.2. Antioxidant Activity and Oxidative Cell Damage
2.5. Measurement of Mitochondrial Markers
2.5.1. Mitochondrial Membrane Potential
2.5.2. Mitochondrial Oxygen Consumption Rate
2.6. Dopamine Marker and Neurodegeneration
2.7. Intracellular Signalling Pathways
2.8. Statistical Analysis
3. Results
3.1. Cell Viability and Morphology
3.2. Antioxidant Activity and Oxidative Cell Damage
3.3. ROS Production and Measurement of Mitochondrial Function
3.3.1. Mitochondrial ROS Production and Mitochondrial Membrane Potential
3.3.2. Mitochondrial Oxygen Consumption
3.4. Dopamine Markers and Neurodegeneration
3.4.1. Tyrosine Hydroxylase Expression
3.4.2. Neurodegeneration
3.5. Intracellular Signalling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 559, 1–7. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Djamshidian, A.; Lees, A.J. Can stress trigger Parkinson’s disease? J. Neurol. Neurosurg. Psychiatry 2014, 85, 879–882. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Kopin, I.J. Linking Stress, Catecholamine Autotoxicity, and Allostatic Load with Neurodegenerative Diseases: A Focused Review in Memory of Richard Kvetnansky. Cell Mol. Neurobiol. 2018, 38, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Soares, N.M.; Pereira, G.M.; Altmann, V.; de Almeida, R.M.M.; Rieder, C.R.M. Cortisol levels, motor, cognitive and behavioral symptoms in Parkinson’s disease: A systematic review. J. Neural Transm. 2019, 126, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.V.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef]
- Herman, J.P.; Cullinan, W.E. Neurocircuitry of stress: Central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997, 20, 78–84. [Google Scholar] [CrossRef]
- Ábrahám, I.M.; Meerlo, P.; Luiten, P.G. Concentration dependent actions of glucocorticoids on neuronal viability and survival. Dose Response 2006, 4, 38–54. [Google Scholar] [CrossRef]
- Choi, G.E.; Han, H.J. Glucocorticoid impairs mitochondrial quality control in neurons. Neurobiol. Dis. 2021, 152, 105301. [Google Scholar] [CrossRef]
- Martin-Montañez, E.; Pavia, J.; Santin, L.J.; Boraldi, F.; Estivill-Torrus, G.; Aguirre, J.A.; Garcia-Fernandez, M. Involvement of IGF-II receptors in the antioxidant and neuroprotective effects of IGF-II on adult cortical neuronal cultures. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1041–1051. [Google Scholar] [CrossRef]
- Martín-Montañez, E.; Millon, C.; Boraldi, F.; Garcia-Guirado, F.; Pedraza, C.; Lara, E.; Santin, L.J.; Pavia, J.; Garcia-Fernandez, M. IGF-II promotes neuroprotection and neuroplasticity recovery in a long-lasting model of oxidative damage induced by glucocorticoids. Redox Biol. 2017, 13, 69–81. [Google Scholar] [CrossRef]
- Smith, A.D.; Castro, S.L.; Zigmond, M.J. Stress-induced Parkinson’s disease: A working hypothesis. Physiol. Behav. 2002, 77, 527–531. [Google Scholar] [CrossRef]
- Vyas, S.; Rodrigues, A.J.; Silva, J.M.; Tronche, F.; Almeida, O.F.; Sousa, N.; Sotiropoulos, I. Chronic Stress and Glucocorticoids: From Neuronal Plasticity to Neurodegeneration. Neural Plast. 2016, 2016, 6391686. [Google Scholar] [CrossRef] [PubMed]
- Maatouk, L.; Yi, C.; Carrillo-de Sauvage, M.A.; Compagnion, A.C.; Hunot, S.; Ezan, P.; Hirsch, E.C.; Koulakoff, A.; Pfrieger, F.W.; Tronche, F.; et al. Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. 2019, 26, 580–596. [Google Scholar] [CrossRef] [PubMed]
- Claros, S.; Gil, A.; Martinelli, M.; Valverde, N.; Lara, E.; Boraldi, F.; Pavia, J.; Martín-Montañez, E.; Garcia-Fernandez, M. Impact of Glucocorticoid on a Cellular Model of Parkinson’s Disease: Oxidative Stress and Mitochondrial Function. Brain Sci. 2021, 11, 1106. [Google Scholar] [CrossRef] [PubMed]
- Ros-Bernal, F.; Hunot, S.; Herrero, M.T.; Parnadeau, S.; Corvol, J.C.; Lu, L.; Alvarez-Fischer, D.; De Sauvage, M.A.C.; Saurini, F.; Coussieu, C.; et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc. Natl. Acad. Sci. USA. 2011, 108, 6632–6637. [Google Scholar] [CrossRef]
- Hawkes, C.; Kar, S. The insulin-like growth factor-II/mannose-6-phosphate receptor: Structure, distribution and function in the central nervous system. Brain Res. Rev. 2004, 44, 117–140. [Google Scholar] [CrossRef]
- Hawkes, C.; Kar, S. Insulin-like growth factor-II/mannose-6-phosphate receptor: Widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J. Comp. Neurol. 2003, 458, 113–127. [Google Scholar] [CrossRef]
- Beletskiy, A.; Chesnokova, E.; Bal, N. Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer—Its Comparative Expression, Processing and Signaling in Mammalian CNS. Int. J. Mol. Sci. 2021, 22, 1849. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; LeRoith, D. Insulin and insulin-like growth factor receptors in the brain: Physiological and pathological aspects. Eur. Neuropsychopharmacol. 2014, 24, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; MacDonald, R.G.; Thinakaran, G.; Kar, S. Insulin-Like Growth Factor-II/Cation-Independent Mannose 6-Phosphate Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 2636–2658. [Google Scholar] [CrossRef]
- Martín-Montañez, E.; Valverde, N.; Ladrón de Guevara-Miranda, D.; Lara, E.; Romero-Zerbo, Y.S.; Millon, C.; Boraldi, F.; Ávila-Gámiz, F.; Pérez-Cano, A.M.; Garrido-Gil, P.; et al. Insulin-like growth factor II prevents oxidative and neuronal damage in cellular and mice models of Parkinson’s disease. Redox Biol. 2021, 46, 102095. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Lucas, M.; Viana da Silva, S.; Di Scala, M.; Garcia-Barroso, C.; Gonzalez-Aseguinolaza, G.; Mulle, C.; Alberini, C.M.; Cuadrado-Tejedor, M.; Garcia-Osta, A. Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol. Med. 2014, 6, 1246–1262. [Google Scholar] [CrossRef]
- Garcia-Fernandez, M.; Sierra, I.; Puche, J.E.; Guerra, L.; Castilla-Cortazar, I. Liver mitochondrial dysfunction is reverted by insulin-like growth factor II (IGF-II) in aging rats. J. Transl. Med. 2011, 9, 1–9. [Google Scholar] [CrossRef]
- Dong, W.; Hu, L.; Xu, X.W. Neuroprotective Effect of Insulin-like Growth Factor-II on 1- Methyl-4-Phenyl Pyridinium-Induced Oxidative Damage in Cortical Neuronal Cells. Trop. J. Pharm. Res. 2015, 14, 1191. [Google Scholar] [CrossRef]
- Castilla-Cortazar, I.; Garcia-Fernandez, M.; Delgado, G.; Puche, J.E.E.; Sierra, I.; Barhoum, R.; Gonzalez-Baron, S. Hepatoprotection and neuroprotection induced by low doses of IGF-II in aging rats. J. Transl. Med. 2011, 9, 103. [Google Scholar] [CrossRef]
- Pardo, M.; Cheng, Y.; Sitbon, Y.H.; Lowell, J.A.; Grieco, S.F.; Worthen, R.J.; Desse, S.; Barreda-Diaz, A. Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review. Neurosci. Res. 2019, 149, 1–13. [Google Scholar] [CrossRef]
- Ouchi, Y.; Banno, Y.; Shimizu, Y.; Ando, S.; Hasegawa, H.; Adachi, K.; Iwamoto, T. Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J. Neurosci. 2013, 33, 9408–9419. [Google Scholar] [CrossRef]
- Steinmetz, A.B.; Stern, S.A.; Kohtz, A.S.; Descalzi, G.; Alberini, C.M. Insulin-like growth factor II targets the mTOR pathway to reverse autism-like phenotypes in mice. J. Neurosci. 2018, 38, 1015–1029. [Google Scholar] [CrossRef]
- Son, J.H.; Chun, H.S.; Joh, T.H.; Cho, S.; Conti, B.; Lee, J.W. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. 1999, 19, 10–20. [Google Scholar] [CrossRef]
- Koh, J.Y.; Choi, D.W. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J. Neurosci. Methods 1987, 20, 83–90. [Google Scholar] [CrossRef]
- Strober, W. Wright-Giemsa and nonspecific esterase staining of cells. Curr. Protoc. Cytom. 2001, 3, 1–5. [Google Scholar] [CrossRef]
- Martín-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef]
- Pasquali-Ronchetti, I.; Garcia-Fernandez, M.I.; Boraldi, F.; Quaglino, D.; Gheduzzi, D.; De Vincenzi Paolinelli, C.; Tiozzo, R.; Bergamini, S.; Ceccarelli, D.; Muscatello, U. Oxidative stress in fibroblasts from patients with pseudoxanthoma elasticum: Possible role in the pathogenesis of clinical manifestations. J. Pathol. 2006, 208, 54–61. [Google Scholar] [CrossRef]
- Arab, K.; Steghens, J.P. Plasma lipid hydroperoxides measurement by an automated xylenol orange method. Anal. Biochem. 2004, 325, 158–163. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, 125–136. [Google Scholar] [CrossRef]
- Gil, A.; Martín-Montañez, E.; Valverde, N.; Lara, E.; Boraldi, F.; Claros, S.; Romero-Zerbo, S.Y.; Fernández, O.; Pavia, J.; Garcia-Fernandez, M. Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage. Cells 2020, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Yoshiya, M.; Komatsuzaki, Y.; Hojo, Y.; Ikeda, M.; Mukai, H.; Hatanaka, Y.; Murakami, G.; Kawata, M.; Kimoto, T.; Kawato, S. Corticosterone rapidly increases thorns of CA3 neurons via synaptic/extranuclear glucocorticoid receptor in rat hippocampus. Front. Neural Circuits 2013, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Shabanpoor, A.; Metz, G.A. Stress and corticosterone alter synaptic plasticity in a rat model of Parkinson’s disease. Neurosci. Lett. 2017, 651, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Håglin, L.; Bäckman, L. Covariation between plasma phosphate and daytime cortisol in early Parkinson’s disease. Brain Behav. 2016, 6, e00556. [Google Scholar] [CrossRef]
- Zou, K.; Guo, W.; Tang, G.; Zheng, B.; Zheng, Z. A Case of early onset Parkinson’s disease after major stress. Neuropsychiatr. Dis. Treat. 2013, 9, 1067–1069. [Google Scholar] [CrossRef]
- Burtscher, J.; Copin, J.C.; Rodrigues, J.; Kumar, S.T.; Chiki, A.; Guillot de Suduiraut, I.; Sandi, C.; Lashuel, H.A. Chronic corticosterone aggravates behavioral and neuronal symptomatology in a mouse model of alpha-synuclein pathology. Neurobiol. Aging 2019, 83, 11–20. [Google Scholar] [CrossRef]
- Hemmerle, A.M.; Dickerson, J.W.; Herman, J.P.; Seroogy, K.B. Stress exacerbates experimental Parkinson’s disease. Mol. Psychiatry 2014, 19, 638–640. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, X.; Zhang, Y.; Zhang, L.; Feng, L. Chronic mild stress accelerates the progression of Parkinson’s disease in A53T alpha-synuclein transgenic mice. Exp. Neurol. 2016, 285, 61–71. [Google Scholar] [CrossRef]
- Van Craenenbroeck, K.; De Bosscher, K.; Berghe, W.V.; Vanhoenacker, P.; Haegeman, G. Role of glucocorticoids in dopamine-related neuropsychiatric disorders. Mol. Cell Endocrinol. 2005, 245, 10–22. [Google Scholar] [CrossRef]
- Doric, Z.; Nakamura, K. Mice with disrupted mitochondria used to model Parkinson’s disease. Nature 2021, 599, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Holley, A.E.; Flitter, W.D.; Slater, T.F.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: An HPLC and ESR study. Mov. Disord. 1994, 9, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Guardia-Laguarta, C.; Schon, E.A.; Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019, 129, 34–45. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The Next (Neurode)Generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gui, W.; Tan, B.; Du, Y.; Zhou, J.; Wu, F.; Li, H.; Lin, X. IGF2 deficiency causes mitochondrial defects in skeletal muscle. Clin. Sci. 2021, 135, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Gui, W.; Zhu, Y.; Sun, S.; Zhu, W.; Tan, B.; Zhao, H.; Shang, C.; Zheng, F.; Lin, X.; Li, H. Knockdown of insulin-like growth factor 2 gene disrupts mitochondrial functions in the liver. J. Mol. Cell Biol. 2021, 13, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Lee, J.; Darley-Usmar, V.M.; Zhang, J. Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS ONE 2012, 7, e044610. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Andrianova, N.V.; Babenko, V.A.; Pevzner, I.B.; Popkov, V.A.; Zorov, S.D.; Zorova, L.D.; Plotnikov, E.Y.; Sukhikh, G.T.; Silachev, D.N. Neuroprotective Potential of Mild Uncoupling in Mitochondria. Pros and Cons. Brain Sci. 2021, 11, 1050. [Google Scholar] [CrossRef]
- Wu, P.Y.; Lai, B.; Dong, Y.; Wang, Z.M.; Li, Z.C.; Zheng, P. Different oxidants and PKC isozymes mediate the opposite effect of inhibition of Q(i) and Q(o) site of mitochondrial complex III on calcium currents in rat cortical neurons. Biochim. Biophys. Acta 2010, 1803, 1072–1082. [Google Scholar] [CrossRef]
- Kowalczyk, J.E.; Kawalec, M.; Beręsewicz, M.; Dębski, J.; Dadlez, M.; Zabłocka, B. Protein kinase C beta in postischemic brain mitochondria. Mitochondrion 2012, 12, 138–143. [Google Scholar] [CrossRef]
- Buelna-Chontal, M.; Guevara-Chávez, J.-G.G.; Silva-Palacios, A.; Medina-Campos, O.-N.N.; Pedraza-Chaverri, J.; Zazueta, C. Nrf2-regulated antioxidant response is activated by protein kinase C in postconditioned rat hearts. Free Radic. Biol. Med. 2014, 74, 145–156. [Google Scholar] [CrossRef]
- Chu, C.-H.; Tzang, B.-S.; Chen, L.-M.; Kuo, C.-H.; Cheng, Y.-C.; Chen, L.-Y.; Tsai, F.-J.; Tsai, C.-H.; Kuo, W.-W.; Huang, C.-Y. IGF-II/mannose-6-phosphate receptor signaling induced cell hypertrophy and atrial natriuretic peptide/BNP expression via Gαq interaction and protein kinase C-α/CaMKII activation in H9c2 cardiomyoblast cells. J. Endocrinol. 2008, 197, 381–390. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 1–13. [Google Scholar] [CrossRef]
- Du, X.; Pang, T.Y. Is Dysregulation of the HPA-Axis a Core Pathophysiology Mediating Co-Morbid Depression in Neurodegenerative Diseases? Front. Psychiatry 2015, 6, 1–33. [Google Scholar] [CrossRef]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1–23. [Google Scholar] [CrossRef]
- Jayaram, S.; Krishnamurthy, P.T. Role of microgliosis, oxidative stress and associated neuroinflammation in the pathogenesis of Parkinson’s disease: The therapeutic role of Nrf2 activators. Neurochem. Int. 2021, 145, 105014. [Google Scholar] [CrossRef]
- Meng, F.; Wang, J.; Ding, F.; Xie, Y.; Zhang, Y.; Zhu, J. Neuroprotective effect of matrine on MPTP-induced Parkinson’s disease and on Nrf2 expression. Oncol. Lett. 2017, 13, 296–300. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, B.; Zhang, C.; Su, Z.; Guo, B.; Zhao, Y.; Zheng, R. The DJ1-Nrf2-STING axis mediates the neuroprotective effects of Withaferin A in Parkinson’s disease. Cell Death Differ. 2021, 28, 2517–2535. [Google Scholar] [CrossRef]
- Li, Q.; Niu, C.; Zhang, X.; Dong, M. Gastrodin and Isorhynchophylline Synergistically Inhibit MPP+-Induced Oxidative Stress in SH-SY5Y Cells by Targeting ERK1/2 and GSK-3β Pathways: Involvement of Nrf2 Nuclear Translocation. ACS Chem. Neurosci. 2018, 9, 482–493. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Zhang, X.; Dong, M. Puerarin suppresses MPP+/MPTP-induced oxidative stress through an Nrf2-dependent mechanism. Food Chem. Toxicol. 2020, 144, 111644. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Okazaki, K.; Nguyen, L.T.T.; Ota, N.; Kitamura, H.; Murakami, S.; Shima, H.; Igarashi, K.; Sekine, H.; Motohashi, H. Glucocorticoid receptor signaling represses the antioxidant response by inhibiting histone acetylation mediated by the transcriptional activator NRF2. J. Biol. Chem. 2017, 292, 7519–7530. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claros, S.; Cabrera, P.; Valverde, N.; Romero-Zerbo, S.Y.; López-González, M.V.; Shumilov, K.; Rivera, A.; Pavia, J.; Martín-Montañez, E.; Garcia-Fernandez, M. Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons. Antioxidants 2022, 11, 41. https://doi.org/10.3390/antiox11010041
Claros S, Cabrera P, Valverde N, Romero-Zerbo SY, López-González MV, Shumilov K, Rivera A, Pavia J, Martín-Montañez E, Garcia-Fernandez M. Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons. Antioxidants. 2022; 11(1):41. https://doi.org/10.3390/antiox11010041
Chicago/Turabian StyleClaros, Silvia, Pablo Cabrera, Nadia Valverde, Silvana Y. Romero-Zerbo, Manuel Víctor López-González, Kirill Shumilov, Alicia Rivera, Jose Pavia, Elisa Martín-Montañez, and María Garcia-Fernandez. 2022. "Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons" Antioxidants 11, no. 1: 41. https://doi.org/10.3390/antiox11010041
APA StyleClaros, S., Cabrera, P., Valverde, N., Romero-Zerbo, S. Y., López-González, M. V., Shumilov, K., Rivera, A., Pavia, J., Martín-Montañez, E., & Garcia-Fernandez, M. (2022). Insulin-like Growth Factor II Prevents MPP+ and Glucocorticoid Mitochondrial-Oxidative and Neuronal Damage in Dopaminergic Neurons. Antioxidants, 11(1), 41. https://doi.org/10.3390/antiox11010041