

Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts


 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fibroblasts Cultures
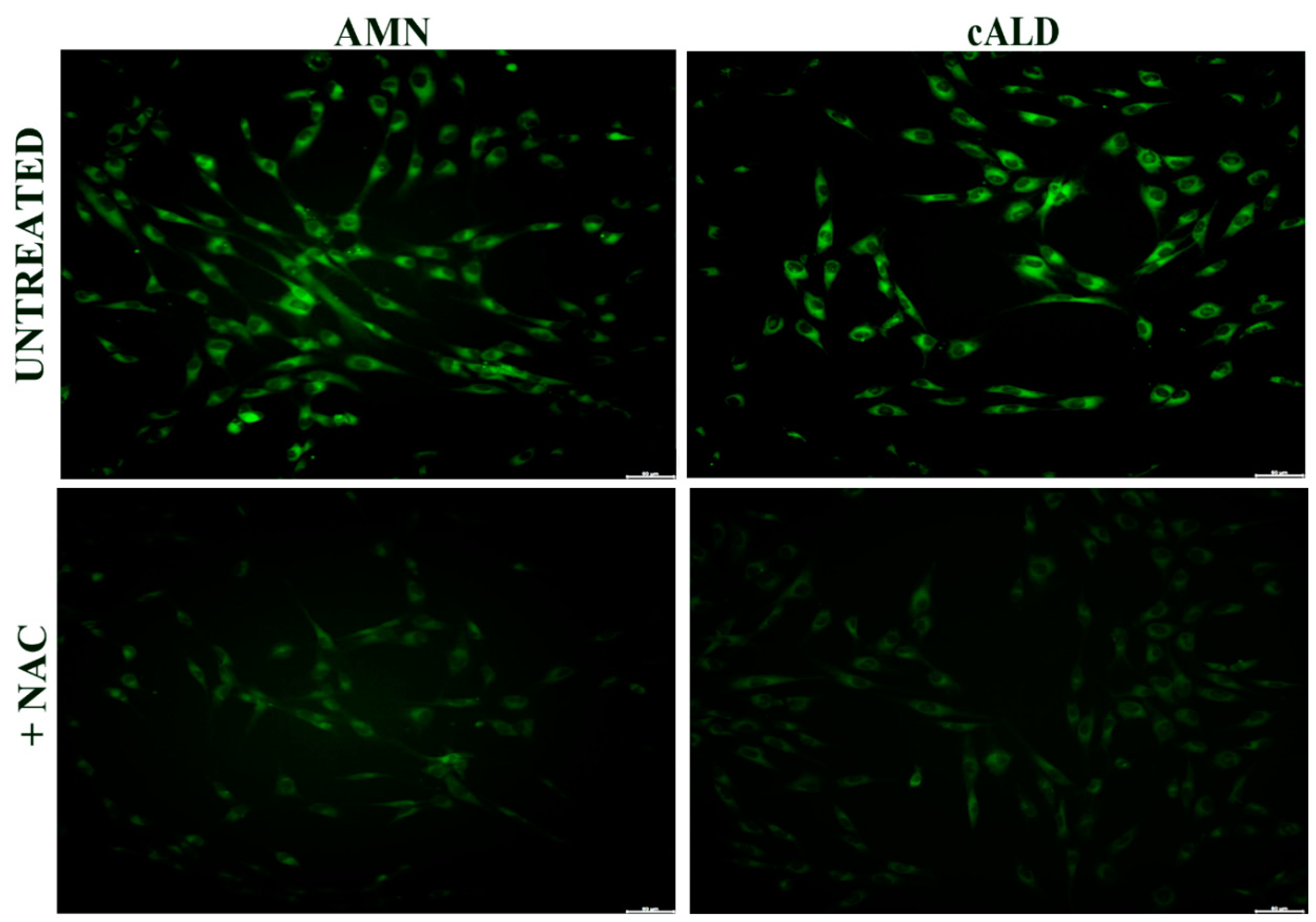
2.2. Assessment of Lipid Peroxidation by C11-Bodipy (581/591) Fluorescent Staining
2.3. Quantitative Real Time PCR (qRT-PCR)
2.4. Western Blot Analysis
2.5. NQO1 Enzyme Activity Assay
2.6. GPX4 Enzyme Activity Assay
2.7. GSH Assay
2.8. Statistical Analysis
3. Results
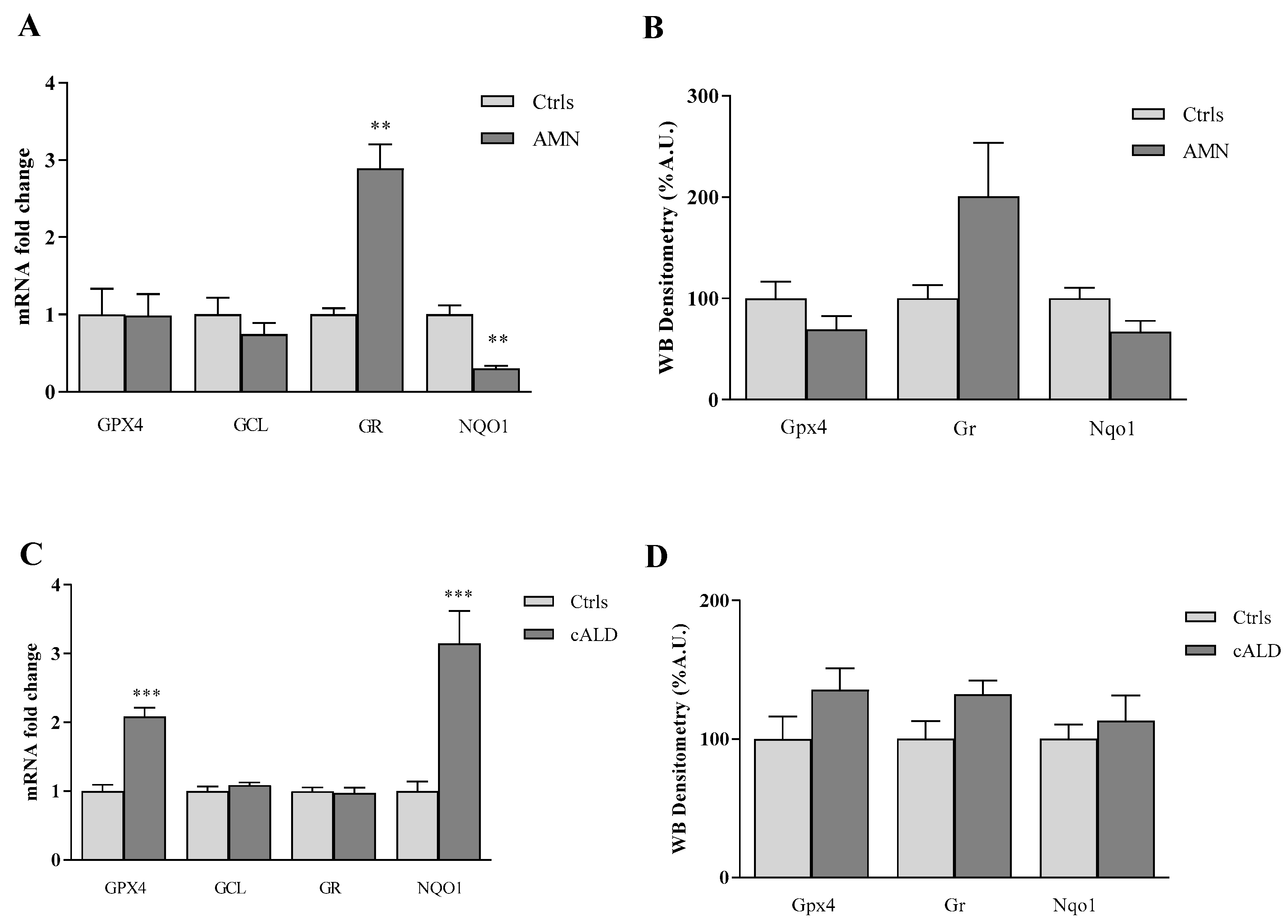
3.1. The Antioxidant Response Is Differently Modulated in Fibroblasts of Patients with AMN and cALD
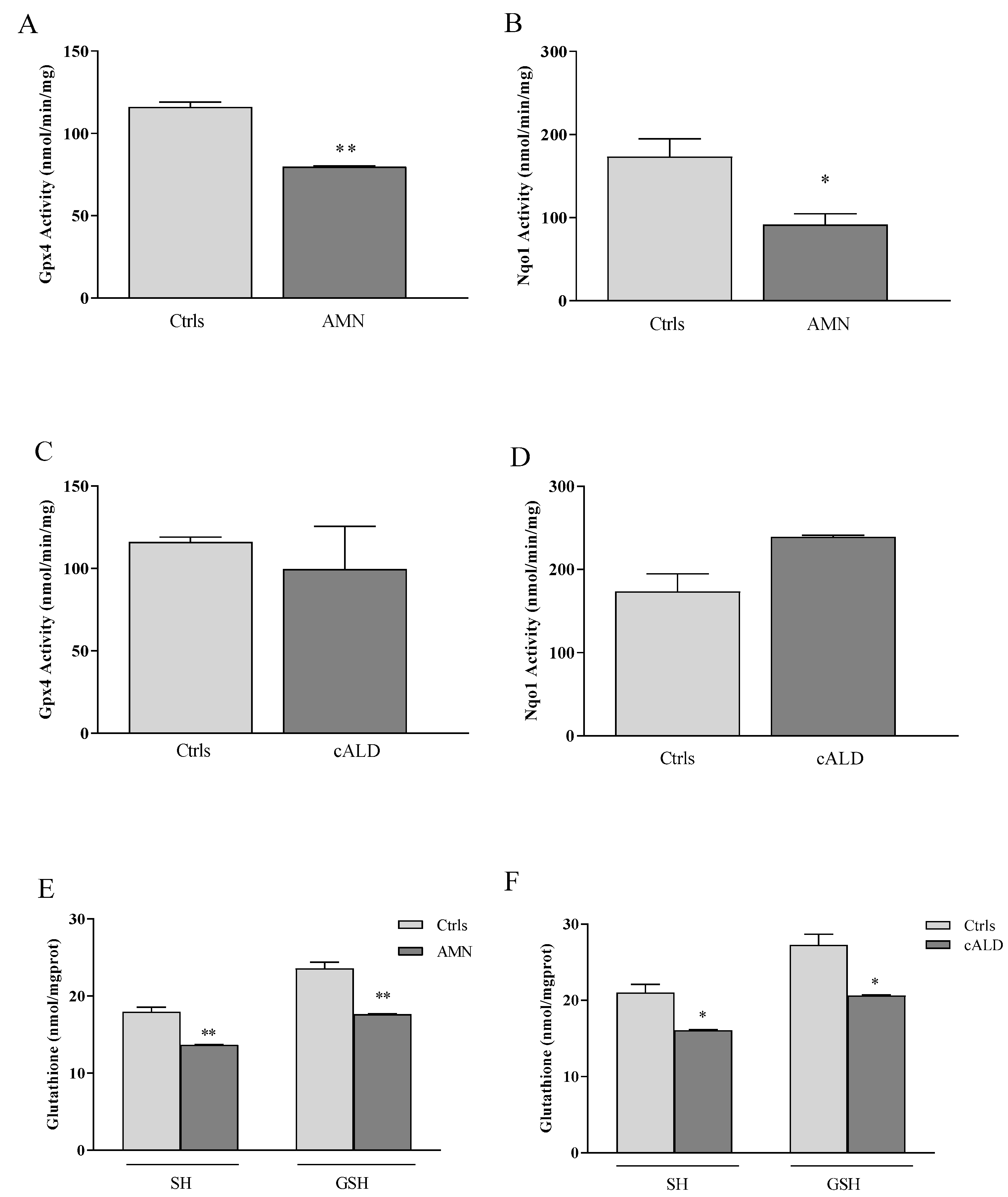
3.2. GPX4, NQO1 and GSH: A Pathological Triad in X-ALD?
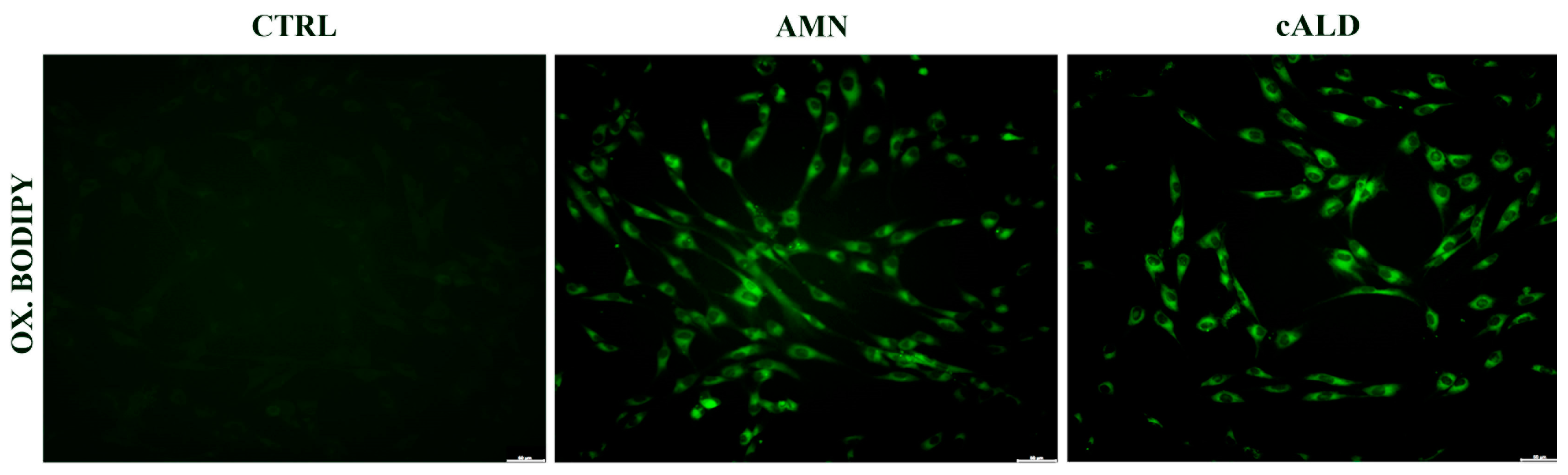
3.3. Lipid Peroxidation Increases in Patients with X-ALD
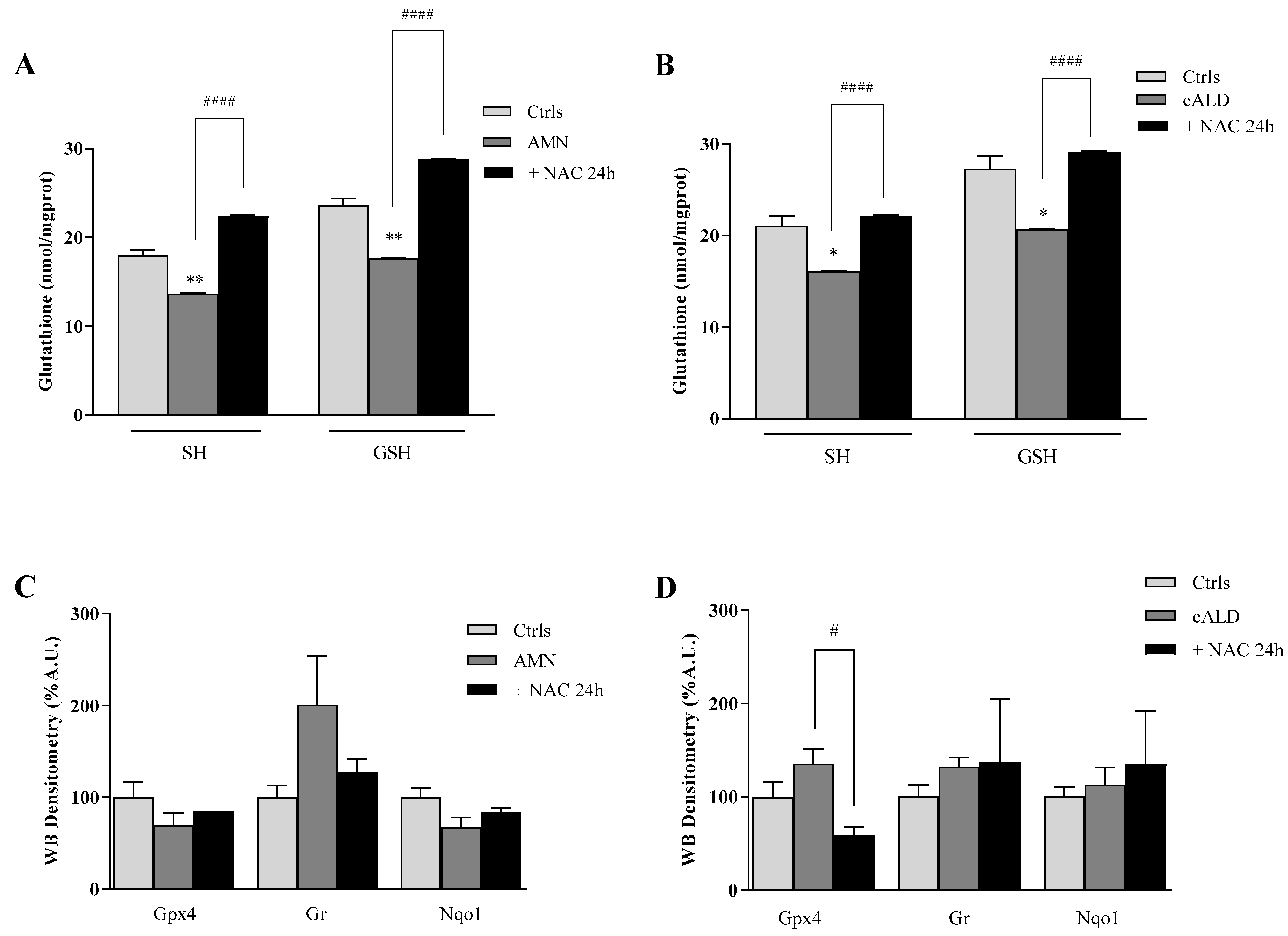
3.4. The Effect of NAC on the Antioxidant Response in Fibroblasts of Patients with X-ALD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fourcade, S.; Lopez-Erauskin, J.; Galino, J.; Duval, C.; Naudi, A.; Jove, M.; Kemp, S.; Villarroya, F.; Ferrer, I.; Pamplona, R.; et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet. 2008, 1, 1762–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Erauskin, J.; Fourcade, S.; Galino, J.; Ruiz, M.; Schlüter, A.; Naudi, A.; Jove, M.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann. Neurol. 2011, 70, 84–92. [Google Scholar] [CrossRef] [PubMed]
- López-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.M.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martínez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrillo, S.; Piemonte, F.; Pastore, A.; Tozzi, G.; Aiello, C.; Pujol, A.; Cappa, M.; Bertini, E. Glutathione imbalance in patients with X-linked adrenoleukodystrophy. Mol. Genet. Metab. 2013, 109, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Deon, M.; Marchetti, D.P.; Donida, B.; Wajner, M.; Vargas, C. Oxidative Stress in Patients with X-Linked Adrenoleukodystrophy. Cell Mol. Neurobiol. 2016, 36, 497–512. [Google Scholar] [CrossRef]
- Yu, J.; Chen, T.; Guo, X.; Zafar, M.I.; Li, H.; Wang, Z.; Zheng, J. The Role of Oxidative Stress and Inflammation in X-Link Adrenoleukodystrophy. Front. Nutr. 2022, 9, 864358. [Google Scholar] [CrossRef]
- van Roermund, C.W.; Visser, W.F.; Ijlst, L.; van Cruchten, A.; Boek, M.; Kulik, W.; Waterham, H.R.; Wanders, R.J. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008, 22, 4201–4208. [Google Scholar] [CrossRef] [Green Version]
- Wiesinger, C.; Kunze, M.; Regelsberger, G.; Forss-Petter, S.; Berger, J. Impaired very long-chain acyl-CoA β-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J. Biol. Chem. 2013, 288, 19269–19279. [Google Scholar] [CrossRef] [Green Version]
- Forss-Petter, S.; Werner, H.; Berger, J.; Lassmann, H.; Molzer, B.; Schwab, M.H.; Bernheimer, H.; Zimmermann, F.; Nave, K.A. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosc. Res. 1997, 50, 829–843. [Google Scholar] [CrossRef]
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2018, 40, 382–395. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Kang, R.; Yang, M.; Tang, D. Lipid Metabolism in Ferroptosis. Adv. Biol. 2021, 20, e2100396. [Google Scholar] [CrossRef]
- Vargas, C.R.; Wajner, M.; Sirtori, L.R.; Goulart, L.; Chiochetta, M.; Coelho, D.; Latini, A.; Llesuy, S.; Bello-Klein, A.; Giugliani, R.; et al. Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochim. Biophys. Acta 2004, 1688, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Powers, J.M.; Pei, Z.; Heinzer, A.K.; Deering, R.; Moser, A.B.; Moser, H.W.; Watkins, P.A.; Smith, K.D. Adreno-leukodystrophy: Oxidative stress of mice and men. J. Neuropathol. Exp. Neurol. 2005, 64, 1067–1079. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Schonfeld, P.; Kahlert, S.; Reiser, G. Toxic effects of X-linked adrenoleukodystrophy associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum. Mol. Genet. 2008, 17, 1750–1761. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, S.; Ferrer, I.; Pujol, A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: A paradigm for axonal degeneration. Free Radic. Biol. Med. 2015, 88, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Moser, H.W.; Moser, A.B.; Kawamura, N.; Murphy, J.; Suzuki, K.; Schaumburg, H.; Kishimoto, Y. Adrenoleukodystrophy: Elevated C26 fatty acid in cultured skin fibroblasts. Ann. Neurol. 1980, 7, 542–549. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Galino, J.; Bianchi, P.; Fourcade, S.; Andreu, A.L.; Ferrer, I.; Muñoz-Pinedo, C.; Pujol, A. Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain 2012, 135, 3584–3598. [Google Scholar] [CrossRef] [Green Version]
- Launay, N.; Aguado, C.; Fourcade, S.; Ruiz, M.; Grau, L.; Riera, J.; Guilera, C.; Giròs, M.; Ferrer, I.; Knecht, E.; et al. Autophagy induction halts axonal degeneration in a mouse model of X-adrenoleukodystrophy. Acta Neuropathol. 2015, 129, 399–415. [Google Scholar] [CrossRef] [Green Version]
- Launay, N.; Ruiz, M.; Grau, L.; Ortega, F.J.; Ilieva, E.V.; Martínez, J.J.; Galea, E.; Ferrer, I.; Knecht, E.; Pujol, A.; et al. Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 2017, 133, 283–301. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Casasnovas, C.; Ruiz, M.; Schlüter, A.; Naudí, A.; Fourcade, S.; Veciana, M.; Castañer, S.; Albertí, A.; Bargalló, N.; Johnson, M.; et al. Biomarker Identification, Safety, and Efficacy of High-Dose Antioxidants for Adrenomyeloneuropathy: A Phase II Pilot Study. Neurotherapeutics 2019, 16, 1167–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semeraro, M.; Rizzo, C.; Boenzi, S.; Cappa, M.; Bertini, E.; Antonetti, G.; Dionisi-Vici, C. A new multiplex method for the diagnosis of peroxisomal disorders allowing simultaneous determination of plasma very-long-chain fatty acids, phytanic, pristanic, docosahexaenoic and bile acids by high-performance liquid chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry. Clin. Chim. Acta 2016, 458, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummen, G.P.; van Liebergen, L.C.; Op den Kamp, J.A.; Post, J.A. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Galino, J.; Ruiz, M.; Fourcade, S.; Schlüter, A.; López-Erauskin, J.; Guilera, C.; Jove, M.; Naudi, A.; García-Arumí, E.; Andreu, A.L.; et al. Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X-adrenoleukodystrophy. Antioxid Redox Signal. 2011, 15, 2095–2107. [Google Scholar] [CrossRef]
- Doria, M.; Nury, T.; Delmas, D.; Moreau, T.; Lizard, G.; Vejux, A. Protective function of autophagy during VLCFA-induced cytotoxicity in a neurodegenerative cell model. Free Radic. Biol. Med. 2019, 137, 46–58. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow up and management. Orphanet J. Rare Dis. 2012, 13, 51. [Google Scholar] [CrossRef]
- Turk, B.R.; Theda, C.; Fatemi, A.; Moser, A.B. X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int. J. Dev. Neurosci. 2020, 80, 52–72. [Google Scholar] [CrossRef] [Green Version]
- Castellano, A.; Papinutto, N.; Cadioli, M.; Brugnara, G.; Iadanza, A.; Scigliuolo, G.; Pareyson, D.; Uziel, G.; Köhler, W.; Aubourg, P.; et al. Quantitative MRI of the spinal cord and brain in adrenomyeloneuropathy: In vivo assessment of structural changes. Brain 2016, 139, 1735–1746. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir Kutbay, N.; Ozbek, M.N.; Sarer Yurekli, B.; Demirbilek, H. A Distinct Clinical Phenotype in Two Siblings with X-linked Adrenoleukodystrophy. Neuro Endocrinol. Lett. 2019, 40, 36–40. [Google Scholar]
- Korenke, G.C.; Fuchs, S.; Krasemann, E.; Doerr, H.G.; Wilichowski, E.; Hunneman, D.H.; Hanefeld, F. Cerebral adrenoleukodystrophy (ALD) in only one of monozygotic twins with an identical ALD genotype. Ann. Neurol. 1996, 40, 254–257. [Google Scholar] [CrossRef]
- Di Rocco, M.; Doria-Lamba, L.; Caruso, U. Monozygotic twins with X-linked adrenoleukodystrophy and different phenotypes. Ann. Neurol. 2001, 50, 424. [Google Scholar] [CrossRef]
- Galea, E.; Launay, N.; Portero-Otin, M.; Ruiz, M.; Pamplona, R.; Aubourg, P.; Ferrer, I.; Pujol, A. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: A paradigm for multifactorial neurodegenerative diseases? Biochim. Biophys. Acta 2012, 1822, 1475–1488. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Singh, J.; Singh, I. Plasmalogen deficiency in cerebral adrenoleukodystrophy and its modulation by lovastatin. J. Neurochem. 2008, 106, 1766–1779. [Google Scholar] [CrossRef] [Green Version]
- Brites, P.; Mooyer, P.A.; El Mrabet, L.; Waterham, H.R.; Wanders, R.J. Plasmalogens participate in very-long-chain fatty acid-induced pathology. Brain 2009, 132, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Pujol, A. Pathomechanisms underlying X-adrenoleukodystrophy: A three-hit hypothesis. Brain Pathol. 2010, 20, 838–844. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. How the brain fights fatty acids’ toxicity. Neurochem. Int. 2021, 1, 105050. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, S.; Huffnagel, I.C.; Linthorst, G.E.; Wanders, R.J.; Engelen, M. Adrenoleukodystrophy-neuroendocrine pathogenesis and redefinition of natural history. Nat. Rev. Endocrinol. 2016, 12, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Miller, W.P.; Rothman, S.M.; Nascene, D.; Kivisto, T.; DeFor, T.E.; Ziegler, R.S.; Eisengart, J.; Leiser, K.; Raymond, G.; Lund, T.C.; et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: The largest single-institution cohort report. Blood 2011, 118, 1971–1978. [Google Scholar] [CrossRef] [Green Version]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Espinós, C.; Galindo, M.I.; García-Gimeno, M.A.; Ibáñez-Cabellos, J.S.; Martínez-Rubio, D.; Millán, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Kim, W.K.; Bae, K.H.; Lee, S.C.; Lee, E.W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age at Skin Biopsy (Yrs) | Mutation | C26:0 (µM) | C26:0/C22:0 (µM) |
|---|---|---|---|---|
| AMN | 56 | [c.1661G > A; p.Arg554His] | 2.62 | 0.062 |
| cALD #1 | 9 | [c.1817C > T; p.(S606L)] | 3.47 | 0.144 |
| cALD #2 | 3 | [c.1817C > T; p.(S606L)] | 3.20 | 0.094 |
| Controls (range) | --- | --- | 0.01–0.9 | 0.006–0.020 |
| Gene Expression (qRT-PCR) | Protein Amounts (WB) | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient | GPX4 (%) | GCL (%) | GR (%) | NQO1 (%) | Patient | Gpx4 (%) | Gr (%) | Nqo1 (%) |
| AMN | 98 | 74 | 290 | 30 | AMN | 69 | 201 | 67 |
| cALD #1 | 195 | 105 | 89 | 266 | cALD #1 | 121 | 122 | 95 |
| cALD #2 | 221 | 113 | 105 | 362 | cALD #2 | 151 | 142 | 131 |
| Ctrls | 100 | 100 | 100 | 100 | Ctrls | 100 | 100 | 100 |
| Patient | Gpx4 (%) | Nqo1 (%) | SH (%) | GSH (%) |
|---|---|---|---|---|
| AMN | 68 | 53 | 78 | 75 |
| cALD #1 | 64 | 136 | 76 | 76 |
| cALD #2 | 109 | 138 | 77 | 77 |
| Ctrls | 100 | 100 | 100 | 100 |
| SH (%) | GSH (%) | Gpx4 (%) | Gr (%) | Nqo1 (%) | |
|---|---|---|---|---|---|
| Patient | +NAC | +NAC | +NAC | +NAC | +NAC |
| AMN | 157 | 161 | 123 | 63 | 124 |
| cALD #1 | 138 | 141 | 40 | 57 | 82 |
| cALD #2 | 137 | 141 | 45 | 144 | 146 |
| Untreated | 100 | 100 | 100 | 100 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrillo, S.; D’Amico, J.; Nicita, F.; Torda, C.; Vasco, G.; Bertini, E.S.; Cappa, M.; Piemonte, F. Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts. Antioxidants 2022, 11, 2125. https://doi.org/10.3390/antiox11112125
Petrillo S, D’Amico J, Nicita F, Torda C, Vasco G, Bertini ES, Cappa M, Piemonte F. Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts. Antioxidants. 2022; 11(11):2125. https://doi.org/10.3390/antiox11112125
Chicago/Turabian StylePetrillo, Sara, Jessica D’Amico, Francesco Nicita, Caterina Torda, Gessica Vasco, Enrico S. Bertini, Marco Cappa, and Fiorella Piemonte. 2022. "Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts" Antioxidants 11, no. 11: 2125. https://doi.org/10.3390/antiox11112125
APA StylePetrillo, S., D’Amico, J., Nicita, F., Torda, C., Vasco, G., Bertini, E. S., Cappa, M., & Piemonte, F. (2022). Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts. Antioxidants, 11(11), 2125. https://doi.org/10.3390/antiox11112125