
Gallic Acid Improves Diabetic Steatosis by Downregulating MicroRNA-34a-5p through Targeting NFE2L2 Expression in High-Fat Diet-Fed db/db Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animal Maintenance and Treatment
2.3. Determination of Plasma Biomarkers for Liver Steatosis and Relevant Biochemical Analysis
2.4. Measurement of Lipid Peroxidation and Antioxidant Enzymes
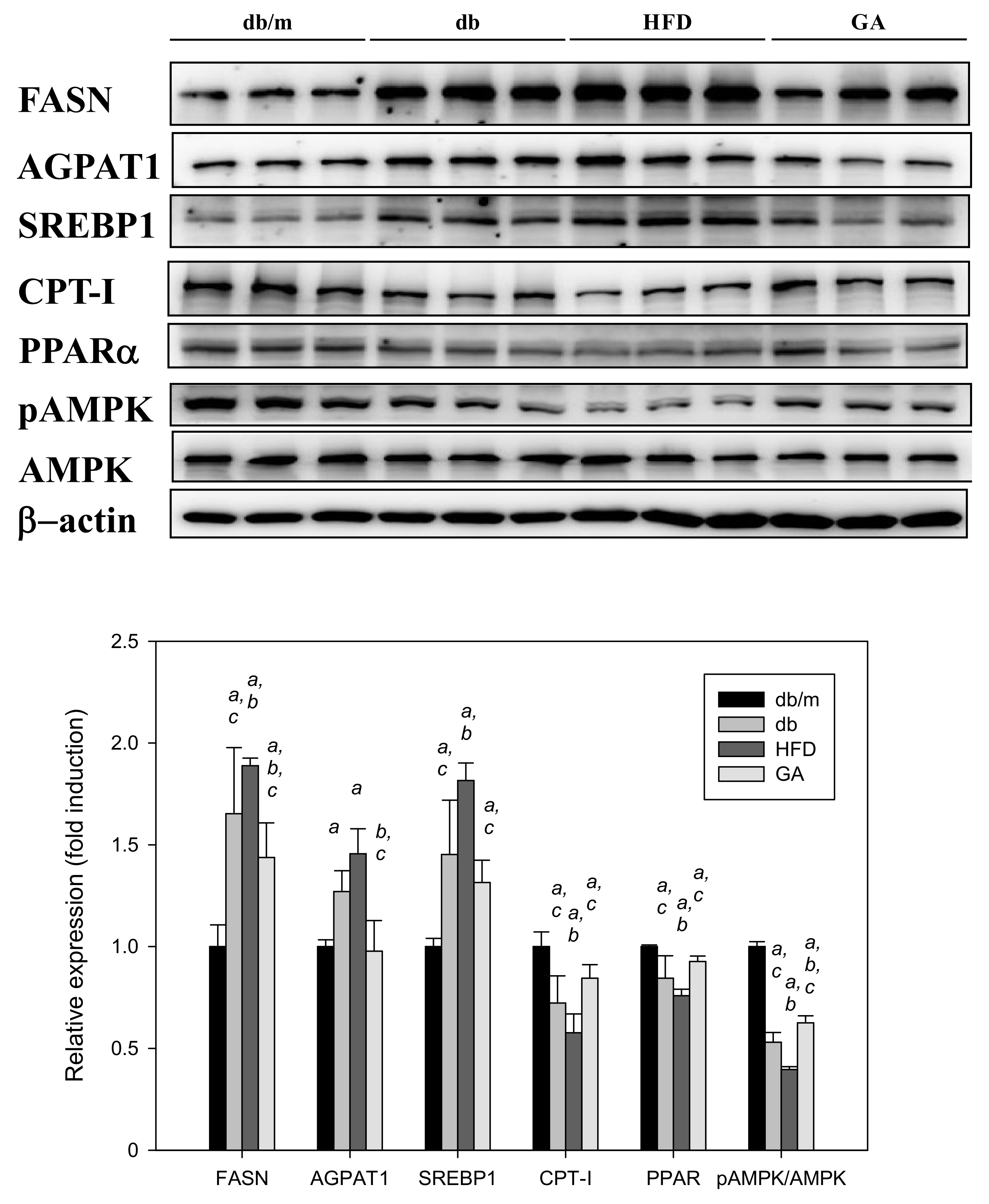
2.5. Western Blotting
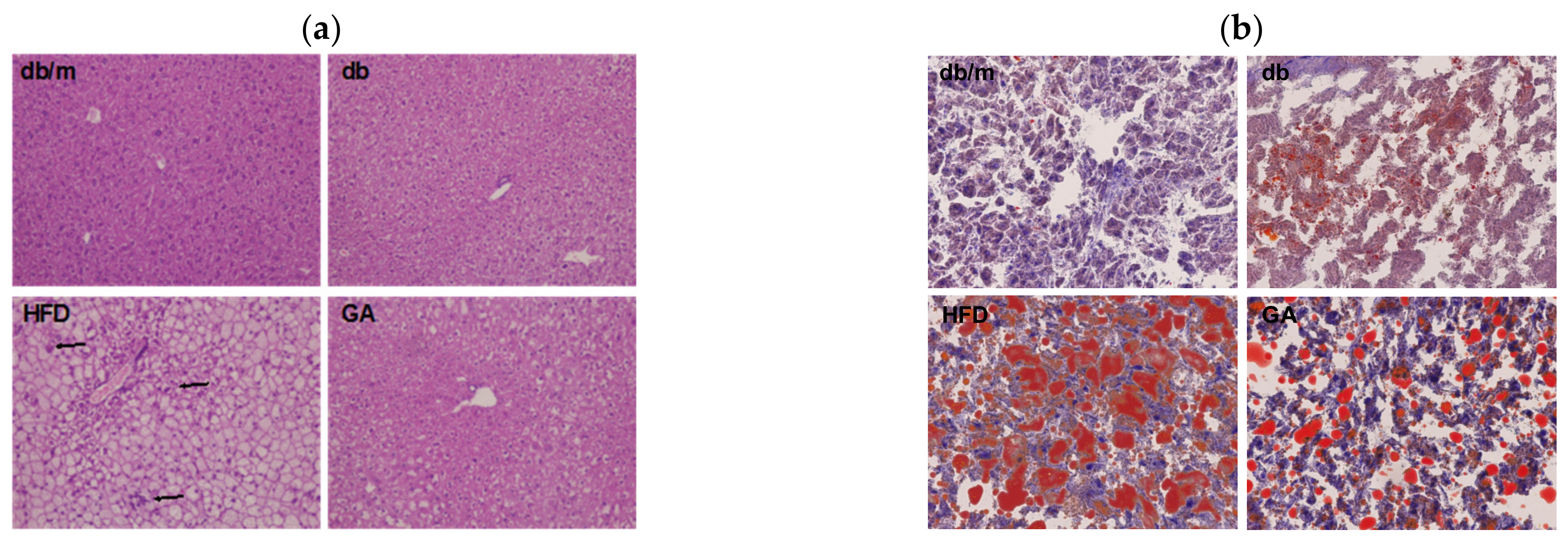
2.6. Histological Analysis of Tissues
2.7. Cell Treatment
2.8. miRNA Isolation and Real-Time Polymerase Chain Reaction
2.9. Luciferase Reporter Assay
2.10. Statistical Analysis
3. Results
3.1. GA Improved Liver Function, Hyperlipidemia, Hyperinsulinemia, and Hepatic Steatosis in Diabetic Mice
3.2. GA Ameliorated Hepatic Lipid Peroxidation and Enhanced the Activities of Hepatic Antioxidant Enzymes in Diabetic Mice
3.3. GA Inhibited the Expression of Hepatic Lipid Metabolism–Related Proteins in Diabetic Mice
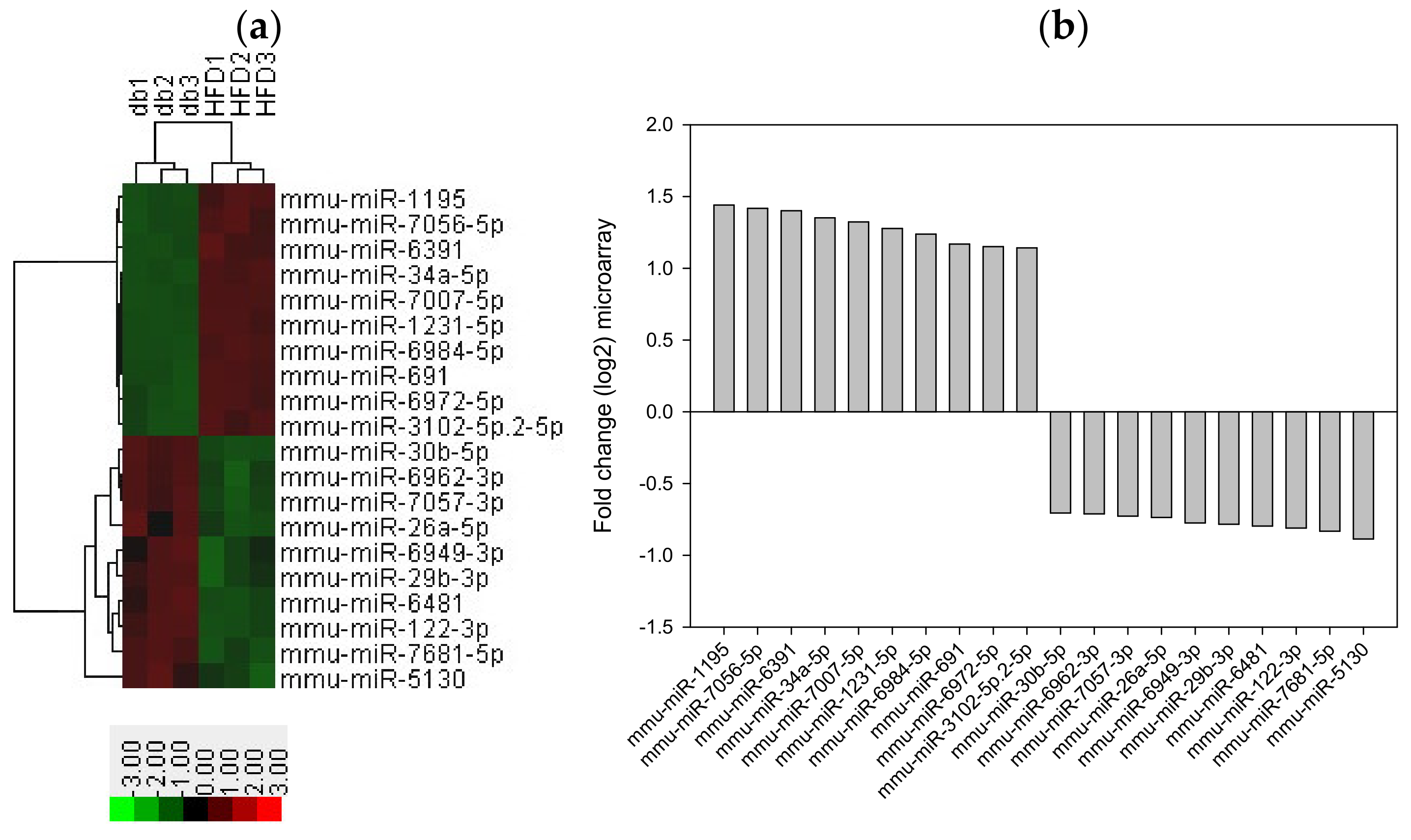
3.4. HFD Induced miRNA Expression in Diabetic Mice
3.5. Search for Predicted miRNA Target Genes through Web-Based Bioinformatic Analysis
3.6. miR-34a-5p Inhibitor Ameliorated Lipid Accumulation in HepG2 Cells
3.7. miR-34a-5p Mimics Affected Lipid Accumulation in HepG2 Cells
3.8. miR-34a-5p Mediated Cellular Lipid Accumulation by Directly Targeting NFE2L2 in HepG2 Cells
3.9. GA Improved HG + O/P-Induced Lipid Accumulation by Downregulating miR-34a-5p in HepG2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, S.K.; Seo, M.H.; Shin, H.C.; Ryoo, J.H. Clinical availability of nonalcoholic fatty liver disease as an early predictor of type 2 diabetes mellitus in Korean men: 5-year prospective cohort study. Hepatology 2013, 57, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus—Mechanisms and treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Karolina, D.S.; Tavintharan, S.; Armugam, A.; Sepramaniam, S.; Pek, S.L.; Wong, M.T.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. Circulating miRNA profiles in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E2271–E2276. [Google Scholar] [CrossRef] [Green Version]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- López-Pastor, A.R.; Infante-Menéndez, J.; Escribano, Ó.; Gómez-Hernández, A. miRNA Dysregulation in the Development of Non-Alcoholic Fatty Liver Disease and the Related Disorders Type 2 Diabetes Mellitus and Cardiovascular Disease. Front. Med. 2020, 7, 527059. [Google Scholar] [CrossRef]
- Meng, X.; Guo, J.; Fang, W.; Dou, L.; Li, M.; Huang, X.; Zhou, S.; Man, Y.; Tang, W.; Yu, L.; et al. Liver MicroRNA-291b-3p Promotes Hepatic Lipogenesis through Negative Regulation of Adenosine 5′-Monophosphate (AMP)-activated Protein Kinase α1. J. Biol. Chem. 2016, 291, 10625–10634. [Google Scholar] [CrossRef] [Green Version]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Doss, C.G.; Bandyopadhyay, S.; Agoramoorthy, G. Influence of miRNA in insulin signaling pathway and insulin resistance: Micro-molecules with a major role in type-2 diabetes. Wiley Interdiscip. Rev. RNA 2014, 5, 697–712. [Google Scholar] [CrossRef]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, D.; Jude, B.; Morand, C. miRNA as molecular target of polyphenols underlying their biological effects. Free Radic. Biol. Med. 2013, 64, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Huo, T.I.; Cheng, H.Y.; Tsai, J.C.; Liao, J.W.; Lee, M.S.; Qin, X.M.; Hsieh, M.T.; Pao, L.H.; Peng, W.H. Gallic acid ameliorated impaired glucose and lipid homeostasis in high fat diet-induced NAFLD mice. PLoS ONE 2014, 9, e96969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Jun, C.D.; Suk, K.; Choi, B.J.; Lim, H.; Park, S.; Lee, S.H.; Shin, H.Y.; Kim, D.K.; Shin, T.Y. Gallic acid inhibits histamine release and pro-inflammatory cytokine production in mast cells. Toxicol. Sci. 2006, 91, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Paolini, A.; Curti, V.; Pasi, F.; Mazzini, G.; Nano, R.; Capelli, E. Gallic acid exerts a protective or an anti-proliferative effect on glioma T98G cells via dose-dependent epigenetic regulation mediated by miRNAs. Int. J. Oncol. 2015, 46, 1491–1497. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, S.A.; Wilson, D.L.; LeBoeuf, R.C. C57BL/6 mice fed high fat diets as models for diabetes-accelerated atherosclerosis. Atherosclerosis 1998, 136, 17–24. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. S2), S262–S268. [Google Scholar] [CrossRef] [Green Version]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Lawrence, R.A.; Burk, R.F. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef]
- Jeon, M.J.; Kim, E.J.; Lee, M.; Kim, H.; Choi, J.R.; Chae, H.D.; Moon, Y.J.; Kim, S.K.; Bai, S.W. MicroRNA-30d and microRNA-181a regulate HOXA11 expression in the uterosacral ligaments and are overexpressed in pelvic organ prolapse. J. Cell. Mol. Med. 2015, 19, 501–509. [Google Scholar] [CrossRef]
- Festing, M.F.; Altman, D.G. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J. 2002, 43, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Rau, C.S.; Jeng, J.C.; Chen, Y.C.; Lu, T.H.; Wu, C.J.; Wu, Y.C.; Tzeng, S.L.; Yang, J.C. Whole blood-derived microRNA signatures in mice exposed to lipopolysaccharides. J. Biomed. Sci. 2012, 19, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Hu, T.; Han, L.; Liu, B.; Tang, X.; Chen, H.; Chen, X.; Wan, M. miRNA microarray profiling in patients with androgenic alopecia and the effects of miR-133b on hair growth. Exp. Mol. Pathol. 2021, 118, 104589. [Google Scholar] [CrossRef] [PubMed]
- Sueta, A.; Fujiki, Y.; Goto-Yamaguchi, L.; Tomiguchi, M.; Yamamoto-Ibusuki, M.; Iwase, H.; Yamamoto, Y. Exosomal miRNA profiles of triple-negative breast cancer in neoadjuvant treatment. Oncol. Lett. 2021, 22, 819. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. Biomed. Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.J.; Sul, H.S. Insulin regulation of fatty acid synthase gene transcription: Roles of USF and SREBP-1c. IUBMB Life 2004, 56, 595–600. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, L.; Liao, P.; Xiao, Z.; Zhang, F.; Sindaye, D.; Xin, Z.; Tan, C.; Deng, J.; Yin, Y.; et al. Impact of Gallic Acid on Gut Health: Focus on the Gut Microbiome, Immune Response, and Mechanisms of Action. Front. Immunol. 2020, 11, 580208. [Google Scholar] [CrossRef]
- Hsu, C.L.; Yen, G.C. Effect of gallic acid on high fat diet-induced dyslipidaemia, hepatosteatosis and oxidative stress in rats. Br. J. Nutr. 2007, 98, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Chao, J.; Cheng, H.Y.; Chang, M.L.; Huang, S.S.; Liao, J.W.; Cheng, Y.C.; Peng, W.H.; Pao, L.H. Gallic Acid Ameliorated Impaired Lipid Homeostasis in a Mouse Model of High-Fat Diet-and Streptozotocin-Induced NAFLD and Diabetes through Improvement of β-oxidation and Ketogenesis. Front. Pharmacol. 2020, 11, 606759. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Angulo, P.; Lindor, K.D. Nonalcoholic fatty liver disease. CMAJ. 2005, 172, 899–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dludla, P.V.; Nkambule, B.B.; Jack, B.; Mkandla, Z.; Mutize, T.; Silvestri, S.; Orlando, P.; Tiano, L.; Louw, J.; Mazibuko-Mbeje, S.E. Inflammation and Oxidative Stress in an Obese State and the Protective Effects of Gallic Acid. Nutrients 2018, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in beta-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Liu, Y.; Mao, Y.; Chen, W.; Xiao, Z.; Yu, Y. Relationship between glycated haemoglobin concentration and erythrocyte survival in type 2 diabetes mellitus determined by a modified carbon monoxide breath test. J. Breath Res. 2018, 12, 026004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramezani Ali Akbari, F.; Badavi, M.; Dianat, M.; Mard, S.A.; Ahangarpour, A. Gallic acid improves oxidative stress and inflammation through regulating micrornas expressions in the blood of diabetic rats. Acta Endocrinol. 2019, 15, 187–194. [Google Scholar]
- Liu, C.H.; Ampuero, J.; Gil-Gómez, A.; Montero-Vallejo, R.; Rojas, Á.; Muñoz-Hernández, R.; Gallego-Durán, R.; Romero-Gómez, M. miRNAs in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2018, 69, 1335–1348. [Google Scholar] [CrossRef]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.M.; Parker, J.S.; Hammond, S.M. Microarray analysis of miRNA gene expression. Methods Enzymol. 2007, 427, 107–122. [Google Scholar]
- Ranade, A.R.; Weiss, G.J. Methods for microRNA microarray profiling. Methods Mol. Biol. 2011, 700, 145–152. [Google Scholar]
- Tagne, J.B.; Mohtar, O.R.; Campbell, J.D.; Lakshminarayanan, M.; Huang, J.; Hinds, A.C.; Lu, J.; Ramirez, M.I. Transcription factor and microRNA interactions in lung cells: An inhibitory link between NK2 homeobox 1, miR-200c and the developmental and oncogenic factors Nfib and Myb. Respir. Res. 2015, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Hao, L.; Li, S.; Lin, S.; Lv, L.; Chen, Y.; Cui, H.; Zi, T.; Chu, X.; Na, L.; et al. Elevated circulating stearic acid leads to a major lipotoxic effect on mouse pancreatic beta cells in hyperlipidaemia via a miR-34a-5p-mediated PERK/p53-dependent pathway. Diabetologia 2016, 59, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Thong, F.S.; Bilan, P.J.; Klip, A. The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 2007, 56, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Xue, M.; Li, Y.; Hu, F.; Jia, Y.J.; Zheng, Z.J.; Wang, L.; Xue, Y.M. High glucose up-regulates microRNA-34a-5p to aggravate fibrosis by targeting SIRT1 in HK-2 cells. Biochem. Biophys. Res. Commun. 2018, 498, 38–44. [Google Scholar] [CrossRef]
- Lin, H.Y.; Yang, Y.L.; Wang, P.W.; Wang, F.S.; Huang, Y.H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef]
- Lee, S.; Park, Y.; Zuidema, M.Y.; Hannink, M.; Zhang, C. Effects of interventions on oxidative stress and inflammation of cardiovascular diseases. World J. Cardiol. 2011, 3, 18–24. [Google Scholar] [CrossRef]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Gao, Y.; Qin, J.; Lu, S. The role of miR-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS ONE 2014, 9, e113305. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Gao, Y.; Straub, K.D. Oxidation of Hydrogen Sulfide by Quinones: How Polyphenols Initiate Their Cytoprotective Effects. Int. J. Mol. Sci. 2021, 22, 961. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Briggs, A.; Devireddy, M.; Iovino, N.A.; Skora, N.C.; Whelan, J.; Villa, B.P.; Yuan, X.; Mannam, V.; Howard, S.; et al. Green tea polyphenolic antioxidants oxidize hydrogen sulfide to thiosulfate and polysulfides: A possible new mechanism underpinning their biological action. Redox Biol. 2020, 37, 101731. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z. Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes. Oxid. Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| db/m | db | HFD | GA | |
|---|---|---|---|---|
| AST (U/L) | 139.67 ± 14.29 | 160.83 ± 21.85 | 312.00 ± 30.73 ab | 212.00 ± 19.06 abc |
| ALT (U/L) | 34.17 ± 7.70 | 87.67 ± 10.17 a | 411.50 ± 30.38 ab | 209.33 ± 23.82 abc |
| Cholesterol (mg/dL) | 102.67 ± 12.16 | 139.17 ± 11.13 | 415.17 ± 52.13 ab | 367.33 ± 44.08 abc |
| TG (mg/dL) | 75.17 ± 12.14 | 98.00 ± 25.61 | 259.00 ± 81.84 ab | 194.50 ± 22.20 abc |
| HbA1c (%) | 4.47 ± 0.71 | 8.23 ± 1.40 a | 8.30 ± 0.88 a | 8.10 ± 1.33 a |
| Insulin (pg/L) | 1.03 ± 0.09 | 5.24 ± 0.37 a | 7.26 ± 1.83 ab | 5.20 ± 0.51 ac |
| Liver weight (g) | 1.30 ± 0.20 | 2.27 ± 0.07 a | 4.75 ± 0.26 ab | 4.69 ± 0.26 ab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.-T.; Yang, M.-Y.; Lee, Y.-J.; Yang, T.-W.; Wang, C.-C.; Wang, C.-J. Gallic Acid Improves Diabetic Steatosis by Downregulating MicroRNA-34a-5p through Targeting NFE2L2 Expression in High-Fat Diet-Fed db/db Mice. Antioxidants 2022, 11, 92. https://doi.org/10.3390/antiox11010092
Lee A-T, Yang M-Y, Lee Y-J, Yang T-W, Wang C-C, Wang C-J. Gallic Acid Improves Diabetic Steatosis by Downregulating MicroRNA-34a-5p through Targeting NFE2L2 Expression in High-Fat Diet-Fed db/db Mice. Antioxidants. 2022; 11(1):92. https://doi.org/10.3390/antiox11010092
Chicago/Turabian StyleLee, Ang-Tse, Mon-Yuan Yang, Yi-Ju Lee, Tzu-Wei Yang, Chi-Chih Wang, and Chau-Jong Wang. 2022. "Gallic Acid Improves Diabetic Steatosis by Downregulating MicroRNA-34a-5p through Targeting NFE2L2 Expression in High-Fat Diet-Fed db/db Mice" Antioxidants 11, no. 1: 92. https://doi.org/10.3390/antiox11010092
APA StyleLee, A.-T., Yang, M.-Y., Lee, Y.-J., Yang, T.-W., Wang, C.-C., & Wang, C.-J. (2022). Gallic Acid Improves Diabetic Steatosis by Downregulating MicroRNA-34a-5p through Targeting NFE2L2 Expression in High-Fat Diet-Fed db/db Mice. Antioxidants, 11(1), 92. https://doi.org/10.3390/antiox11010092