
Low-Dose Dioxin Reduced Glucose Uptake in C2C12 Myocytes: The Role of Mitochondrial Oxidative Stress and Insulin-Dependent Calcium Mobilization


Abstract
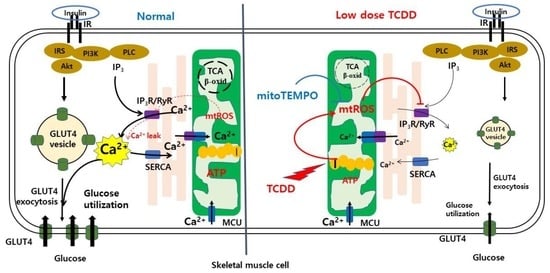
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Plasmids and Transfection
2.3. Enzymatic Assay for 2-Deoxyglucose Uptake
2.4. Assays for GLUT4 Translocation
2.5. Western Blot
2.6. Real-Time Quantitative RT-PCR
2.7. Assays for Mitochondrial Activity
2.8. Measurement of Oxygen Consumption Rate
2.9. OXPHOS Complex I Activity
2.10. Calcium Monitoring
2.11. Measurement of Glucose Uptake in Mouse Muscles
2.12. Statistical Analysis
3. Results
3.1. Inhibition of Insulin-Triggered Glucose Uptake by TCDD
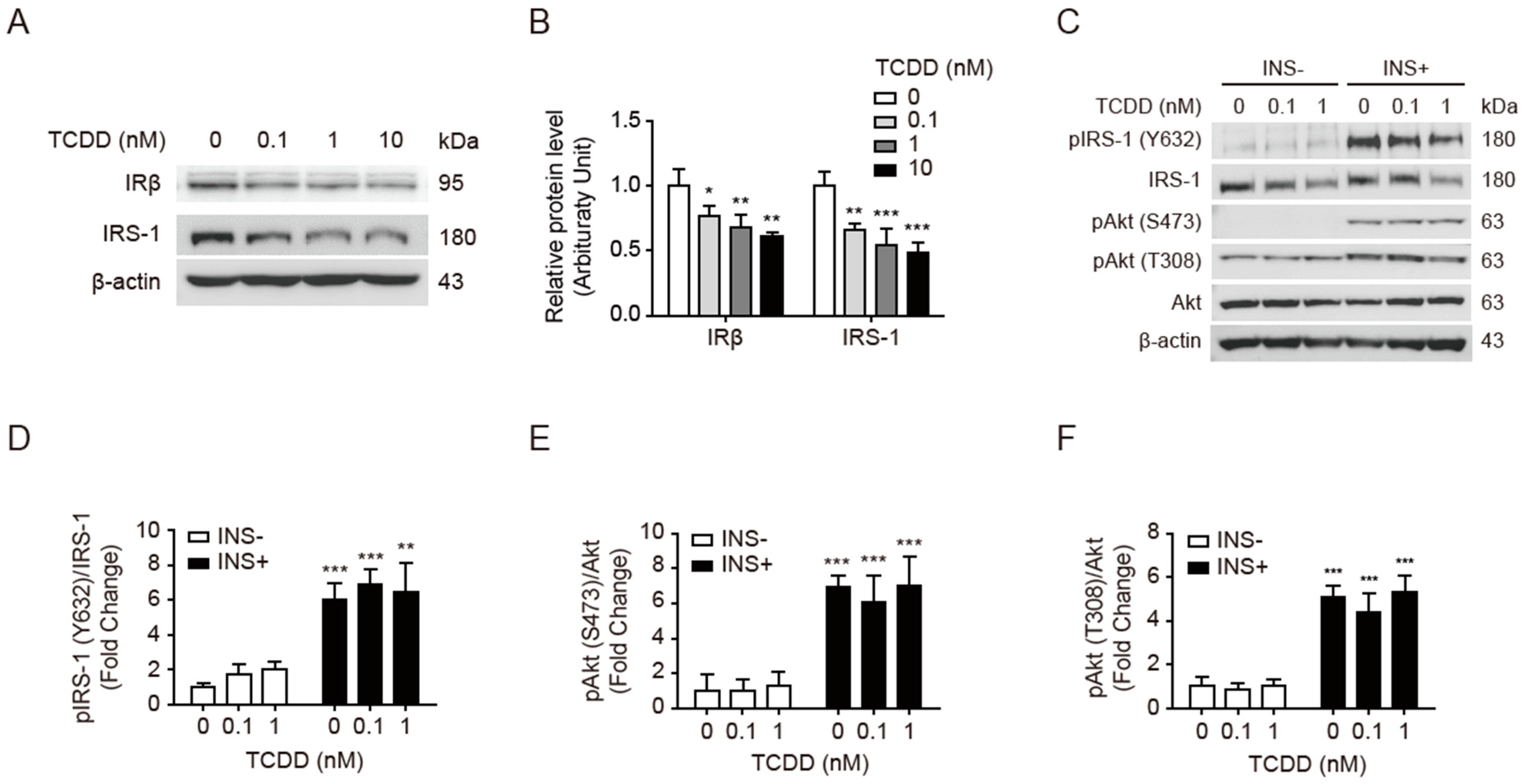
3.2. Low-Dose TCDD Reduced IRβ and IRS-1 Protein Expression without Affecting Akt Phosphorylation
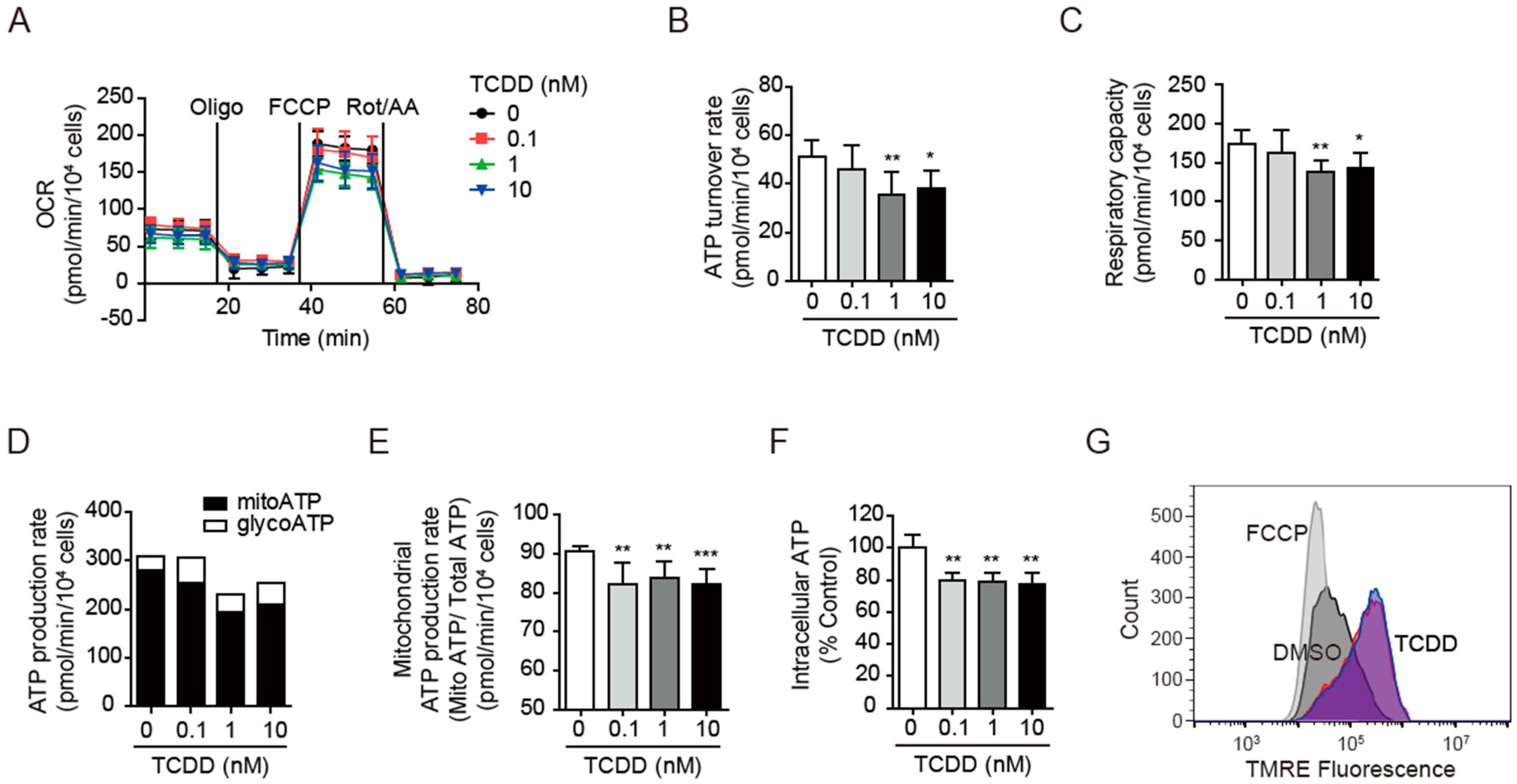
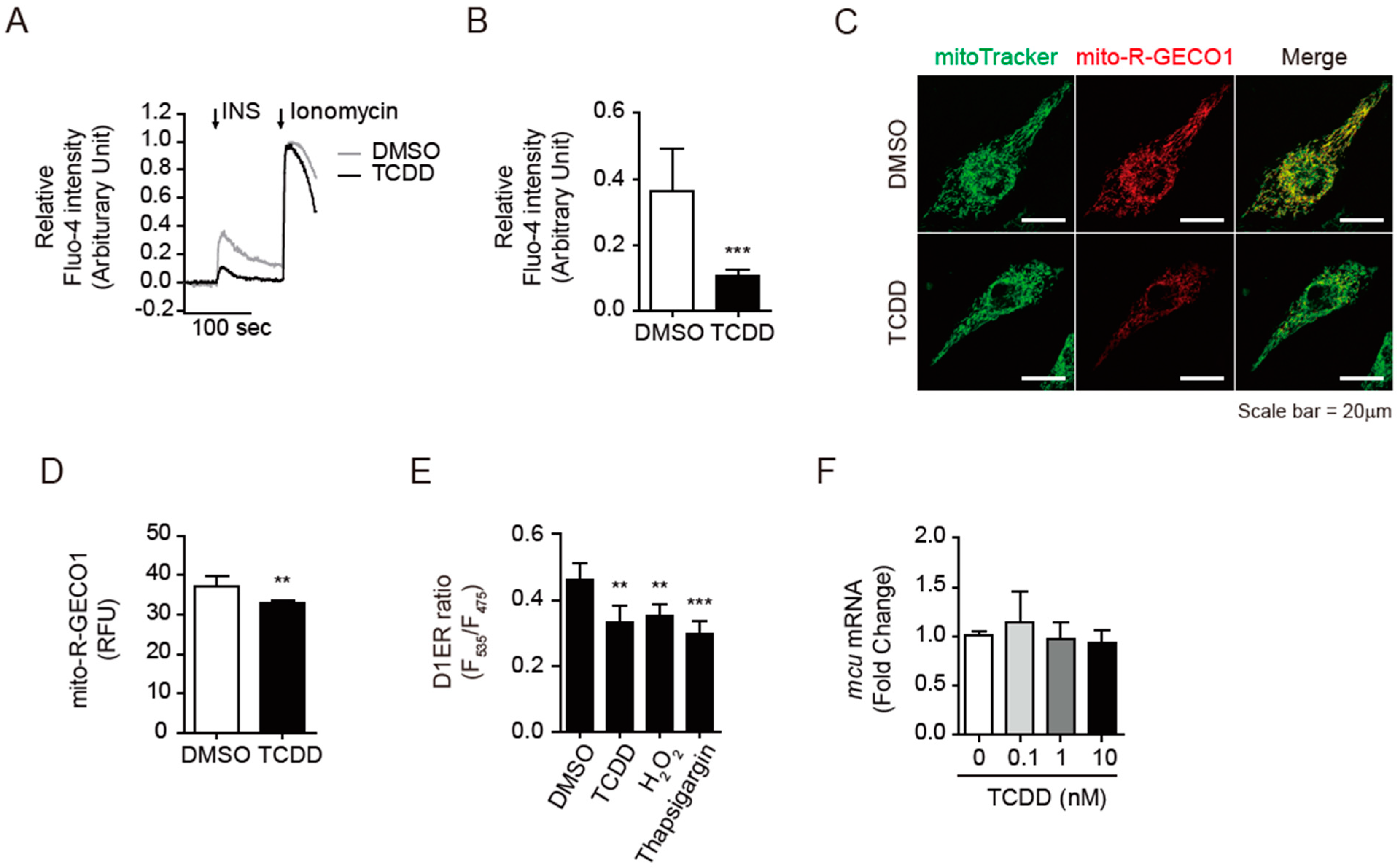
3.3. TCDD Impaired Mitochondrial Activity
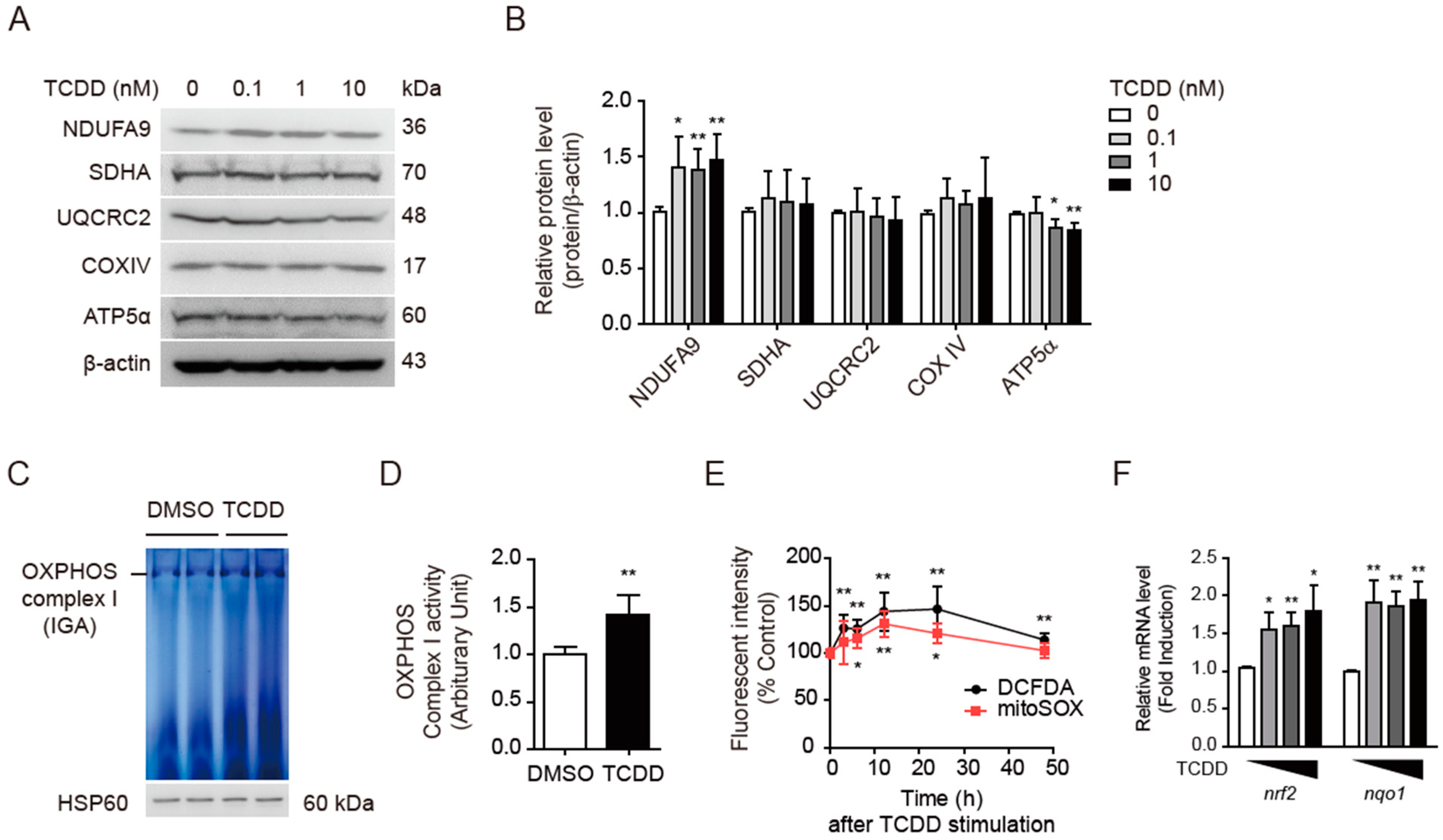
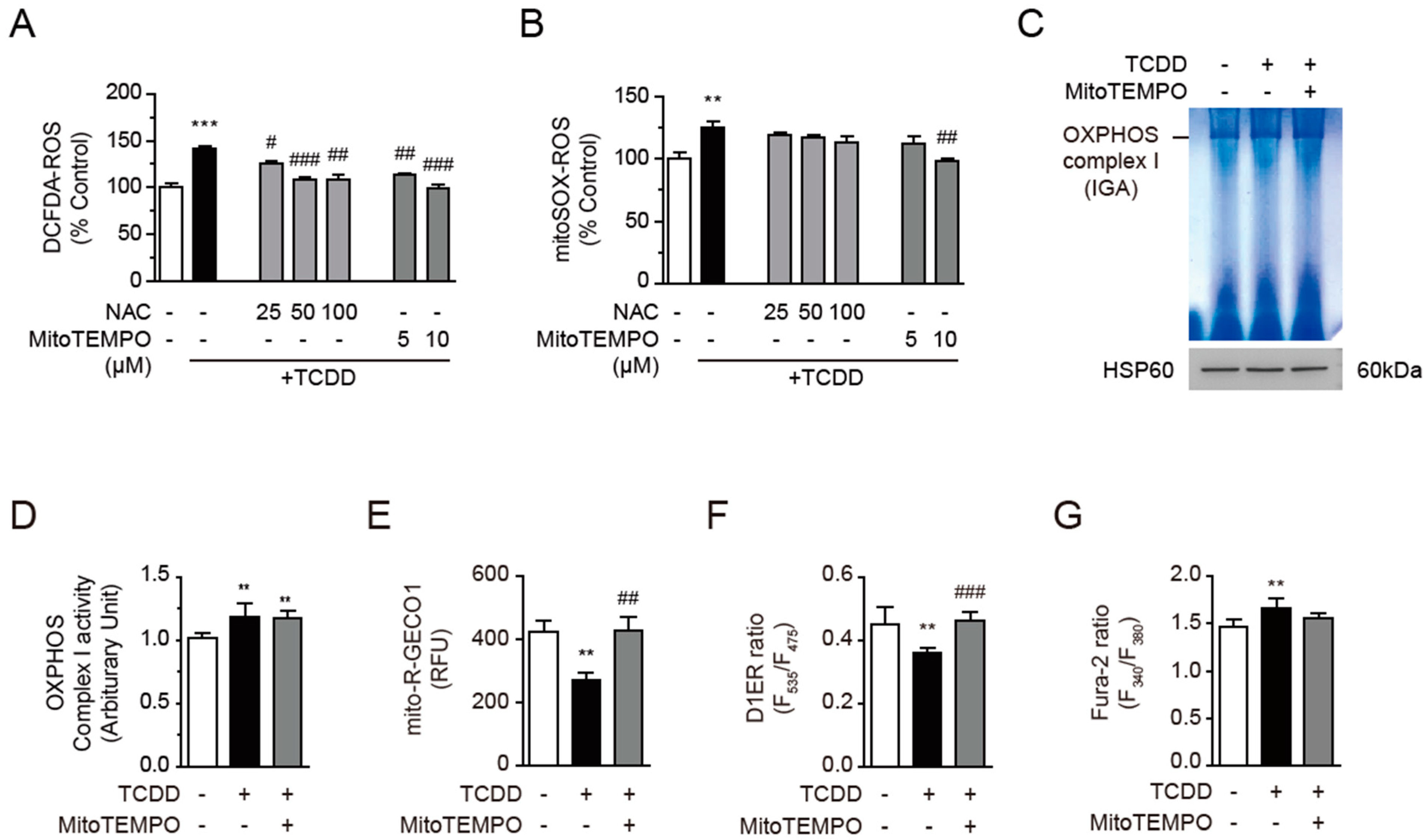
3.4. TCDD Increased OXPHOS Complex I Activity and Mitochondrial ROS
3.5. TCDD Disturbed the Insulin-Triggered Calcium Release into Cytosol
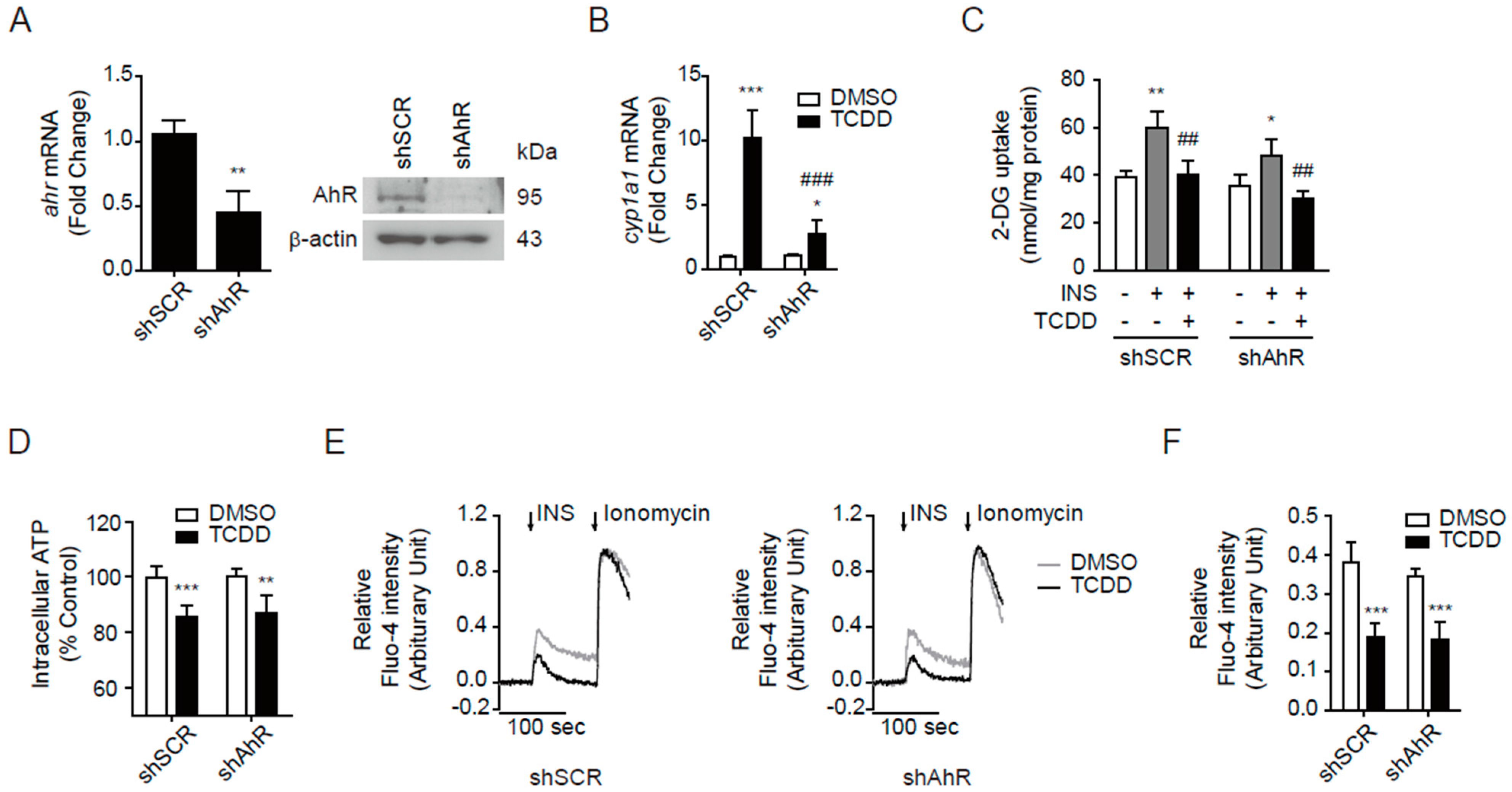
3.6. AhR-Independent Effects of TCDD in Skeletal Muscle
3.7. Calcium Dysregulation Resulted from Mitochondrial ROS
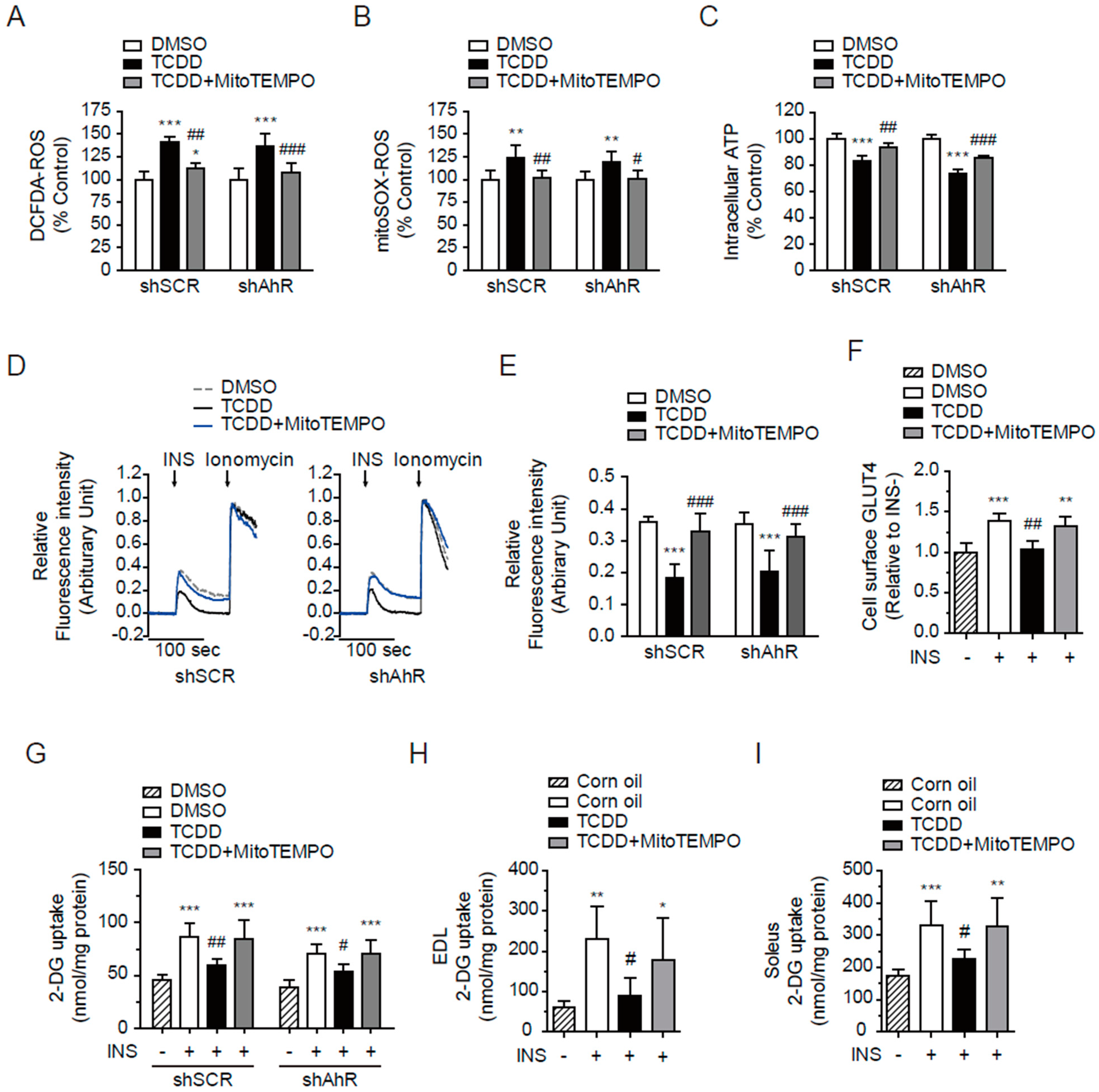
3.8. MitoTEMPO Protected from TCDD-Induced Insulin Resistance in the Absence of AhR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, D.H.; Lee, I.K.; Jin, S.H.; Steffes, M.; Jacobs, D.R., Jr. Association between serum concentrations of persistent organic pollutants and insulin resistance among nondiabetic adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetes Care 2007, 30, 622–628. [Google Scholar] [CrossRef] [Green Version]
- Lind, P.M.; Riserus, U.; Salihovic, S.; Bavel, B.; Lind, L. An environmental wide association study (EWAS) approach to the metabolic syndrome. Environ. Int. 2013, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sargis, R.M.; Simmons, R.A. Environmental neglect: Endocrine disruptors as underappreciated but potentially modifiable diabetes risk factors. Diabetologia 2019, 62, 1811–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.; Lyche, J.L.; Polder, A.; Aaseth, J.; Skaug, M.A. Increased blood levels of persistent organic pollutants (POP) in obese individuals after weight loss-A review. J. Toxicol. Environ. Health B Crit. Rev. 2017, 20, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Heindel, J.J.; Newbold, R.; Schug, T.T. Endocrine disruptors and obesity. Nat. Rev. Endocrinol. 2015, 11, 653–661. [Google Scholar] [CrossRef]
- Lee, D.H.; Porta, M.; Jacobs, D.R., Jr.; Vandenberg, L.N. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr. Rev. 2014, 35, 557–601. [Google Scholar] [CrossRef] [Green Version]
- Neel, B.A.; Sargis, R.M. The paradox of progress: Environmental disruption of metabolism and the diabetes epidemic. Diabetes 2011, 60, 1838–1848. [Google Scholar] [CrossRef] [Green Version]
- Ruzzin, J.; Petersen, R.; Meugnier, E.; Madsen, L.; Lock, E.J.; Lillefosse, H.; Ma, T.; Pesenti, S.; Sonne, S.B.; Marstrand, T.T.; et al. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ. Health Perspect. 2010, 118, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiumi, S.; Yoshida, M.; Azuma, T.; Yoshida, K.; Ashida, H. 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs an insulin signaling pathway through the induction of tumor necrosis factor-alpha in adipocytes. Toxicol. Sci. 2010, 115, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grun, F.; Blumberg, B. Endocrine disrupters as obesogens. Mol. Cell Endocrinol. 2009, 304, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Park, W.H.; Kang, Y.C.; Kang, S.; Im, S.; Park, S.; Kim, J.T.; Lee, M.; Seok, J.; Oh, M.S.; et al. Serum biomarkers from cell-based assays for AhRL and MIS strongly predicted the future development of diabetes in a large community-based prospective study in Korea. Sci. Rep. 2020, 10, 6339. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K. Mitochondrial dysfunction and insulin resistance: The contribution of dioxin-like substances. Diabetes Metab. J. 2011, 35, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Feldman, E.L. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp. Mol. Med. 2015, 47, e149. [Google Scholar] [CrossRef] [Green Version]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C.; American Heart, A.; National Heart, L.; Blood, I. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141. [Google Scholar] [CrossRef]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Choi, E.M.; Suh, K.S.; Rhee, S.Y.; Oh, S.; Woo, J.T.; Kim, S.W.; Kim, Y.S.; Pak, Y.K.; Chon, S. Perfluorooctanoic acid induces mitochondrial dysfunction in MC3T3-E1 osteoblast cells. J. Environ. Sci. Health Part A Tox. Hazard. Subst. Environ. Eng. 2017, 52, 281–289. [Google Scholar] [CrossRef]
- Suh, K.S.; Choi, E.M.; Kim, Y.J.; Hong, S.M.; Park, S.Y.; Rhee, S.Y.; Oh, S.; Kim, S.W.; Pak, Y.K.; Choe, W.; et al. Perfluorooctanoic acid induces oxidative damage and mitochondrial dysfunction in pancreatic beta-cells. Mol. Med. Rep. 2017, 15, 3871–3878. [Google Scholar] [CrossRef]
- Park, W.H.; Kang, S.; Lee, H.K.; Salihovic, S.; Bavel, B.V.; Lind, P.M.; Pak, Y.K.; Lind, L. Relationships between serum-induced AhR bioactivity or mitochondrial inhibition and circulating polychlorinated biphenyls (PCBs). Sci. Rep. 2017, 7, 9383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Monica Lind, P.; Salihovic, S.; van Bavel, B.; Lind, L.; Ingelsson, E. Influence of persistent organic pollutants on oxidative stress in population-based samples. Chemosphere 2014, 114, 303–309. [Google Scholar] [CrossRef]
- Artacho-Cordon, F.; Leon, J.; Saenz, J.M.; Fernandez, M.F.; Martin-Olmedo, P.; Olea, N.; Arrebola, J.P. Contribution of Persistent Organic Pollutant Exposure to the Adipose Tissue Oxidative Microenvironment in an Adult Cohort: A Multipollutant Approach. Environ. Sci. Technol. 2016, 50, 13529–13538. [Google Scholar] [CrossRef]
- Sargis, R.M. The hijacking of cellular signaling and the diabetes epidemic: Mechanisms of environmental disruption of insulin action and glucose homeostasis. Diabetes Metab. J. 2014, 38, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Toledo, F.G.; Goodpaster, B.H. The role of weight loss and exercise in correcting skeletal muscle mitochondrial abnormalities in obesity, diabetes and aging. Mol. Cell. Endocrinol. 2013, 379, 30–34. [Google Scholar] [CrossRef]
- Koliaki, C.; Roden, M. Alterations of Mitochondrial Function and Insulin Sensitivity in Human Obesity and Diabetes Mellitus. Annu. Rev. Nutr. 2016, 36, 337–367. [Google Scholar] [CrossRef]
- Contreras-Ferrat, A.; Lavandero, S.; Jaimovich, E.; Klip, A. Calcium signaling in insulin action on striated muscle. Cell Calcium 2014, 56, 390–396. [Google Scholar] [CrossRef]
- Lanner, J.T.; Bruton, J.D.; Katz, A.; Westerblad, H. Ca2+ and insulin-mediated glucose uptake. Curr. Opin. Pharmacol. 2008, 8, 339–345. [Google Scholar] [CrossRef]
- Contreras-Ferrat, A.; Llanos, P.; Vasquez, C.; Espinosa, A.; Osorio-Fuentealba, C.; Arias-Calderon, M.; Lavandero, S.; Klip, A.; Hidalgo, C.; Jaimovich, E. Insulin elicits a ROS-activated and an IP(3)-dependent Ca2+ release, which both impinge on GLUT4 translocation. J. Cell Sci. 2014, 127, 1911–1923. [Google Scholar] [CrossRef] [Green Version]
- Park, W.H.; Jun, D.W.; Kim, J.T.; Jeong, J.H.; Park, H.; Chang, Y.S.; Park, K.S.; Lee, H.K.; Pak, Y.K. Novel cell-based assay reveals associations of circulating serum AhR-ligands with metabolic syndrome and mitochondrial dysfunction. Biofactors 2013, 39, 494–504. [Google Scholar] [CrossRef]
- Yamamoto, N.; Sato, T.; Kawasaki, K.; Murosaki, S.; Yamamoto, Y. A nonradioisotope, enzymatic assay for 2-deoxyglucose uptake in L6 skeletal muscle cells cultured in a 96-well microplate. Anal. Biochem. 2006, 351, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, M.M.; Lim, C.Y.; Bi, X.; Robinson, R.C.; Han, W. Tmod3 Phosphorylation Mediates AMPK-Dependent GLUT4 Plasma Membrane Insertion in Myoblasts. Front. Endocrinol. (Lausanne) 2021, 12, 653557. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Khayat, Z.; Kishi, K.; Ebina, Y.; Klip, A. GLUT4 translocation by insulin in intact muscle cells: Detection by a fast and quantitative assay. FEBS Lett. 1998, 427, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Piao, Y.; Kang, Y.C.; Lim, S.; Pak, Y.K. Qi-activating quercetin alleviates mitochondrial dysfunction and neuroinflammation in vivo and in vitro. Arch. Pharmacal Res. 2020, 43, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Boytard, L.; Hadi, T.; Koelwyn, G.; Simon, R.; Ouimet, M.; Seifert, L.; Spiro, W.; Yan, B.; Hutchison, S.; et al. Enhanced glycolysis and HIF-1alpha activation in adipose tissue macrophages sustains local and systemic interleukin-1beta production in obesity. Sci. Rep. 2020, 10, 5555. [Google Scholar] [CrossRef] [Green Version]
- Giddings, E.L.; Champagne, D.P.; Wu, M.H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef]
- Gao, Y.; Bai, X.; Zhang, D.; Han, C.; Yuan, J.; Liu, W.; Cao, X.; Chen, Z.; Shangguan, F.; Zhu, Z.; et al. Mammalian elongation factor 4 regulates mitochondrial translation essential for spermatogenesis. Nat. Struct. Mol. Biol. 2016, 23, 441–449. [Google Scholar] [CrossRef]
- Ibarra, C.; Estrada, M.; Carrasco, L.; Chiong, M.; Liberona, J.L.; Cardenas, C.; Diaz-Araya, G.; Jaimovich, E.; Lavandero, S. Insulin-like growth factor-1 induces an inositol 1,4,5-trisphosphate-dependent increase in nuclear and cytosolic calcium in cultured rat cardiac myocytes. J. Biol. Chem. 2004, 279, 7554–7565. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, Y.; Kamiya, A.; Ishii, T.; Ishizuka, M.; Yamashita, Y.; Ashida, H. 5-Aminolevulinic acid combined with ferrous iron improves glucose tolerance in high-fat diet-fed mice via upregulation of glucose transporter 1. Exp. Ther. Med. 2021, 22, 1454. [Google Scholar] [CrossRef]
- Lim, C.Y.; Bi, X.; Wu, D.; Kim, J.B.; Gunning, P.W.; Hong, W.; Han, W. Tropomodulin3 is a novel Akt2 effector regulating insulin-stimulated GLUT4 exocytosis through cortical actin remodeling. Nat. Commun. 2015, 6, 5951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, J.; Schrauwen, P. Muscle mitochondria and insulin resistance: A human perspective. Trends Endocrinol. Metab. 2012, 23, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Forkink, M.; Manjeri, G.R.; Liemburg-Apers, D.C.; Nibbeling, E.; Blanchard, M.; Wojtala, A.; Smeitink, J.A.; Wieckowski, M.R.; Willems, P.H.; Koopman, W.J. Mitochondrial hyperpolarization during chronic complex I inhibition is sustained by low activity of complex II, III, IV and V. Biochim. Biophys. Acta 2014, 1837, 1247–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Harper, M.E. Mitochondrial proticity and ROS signaling: Lessons from the uncoupling proteins. Trends Endocrinol. Metab. 2012, 23, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Cui, W.; Klaassen, C.D. Nrf2 protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced oxidative injury and steatohepatitis. Toxicol. Appl. Pharmacol. 2011, 256, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Xu, C.; Lu, Y.; Hu, H.; Qin, B.; Wang, Y.; He, D.; Li, C.; Yu, X.; et al. MitoTEMPO Prevents Oxalate Induced Injury in NRK-52E Cells via Inhibiting Mitochondrial Dysfunction and Modulating Oxidative Stress. Oxidative Med. Cell. Longev. 2017, 2017, 7528090. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Kang, G.M.; Min, S.H.; Lee, C.H.; Kim, J.Y.; Lim, H.S.; Choi, M.J.; Jung, S.B.; Park, J.W.; Kim, S.; Park, C.B.; et al. Mitohormesis in Hypothalamic POMC Neurons Mediates Regular Exercise-Induced High-Turnover Metabolism. Cell Metab. 2021, 33, 334–349.e6. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Choi, Y.S.; Koo, H.J.; Jeong, J.H.; Park, W.H.; Kim, M.; Piao, Y.; Pak, Y.K. Mitochondrial dysfunction enhances the migration of vascular smooth muscles cells via suppression of Akt phosphorylation. Biochim. Biophys. Acta 2010, 1800, 275–281. [Google Scholar] [CrossRef]
- Lanner, J.T.; Katz, A.; Tavi, P.; Sandstrom, M.E.; Zhang, S.J.; Wretman, C.; James, S.; Fauconnier, J.; Lannergren, J.; Bruton, J.D.; et al. The role of Ca2+ influx for insulin-mediated glucose uptake in skeletal muscle. Diabetes 2006, 55, 2077–2083. [Google Scholar] [CrossRef] [Green Version]
- Park, D.R.; Park, K.H.; Kim, B.J.; Yoon, C.S.; Kim, U.H. Exercise ameliorates insulin resistance via Ca2+ signals distinct from those of insulin for GLUT4 translocation in skeletal muscles. Diabetes 2015, 64, 1224–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef]
- Biswas, G.; Srinivasan, S.; Anandatheerthavarada, H.K.; Avadhani, N.G. Dioxin-mediated tumor progression through activation of mitochondria-to-nucleus stress signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Hernandez, A.; Sanchez-Martin, F.J.; Hortigon-Vinagre, M.P.; Henao, F.; Merino, J.M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptosis by disruption of intracellular calcium homeostasis in human neuronal cell line SHSY5Y. Apoptosis 2012, 17, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Piaggi, S.; Novelli, M.; Martino, L.; Masini, M.; Raggi, C.; Orciuolo, E.; Masiello, P.; Casini, A.; De Tata, V. Cell death and impairment of glucose-stimulated insulin secretion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the beta-cell line INS-1E. Toxicol. Appl. Pharmacol. 2007, 220, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Mongillo, M.; Marks, A.R. Stressed out: The skeletal muscle ryanodine receptor as a target of stress. J. Clin. Investig. 2008, 118, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aracena-Parks, P.; Goonasekera, S.A.; Gilman, C.P.; Dirksen, R.T.; Hidalgo, C.; Hamilton, S.L. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 2006, 281, 40354–40368. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox. Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Dolwick, K.M.; Schmidt, J.V.; Carver, L.A.; Swanson, H.I.; Bradfield, C.A. Cloning and expression of a human Ah receptor cDNA. Mol. Pharmacol. 1993, 44, 911–917. [Google Scholar] [PubMed]
- Li, W.; Donat, S.; Dohr, O.; Unfried, K.; Abel, J. Ah receptor in different tissues of C57BL/6J and DBA/2J mice: Use of competitive polymerase chain reaction to measure Ah-receptor mRNA expression. Arch. Biochem. Biophys. 1994, 315, 279–284. [Google Scholar] [CrossRef]
- Xie, H.Q.; Xia, Y.; Xu, T.; Chen, Y.; Fu, H.; Li, Y.; Luo, Y.; Xu, L.; Tsim, K.W.K.; Zhao, B. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces alterations in myogenic differentiation of C2C12 cells. Environ. Pollut. 2018, 235, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Shertzer, H.G.; Genter, M.B.; Shen, D.; Nebert, D.W.; Chen, Y.; Dalton, T.P. TCDD decreases ATP levels and increases reactive oxygen production through changes in mitochondrial F(0)F(1)-ATP synthase and ubiquinone. Toxicol. Appl. Pharmacol. 2006, 217, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Tappenden, D.M.; Lynn, S.G.; Crawford, R.B.; Lee, K.; Vengellur, A.; Kaminski, N.E.; Thomas, R.S.; LaPres, J.J. The aryl hydrocarbon receptor interacts with ATP5alpha1, a subunit of the ATP synthase complex, and modulates mitochondrial function. Toxicol. Appl. Pharmacol. 2011, 254, 299–310. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Accession Number | Forward Sequence (5′→3′) | Reverse Sequence (5′→3′) |
|---|---|---|---|
| AHR | NM_013464 | GTGTGCAGTTGGACTTCCCT | TGGCTGGCACTGATACATGG |
| CYP1A1 | NM_009992 | TCCGGCATTCATCCTTCGTC | ACAGTTCCCGGTCATGGTTA |
| MCU | NM_001033259 | TGATGACGTGACGGTGGTTT | CGAACGCCATCTGGTGAGTA |
| PKM2 | NM_011099 | AGCACCTGATTGCCCGAGAG | GTGAGCACGATAATGGCCCC |
| LDHA | NM_010699 | AATGAAGGACTTGGCGGATG | ATGACCAGCTTGGAGTTCGC |
| LDHB | NM_008492 | TGGTGGACAGTGCCTATGAAG | CATTGAGGATGCACGGGAGA |
| NRF2 | NM_010902 | 5′-TTCTCCGCTGCTCGGACTA | ATGTCTTGCCTCCAAAGGATGT |
| NQO1 | NM_008706 | TAGCCTGTAGCCAGCCCTAA | GCCTCCTTCATGGCGTAGTT |
| 18S rRNA | NR_003278 | GAGCGAAAGCATTTGCCAAG | GGCATCGTTTATGGTCGGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Im, S.; Kang, S.; Kim, J.H.; Oh, S.J.; Pak, Y.K. Low-Dose Dioxin Reduced Glucose Uptake in C2C12 Myocytes: The Role of Mitochondrial Oxidative Stress and Insulin-Dependent Calcium Mobilization. Antioxidants 2022, 11, 2109. https://doi.org/10.3390/antiox11112109
Im S, Kang S, Kim JH, Oh SJ, Pak YK. Low-Dose Dioxin Reduced Glucose Uptake in C2C12 Myocytes: The Role of Mitochondrial Oxidative Stress and Insulin-Dependent Calcium Mobilization. Antioxidants. 2022; 11(11):2109. https://doi.org/10.3390/antiox11112109
Chicago/Turabian StyleIm, Suyeol, Sora Kang, Ji Hwan Kim, Seung Jun Oh, and Youngmi Kim Pak. 2022. "Low-Dose Dioxin Reduced Glucose Uptake in C2C12 Myocytes: The Role of Mitochondrial Oxidative Stress and Insulin-Dependent Calcium Mobilization" Antioxidants 11, no. 11: 2109. https://doi.org/10.3390/antiox11112109
APA StyleIm, S., Kang, S., Kim, J. H., Oh, S. J., & Pak, Y. K. (2022). Low-Dose Dioxin Reduced Glucose Uptake in C2C12 Myocytes: The Role of Mitochondrial Oxidative Stress and Insulin-Dependent Calcium Mobilization. Antioxidants, 11(11), 2109. https://doi.org/10.3390/antiox11112109