
Thiamine as a Possible Neuroprotective Strategy in Neonatal Hypoxic-Ischemic Encephalopathy

 , ,
, ,
Abstract
:1. Introduction
2. Methods
3. Neonatal Hypoxic-Ischemic Encephalopathy
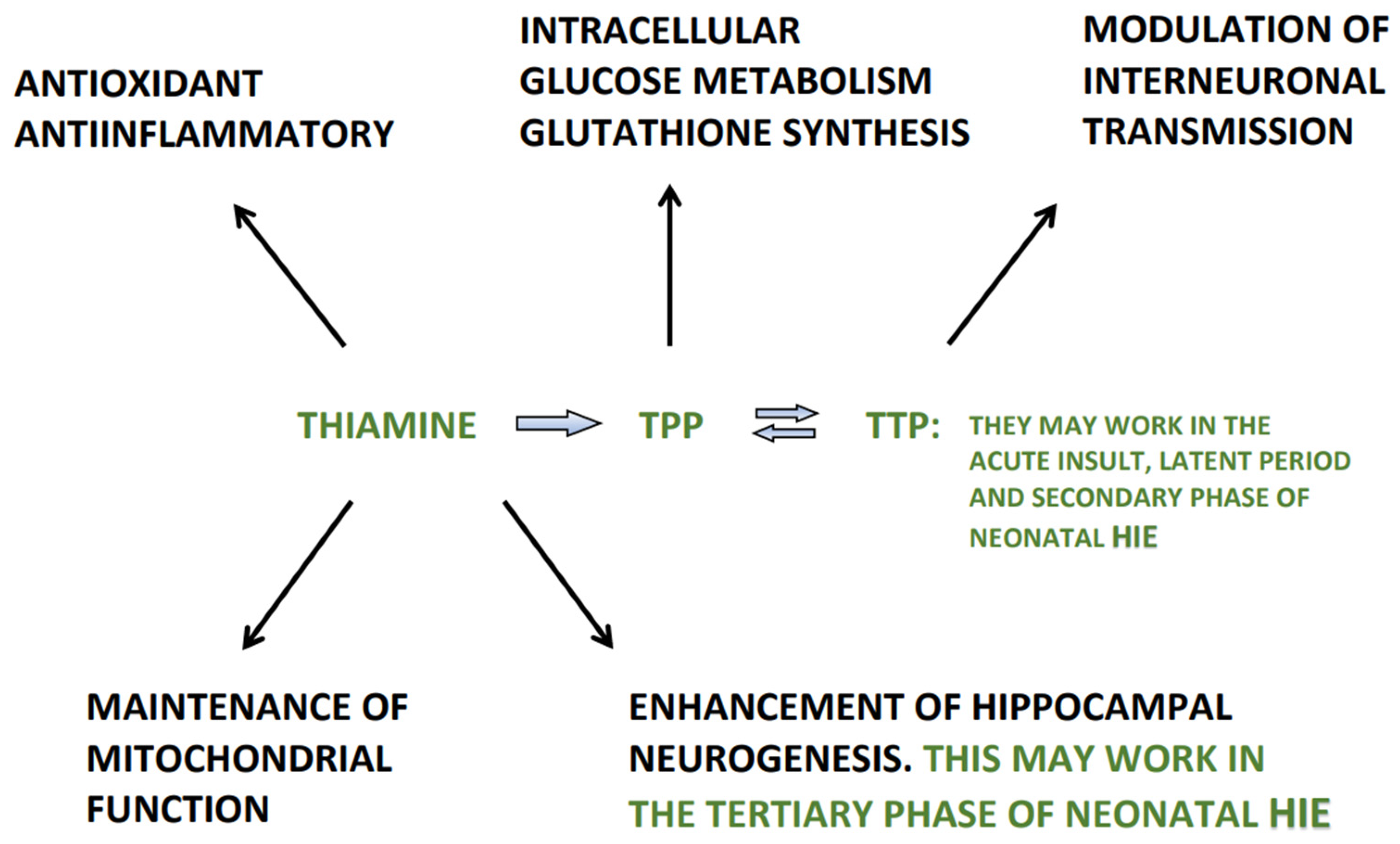
4. Thiamine
5. Hypoxia-Ischemia and Thiamine Deficiency in the Brain: Similar Biochemical and Histological Lesions
6. Thiamine and Its Derivatives in the Management of Neonatal HIE
7. Thiamine Neonatal Requirements
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Low, J.A. Determining the contribution of asphyxia to brain damage in the neonate. J. Obstet. Gynaecol. Res. 2004, 30, 276–286. [Google Scholar] [CrossRef]
- Blair, E.; Watson, L. Epidemiology of cerebral palsy. Semin. Fetal Neonatal Med. 2006, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Locci, E.; Noto, A.; Puddu, M.; Pomero, G.; Demontis, R.; Dalmazzo, C.; Delogu, A.; Fanos, V.; d’Aloja, E.; Gancia, P. A longitudinal 1H-NMR metabolomics analysis of urine from newborns with hypoxic-ischemic encephalopathy undergoing hypothermia therapy. Clinical and medical legal insights. PLoS ONE 2018, 13, e0194267. [Google Scholar] [CrossRef] [Green Version]
- Hassell, K.J.; Ezzati, M.; Alonso-Alconada, D.; Hausenloy, D.G.; Robertson, N.J. New horizons for newborn brain protection: Enhancing endogenous neuroprotection. Arch. Dis. Child. Fetal Neonatal 2015, 100, F541–F552. [Google Scholar] [CrossRef] [PubMed]
- Perlman, J.M. Summary proceedings from the neurology group on hypoxic ischemic encephalopathy. Pediatrics 2006, 117, S28–S33. [Google Scholar] [CrossRef] [PubMed]
- Perlman, J.M. Intervention strategies for neonatal hypoxic-ischemic cerebral injury. Clin. Ther. 2006, 9, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Dean, J.M.; Fraser, M.; Wassink, G.; Andellus, T.C.; Dhillon, S.K.; Bennet, L.; Gunn, A.J. Perinatal brain injury: Mechanisms and therapeutic approaches. Front. Biosci. 2018, 23, 2204–2226. [Google Scholar]
- Amer, A.R.; Oorschot, D.E. Xenon combined with hypothermia in perinatal hypoxic-ischemic encephalopathy: A noble gas, a noble mission. Pediatr. Neurol. 2018, 84, 5–10. [Google Scholar] [CrossRef]
- Broad, K.D.; Fierens, I.; Fleiss, B.; Rocha-Ferreira, E.; Ezzati, M.; Hassell, J.; Alonso-Alconada, D.; Bainbridge, A.; Kawano, G.; Na, D.; et al. Inhaled 45-50% argon augments hypothermic brain protection in a piglet model of perinatal asphyxia. Neurobiol. Dis. 2016, 87, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Koning, G.; Leverin, A.L.; Nair, S.; Schwendimann, L.; Ek, J.; Carlsson, Y.; Gressens, P.; Thornton, C.; Wang, X.; Mallard, C.; et al. Magnesium induces preconditioning of the neonatal brain via profound mitochondrial protection. J. Cereb. Blood Flow Metab. 2019, 39, 1038–1055. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Fanjul, J.; Fernandez-Feijòo, C.D.; Lopez-Abad, M.; Ramos, M.G.L.; Caballè, R.B.; Alcantara-Horillo, S.; Camprubi, M. Neuroprotection with hypothermia and allopurinol in an animal model of hypoxic-ischemic injury: Is it a gender question? PLoS ONE 2017, 12, e0184643. [Google Scholar] [CrossRef] [Green Version]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martinez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT(IA) and CBZ receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef]
- Xiong, T.; Yang, X.; Qu, Y.; Chen, H.; Yue, Y.; Wang, H.; Zhao, F.; Li, S.; Zou, R.; Zhang, L.; et al. Erythropoietin induces synaptogenesis and neurite repair after hypoxia ischemia-mediated brain injury in neonatal rats. NeuroReport 2019, 30, 783–789. [Google Scholar] [CrossRef]
- Robertson, N.J.; Lingam, I.; Mechan, C.; Martinello, K.A.; Avdic-Bectheus, A.; Stein, L.; Tachrount, M.; Price, D.; Sokolska, M.; Bainbridge, A.; et al. High dose melatonin and ethanol excipient combined with therapeutic hypothermia in a newborn piglet asphyxia model. Sci. Rep. 2020, 10, 3898. [Google Scholar] [CrossRef] [Green Version]
- Filippi, L.; Poggi, C.; La Marca, G.; Furlanetto, S.; Fiorini, P.; Cavallaro, G.; Plantulli, A.; Donzelli, G.; Guerrini, R. Oral topiramate in neonates with hypoxic-ischemic encephalopathy: A safety study. J. Pediatr. 2010, 157, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barks, J.D.; Liu, Y.O.; Shangguan, Y.; Silverstein, F.S. Phenobarbital augments hypothermic neuroprotection. Pediatr. Res. 2010, 67, 532–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Svedin, P.; Nie, C.; Lapatto, R.; Zhu, C.; Gustavsson, M.; Sandberg, M.; Karlsson, J.O.; Romero, R.; Hagberg, H.; et al. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann. Neurol. 2007, 61, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Tetorou, K.; Sisa, C.; Iqbal, A.; Dhillon, K.; Hristova, M. Current therapies for neonatal hypoxic-ischaemic and infection-sensitized hypoxix-ischaemic brain damage. Front. Synaptic Neurosci. 2021, 13, 709301. [Google Scholar] [CrossRef] [PubMed]
- West, T.; Atzeva, M.; Holtzman, D.M. Pomegranate polyphenols and resveratrol protect the neonate brain against hypoxic-ischemic injury. Dev. Neurosci. 2007, 29, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Nijboer, C.H.; Groenendaal, F.; Kavellaars, A.; Hagberg, H.H.; van Bell, F.; Heijnen, C.J. Gender-specific neuroprotection by 2-iminobiotin after hypoxia-ischemia in the neonatal rat via a nitric oxide independent patway. J. Cereb. Blood Flow Metab. 2007, 27, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Faviè, L.M.A.; Cox, A.R.; van der Hoogen, A.; Nijboer, C.H.A.; Peeters, C.M.P.C.D.; van Bel, F.; Egberts, T.C.G.; Rademaker, C.M.A.; Groenendaal, F. Nitric oxide synthase inhibition as a neuroprotective strategy following hypoxic-ischemic encephalopathy: Evidence from animal studies. Front. Neurol. 2018, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, C.-Y.; Chen, H.-R.; Gao, N.; Kuo, Y.-M.; Chen, C.-W.; Yang, D.; Kinkaid, M.M.; Hu, E.; Sun, Y.-Y. Brain-targeted hypoxia-inducible factor stabilization reduces neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2021, 148, 105200. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Rocha-Ferreira, E.; Fleiss, B.; Nijboer, C.H.; Gressens, P.; Mallard, C.; Hagberg, H. Neuroprotection offered by mesenchymal stem cells in perinatal brain injury: Role of mitochondria, inflammation, and reactive oxygen species. J. Neurochem. 2021, 158, 59–73. [Google Scholar] [CrossRef]
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455. [Google Scholar] [CrossRef]
- Sechi, G.; Sechi, E.; Fois, C.; Kumar, N. Advances in clinical determinants and neurological manifestations of B vitamin deficiency in adults. Nutr. Rev. 2016, 74, 281–300. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Liu, X.; Kida, K.; Marutani, E.; Hirai, S.; Sakaguchi, M.; Andersen, L.W.; Bagchi, A.; Cocchi, M.N.; Berg, K.M.; et al. Thiamine as a neuroprotective agent after cardiac arrest. Resuscitation 2016, 105, 138–144. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Colmant, H.J. Differentiation between brain lesions in experimental thiamine deficiency. Virchows Arch. A Pathol. Anat. Histopathol. 1988, 414, 61–67. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Hagel, C.; Laas, R. Hypoxia-ischemia and thiamine deficiency. Clin. Neuropathol. 1993, 12, 184–190. [Google Scholar]
- Zera, K.; Zastre, J. Thiamine deficiency activates hypoxia inducible factor-1α to facilitate pro-apoptotic responses in mouse primary astrocytes. PLoS ONE 2017, 12, e0186707. [Google Scholar] [CrossRef] [Green Version]
- Zera, K.; Zastre, J. Stabilization of the hypoxia-inducible transcription Factor-1 alpha (HIF-1α) in thiamine deficiency is mediated by pyruvate accumulation. Toxicol. Appl. Pharmacol. 2018, 355, 180–188. [Google Scholar] [CrossRef]
- Liang, X.; Liu, X.; Lu, F.; Zhang, Y.; Jiang, X.; Ferriero, D.M. HIF1α signalling in the endogenous protective responses after neonatal brain hypoxia-ischemia. Dev. Neurosci. 2018, 40, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.P.; Ramaswamy, V.; Michelson, D.; Barkovich, J.; Holsuser, B.; Wycliffe, N.; Glidden, D.V.; Deming, D.; Partridge, J.C.; Wu, Y.W.; et al. Pattern of brain injury in term neonatal encephalopathy. J. Pediatr. 2005, 146, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.M.T.; Perlman, M. Follow-up of the term infant after hypoxic-ischemic encephalopathy. Paediatr. Child Health 2006, 11, 278–282. [Google Scholar]
- Kurinezuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordì, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic-ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 1, CD003311. [Google Scholar] [CrossRef] [PubMed]
- Pauliah, S.S.; Shankaran, S.; Wade, A.; Cady, E.B.; Thayyl, S. Therapeutic hypothermia for neonatal encephalopathy in low- and middle-income countries: A systematic review and meta-analysis. PLoS ONE 2013, 8, e58834. [Google Scholar] [CrossRef] [Green Version]
- McGready, R.; Simpson, J.A.; Cho, T.; Dubowitz, L.; Changbumrung, S.; Bohm, V.; Munger, R.G.; Sauberlich, H.E.; White, N.J.; Nostem, F. Postpartum thiamine deficiency in a Karen displaced population. Am. J. Clin. Nutr. 2001, 74, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy. A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef]
- Hansford, R.G. Control of mitochondrial substrate oxidation. Curr. Top Bioenerg. 1980, 10, 217–278. [Google Scholar]
- Terwel, D.; Bothmer, J.; Wolf, E.; Meng, F.; Jolles, J. Affected enzyme activities in Alzheimer’s disease are sensitive to antemortem hypoxia. J. Neurol. Sci. 1998, 161, 47–56. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. B 2005, 360, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Reiter, C.D.; Teng, R.-J.; Beckman, J.S. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J. Biol. Chem. 2000, 275, 32460–32466. [Google Scholar] [CrossRef] [Green Version]
- Groenendaal, F.; Lammers, H.; Smit, D.; Nikkels, P.G.J. Nitrotyrosine in brain tissue of neonates after perinatal asphyxia. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, F429–F433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzopardi, D.; Wyatt, T.S.; Cady, E.B.; Delpy, D.T.; Baudin, J.; Stewart, A.L.; Hope, P.L.; Hamilton, P.A.; Reynolds, E.O.R. Prognosis of newborn infants with hypoxia-ischemia brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1989, 25, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapadia, V.S.; Chalak, L.F.; DuPont, T.L.; Rollins, N.K.; Brion, L.P.; Wyckof, M.H. Perinatal asphyxia with hyperoxemia within the first hour of life is associated with moderate to severe hypoxic-ischemic encephalopathy. J. Pediatr. 2013, 163, 949–954. [Google Scholar] [CrossRef]
- Saugstad, O.D. The oxygen paradox in the newborn: Keep oxygen at normal levels. J. Pediatr. 2013, 163, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Wassink, G.; Gunn, E.R.; Drury, P.P.; Bennet, L.; Gunn, A.J. The mechanisms and treatment of asphyxia encephalopathy. Front. Neurosci. 2014, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Otsuguro, K.; Ohta, T.; Ito, S. Adenosine and inosine release during hypoxia in the isolated spinal cord of neonatal rats. Br. J. Pharmacol. 2010, 161, 1806–1816. [Google Scholar] [CrossRef] [Green Version]
- Azzopardi, D.V.; Strohm, B.; Edwards, A.D.; Dyet, L.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; Levene, M.; Marlow, N.; Porter, E.; et al. TOBY Study Group. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 2009, 36, 1349–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, A.J.; Wyatt, J.S.; Whitelaw, A.; Barks, J.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Gluckman, P.D.; Polin, R.A.; et al. Therapeutic hypothermia changes the prognostic value of clinical evaluation of neonatal encephalopathy. J. Pediatr. 2008, 152, 55–58.e1. [Google Scholar] [CrossRef]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, T.A.; Tyson, J.E.; Mcdonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. National Institite of Child Health and Human Development Neonatal research Network. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef]
- Rutherford, M.; Ramenghi, I.A.; Edwards, A.D.; Brocklehurst, P.; Halliday, H.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Assessment of brain tissue injury after moderate hypothermia in neonates with hypoxic-ischaemic encephalopathy: A nested substudy of a randomized controlled trial. Lancet Neurol. 2010, 9, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Shankaran, S.; Barnes, P.D.; Hintz, S.R.; Laptook, A.R.; Zaterka-Baxter, K.M.; McDonald, S.A.; Ehrenkranz, R.A.; Walsh, M.C.; Tyson, J.E.; Donovan, E.F.; et al. Brain injury following trial of hypothermia for neonatal hipoxic-ischaemic encephalopathy. Arch. Dis. Child. Fetal Neonatal Ed. 2012, 97, F398–F404. [Google Scholar] [PubMed] [Green Version]
- Gehrmann, J.; Banati, R.B.; Wiessner, C.; Hossmann, K.A.; Kreutzberg, G.W. Reactive microglia in cerebral ischaemia. An early mediator of tissue damage? Neuropathol. Appl. Neurobiol. 1995, 21, 277–289. [Google Scholar] [CrossRef]
- Sechi, G. Concerning “Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics and functional studies” by Marcé-Grau et al. J. Inherit. Metab. Dis. 2020, 43, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Manzetti, S.; Zhang, J.; van der Spoel, D. Thiamin function, metabolism, uptake, and transport. Biochemistry 2014, 53, 821–835. [Google Scholar] [CrossRef]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Di Salvo, L.M.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-M.; Chen, H.-L.; Gibson, G.E. Thiamine and oxidants interact to modify cellular calcium stores. Neurochem. Res. 2010, 35, 2107–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, A. Metabolic and structural role of thiamine in nervous tissues. Cell. Mol. Neurobiol. 2008, 28, 923–931. [Google Scholar] [CrossRef]
- Zhao, N.; Zhong, C.; Wang, Y.; Zhao, Y.; Gong, N.; Zhou, G.; Xu, T.; Hong, Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiol. Dis. 2008, 29, 176–185. [Google Scholar] [CrossRef]
- Ramalingam, M.; Kim, S.-J. Reactive oxygen/nitrogen species and their functional correlations in neurodegenerative diseases. J. Neural Transm. 2012, 119, 891–910. [Google Scholar] [CrossRef]
- Sechi, G.; Batzu, L.; Agrò, L.; Fois, C. Cancer-related Wernicke-Korsakoff syndrome. Lancet Oncol. 2016, 17, e221–e222. [Google Scholar] [CrossRef] [Green Version]
- Sechi, E.; Addis, A.; Fadda, G.; Minafra, L.; Bravatà, V.; Sechi, G. Teaching neuroimages: Subacute encephalopathy in a young woman with THTR2 gene mutation. Neurology 2015, 85, e108–e109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sechi, G.; Sechi, M.M. Challenges in prediction and diagnosis of alcohol withdrawal syndrome and Wernicke encephalopathy. JAMA Intern. Med. 2020, 180, 1716. [Google Scholar] [CrossRef]
- Schmidtke, K. Wernicke-Korsakoff syndrome following attempted hanging. Rev. Neurol. 1993, 149, 213–216. [Google Scholar]
- Johkura, K.; Naito, M. Wernicke’s encephalopathy-like lesions in global cerebral hypoxia. J. Clin. Neurosci. 2008, 15, 318–319. [Google Scholar] [CrossRef]
- Molavi, M.; Vann, S.D.; de Vries, L.S.; Groenendaal, F.; Lequin, M. Signal change in the mammillary bodies after perinatal asphyxia. AJNR Am. J. Neuroradiol. 2019, 40, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.S.; Groenendaal, F. Patterns of neonatal hypoxic-ischaemic brain injury. Neuroradiology 2010, 52, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, P.; Juul, S.E. Neuroprotective strategies in neonatal brain injury. J. Pediatr. 2018, 192, 22–32. [Google Scholar] [CrossRef]
- Jhala, S.S.; Hazel, A.S. Modeling neurodegenerative disease pathophysiology in thiamine deficiency consequences of impaired oxidative metabolism. Neurochem. Int. 2011, 58, 248–260. [Google Scholar] [CrossRef]
- Johnston, M.V.; Fatemi, A.; Wilson, M.A.; Northington, F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011, 10, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.C.; Baranova, O.; Lin, J.; Pichiule, P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J. Neurosci. 2006, 26, 9471–9481. [Google Scholar] [CrossRef]
- Trollmann, R.; Gassmann, M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev. 2009, 31, 503–509. [Google Scholar] [CrossRef]
- Greijer, A.; Van der Wall, E. The role of hypoxia inducible factor 1(HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 100914. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Wassink, G.; van den Heuj, L.G.; Bennet, L.; Gunn, A.J. Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy: Where to from here? Front. Neurol. 2015, 6, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okai, Y.; Higashi-Okai, K.; Sato, E.F.; Konaka, R.; Inoue, M. Potent radical-scavenging activities of thiamin and thiamin diphosphate. J. Clin. Biochem. Nutr. 2007, 40, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepuro, A.I.; Piletskaya, T.P.; Stepuro, I.I. Role of thiamine thiol form in nitric oxide metabolism. Biochemistry 2005, 70, 339–349. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Gibson, G.E. Vascular endothelium in a site of free radical production and inflammation in areas of neuronal loss in thiamine-deficient brain. Ann. N. Y. Acad. Sci. 2000, 903, 353–356. [Google Scholar] [CrossRef]
- Gong, P.; Hua, R.; Zhang, Y.; Zhao, H.; Tang, Z.; Mei, X.; Zhang, M.; Cui, J.; Li, C. Hypothermia-induced neuroprotection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J. Cereb. Blood Flow Metab. 2013, 33, 928–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, R.R.; Godin, A.M.; Rodrigues, F.F.; Coura, G.M.E.; Melo, I.S.F.; Brito, A.M.S.; Bertollo, C.M.; Paulino, T.P.; Rachid, M.A.; Machado, R.R.; et al. Thiamine and riboflavin inhibit production of cytokines and increase the anti-inflammatory activity of a corticosteroid in a chronic model of inflammation induced by complete Freund’s adjuvant. Pharmacol. Rep. 2017, 69, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.J.; Thoresen, M. Hypothermic neuroprotection. NeuroRx 2006, 3, 154–169. [Google Scholar] [CrossRef]
- Zempleni, J.; Link, G.; Kubler, W. The transport of thiamin, riboflavin and pyridoxal 5-phosphate by human placenta. Int. J. Vitam. Nutr. Res. 1991, 62, 165–172. [Google Scholar]
- Dancis, J.; Wilson, D.; Iffath, A.; Hoskins, I.A.; Levitz, M. Placental transfer of thiamin in the human subject: In vitro perfusion studies and maternal-cord plasma concentrations. Am. J. Obstet. Gynecol. 1988, 159, 1435–1439. [Google Scholar] [CrossRef]
- Smith, T.J.; Hess, S.Y. Infantile thiamine deficiency in South and Southeast Asia: An age-old problem needing new solutions. Nutr. Bull. 2021, 46, 12–25. [Google Scholar] [CrossRef]
- Rakotoambinina, B.; Hiffler, L.; Gomes, F. Pediatric thiamine deficiency disorders in high-income countries between 2000 and 2020: A clinical reappraisal. Ann. N. Y. Acad. Sci. 2021, 1498, 57–76. [Google Scholar] [CrossRef]
- Mimouni-Bloch, A.; Goldberg-Stern, H.; Strausberg, R.; Brezner, A.; Heyman, E.; Inbar, D.; Kivity, S.; Zvulunov, A.; Sztarkier, I.; Fogelman, R.; et al. Thiamine deficiency in infancy: Long-term follow-up. Pediatr. Neurol. 2014, 51, 311–316. [Google Scholar] [CrossRef]
- Harel, Y.; Zuk, L.; Guindy, M.; Nakar, O.; Lotan, D.; Fattal-Valevski, A. The effect of subclinical infantile thiamine deficiency on motor function in preschool children. Matern. Child Nutr. 2017, 13, e12397. [Google Scholar] [CrossRef]
- Fattal-Valenski, A.; Azouri-Fattal, I.; Greenstein, Y.J.; Guindy, M.; Blau, A.; Zelnik, N. Delayed language development due to infantile thiamine deficiency. Dev. Med. Child Neurol. 2009, 51, 629–634. [Google Scholar] [CrossRef]
- Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academy Press: Washington, DC, USA, 1998. [Google Scholar]
- Ilves, P.; Kiisk, M.; Soopold, T.; Talvik, T. Serum total magnesium and ionized calcium concentrations in asphyxiated term newborn infants with hypoxic-ischaemic encephalopathy. Acta Paediatr. 2000, 89, 680–685. [Google Scholar] [CrossRef] [PubMed]
- FAO (Food and Agriculture Organization and World Health Organization). Vitamin and Mineral Requirements in Human Nutrition. Report of a Joint WHO/FAO Expert Consultation; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Rao, S.N.; Chandak, G.R. Cardiac Beriberi: Often a missed diagnosis. J. Trop. Ped. 2010, 56, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.G.; Coats, D.; Fisher, P.R.; Ou, K.; Frank, E.L.; Sreang, P.; Saing, S.; Topazian, M.D.; Enders, F.T.; Cabalka, A.K. Thiamine deficiency and cardiac dysfunction in Cambodian infants. J. Pediatr. 2014, 164, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Hiffler, L.; Rakotoambinina, B.; Lafferty, N.; Garcia, D.M. Thiamine deficiency in tropical pediatrics: New insights into a neglected but vital metabolic challenge. Front. Nutr. 2016, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Agents | Type of Study | Main Mechanisms of Action | References |
|---|---|---|---|
| Xenon/Argon | Prec./Clin. | NMDA-receptor antagonists; anti-inflammatory; improve apoptosis | Amer et al. (2018) [8] Broad et al. (2016) [9] |
| Magnesium | Preclinical | NMDA-receptor antagonist; anti-oxidant; anti-inflammatory | Koning et al. (2019) [10] |
| Allopurinol | Preclinical | Antioxidant; possible gender effect | Rodriguez-Fanjul et al. (2017) [11] |
| Cannabinoids | Preclinical | Modulation excitotoxicity, oxidative stress, inflammation | Pazos et al. (2013) [12] |
| Erythropoietin | Preclinical | Antiapoptotic, antioxidant, anti-inflammatory; neurovascular remodelling; promotes neural stem cell proliferation | Xiong et al. (2019) [13] |
| Melatonin | Preclinical | Antioxidant, anti-inflammatory. | Robertson et al. (2020) [14] |
| Topiramate | Clinical | Anticonvulsant; anti-excitotoxicity | Filippi et al. (2020) [15] |
| Phenobarbital | Preclinical | Anticonvulsant; reduced cerebral metabolic demand; antioxidant | Barks et al. (2010) [16] |
| N-acetylcysteine | Preclinical | Antioxidant, anti-inflammatory; restores intracellular glutathione | Wang et al. (2007) [17] |
| Indometacin | Preclinical | Anticaspase activity and antiapoptotic; restores intracellular glutathione | Tetorou et al. (2021) [18] |
| Pentoxifylline | Preclinical | Phosphodiesterase inhibitor; anti-inflammatory | Tetorou et al. (2021) [18] |
| Quercetin/Coumestrol | Preclinical | Flavonoid/isoflavonoid antioxidants, anti-inflammatory | Tetorou et al. (2021) [18] |
| Polyphenols | Preclinical | Antioxidant, anti-inflammatory, antiapoptotic properties | West et al. (2007) [19] |
| 2-Iminobiotin | Preclinical | Inhibition of cytochrome C-caspase 3 neuronal death pathway in female. Gender specificity | Nijboer et al. (2007) [20] |
| NTR5221/NCT01626934 | Preclinical | Antioxidants by inhibition of nitric oxide synthesis | Faviè et al. (2018) [21] |
| GSK360A | Preclinical | Brain-targeted hypoxia-inducible factor1α-stabilization by inhibition of prolyl-4-hydroxylase | Kuan et al. (2021) [22] |
| Stem Cells | Preclinical | Modulation of immune/inflammatory response, | Nair et al. (2021) [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sechi, G.P.; Bardanzellu, F.; Pintus, M.C.; Sechi, M.M.; Marcialis, M.A.; Fanos, V. Thiamine as a Possible Neuroprotective Strategy in Neonatal Hypoxic-Ischemic Encephalopathy. Antioxidants 2022, 11, 42. https://doi.org/10.3390/antiox11010042
Sechi GP, Bardanzellu F, Pintus MC, Sechi MM, Marcialis MA, Fanos V. Thiamine as a Possible Neuroprotective Strategy in Neonatal Hypoxic-Ischemic Encephalopathy. Antioxidants. 2022; 11(1):42. https://doi.org/10.3390/antiox11010042
Chicago/Turabian StyleSechi, Gian Pietro, Flaminia Bardanzellu, Maria Cristina Pintus, Maria Margherita Sechi, Maria Antonietta Marcialis, and Vassilios Fanos. 2022. "Thiamine as a Possible Neuroprotective Strategy in Neonatal Hypoxic-Ischemic Encephalopathy" Antioxidants 11, no. 1: 42. https://doi.org/10.3390/antiox11010042