
New Players in Neuronal Iron Homeostasis: Insights from CRISPRi Studies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Iron Regulatory Element (IRE) Prediction
2.3. Pathways Analysis
2.4. Analysis and Figures
3. Results and Discussion
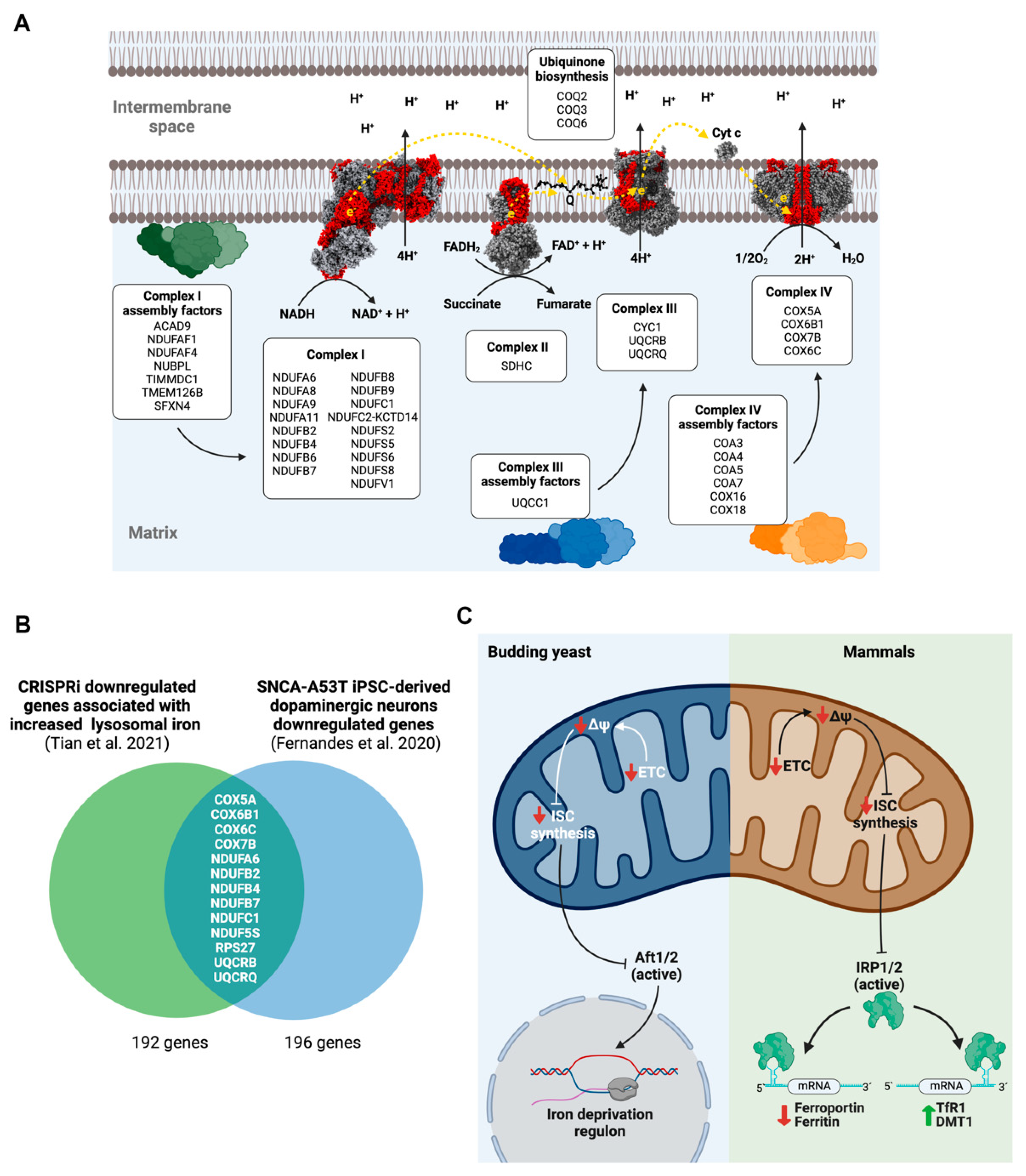
3.1. Mitochondrial Electron Transport Chain (ETC) Dysfunction Disturbs Neuronal Iron Homeostasis
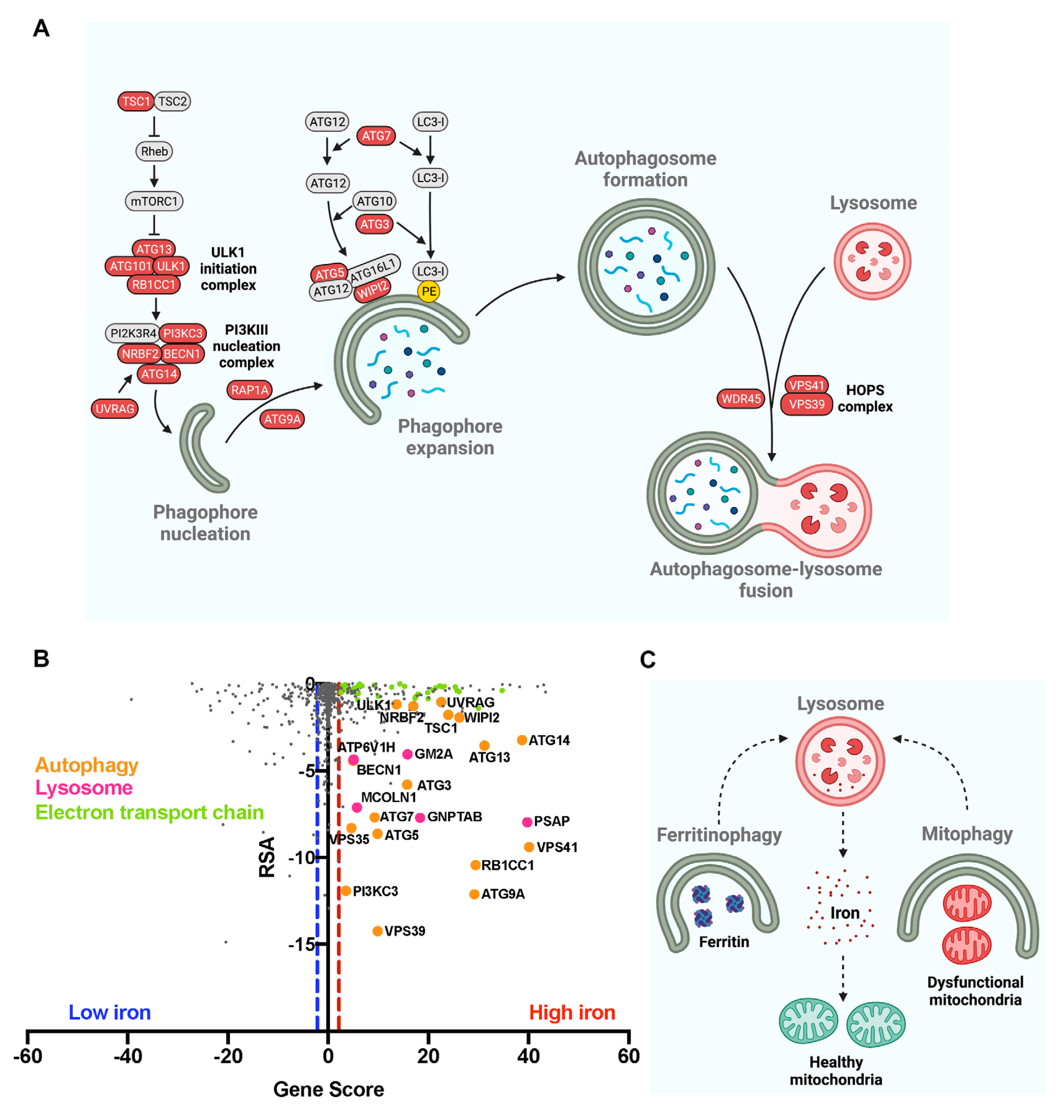
3.2. Autophagy Impairment Results in Lysosomal Iron Overload in Neurons
3.3. Perturbed Glycosylphosphatidylinositol (GPI) Synthesis and GPI-Anchored Protein Trafficking Increases Neuronal Iron Levels
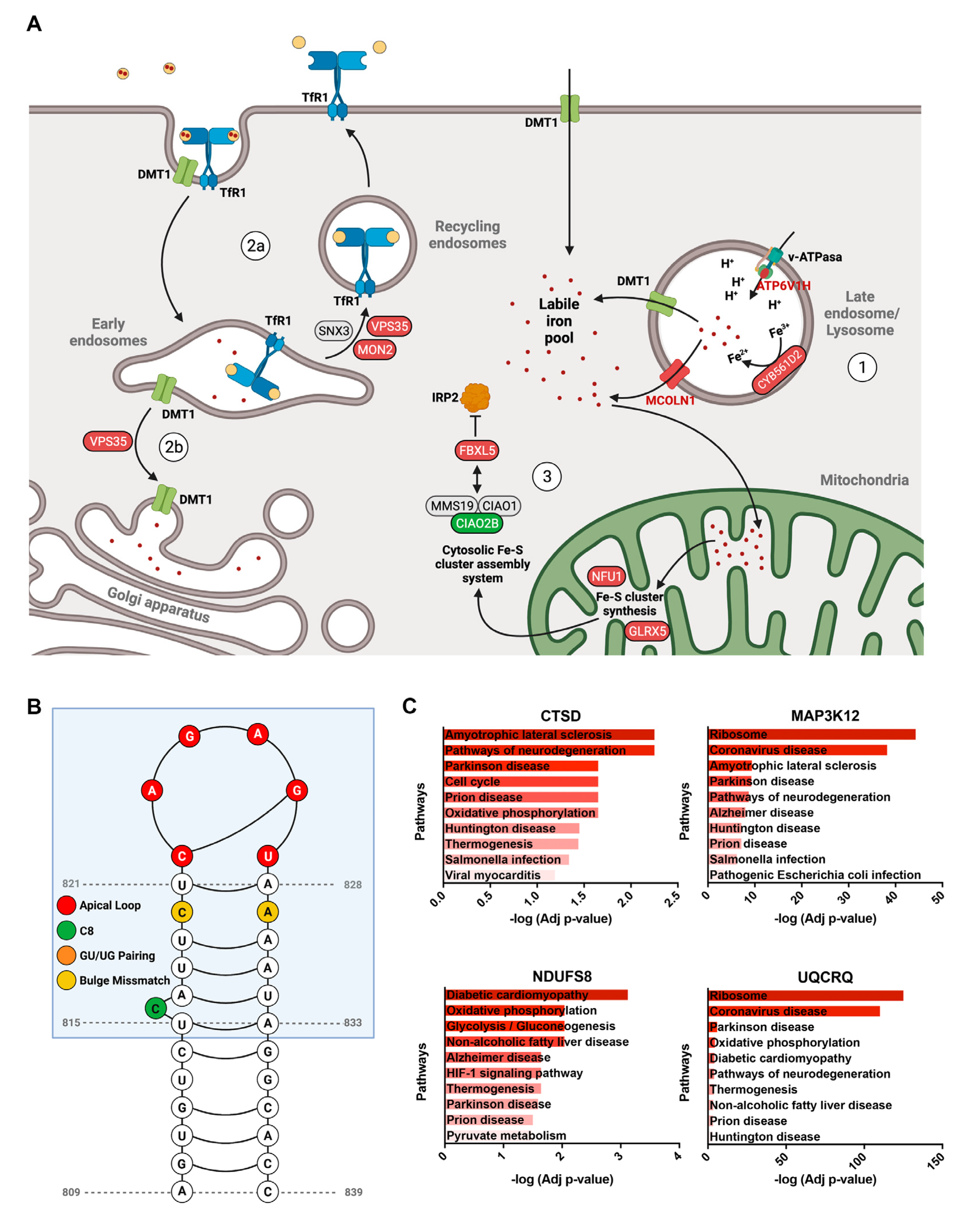
3.4. Poorly Characterized Proteins That Contribute to Maintain Iron Homeostasis in Neurons
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urrutia, P.J.; Borquez, D.A.; Nunez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Moller, H.E.; Bossoni, L.; Connor, J.R.; Crichton, R.R.; Does, M.D.; Ward, R.J.; Zecca, L.; Zucca, F.A.; Ronen, I. Iron, Myelin, and the Brain: Neuroimaging Meets Neurobiology. Trends Neurosci. 2019, 42, 384–401. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, B.; Zhu, J.; Yu, J.; Hui, J.; Xia, S.; Ji, J. Emerging Mechanisms and Targeted Therapy of Ferroptosis in Neurological Diseases and Neuro-oncology. Int. J. Biol. Sci. 2022, 18, 4260–4274. [Google Scholar] [CrossRef]
- Vitalakumar, D.; Sharma, A.; Flora, S.J.S. Ferroptosis: A potential therapeutic target for neurodegenerative diseases. J. Biochem. Mol. Toxicol. 2021, 35, e22830. [Google Scholar] [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron accumulates in Huntington’s disease neurons: Protection by deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef]
- Mechlovich, D.; Amit, T.; Bar-Am, O.; Mandel, S.; Youdim, M.B.; Weinreb, O. The novel multi-target iron chelator, M30 modulates HIF-1alpha-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer’s disease mice. Curr. Alzheimer Res. 2014, 11, 119–127. [Google Scholar] [CrossRef]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, Y.; Lei, Y.; Liu, T.; Wan, Z.; Meng, H.; Wang, H. Ferroptosis Is Regulated by Mitochondria in Neurodegenerative Diseases. Neurodegener. Dis. 2020, 20, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef]
- Tian, R.; Abarientos, A.; Hong, J.; Hashemi, S.H.; Yan, R.; Drager, N.; Leng, K.; Nalls, M.A.; Singleton, A.B.; Xu, K.; et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 2021, 24, 1020–1034. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Halcrow, P.W.; Kumar, N.; Afghah, Z.; Fischer, J.P.; Khan, N.; Chen, X.; Meucci, O.; Geiger, J.D. Heterogeneity of ferrous iron-containing endolysosomes and effects of endolysosome iron on endolysosome numbers, sizes, and localization patterns. J. Neurochem. 2022, 161, 69–83. [Google Scholar] [CrossRef]
- Tian, R.; Gachechiladze, M.A.; Ludwig, C.H.; Laurie, M.T.; Hong, J.Y.; Nathaniel, D.; Prabhu, A.V.; Fernandopulle, M.S.; Patel, R.; Abshari, M.; et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 2019, 104, 239–255.e12. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Patikas, N.; Foskolou, S.; Field, S.F.; Park, J.-E.; Byrne, M.L.; Bassett, A.R.; Metzakopian, E. Single-Cell Transcriptomics of Parkinson’s Disease Human In Vitro Models Reveals Dopamine Neuron-Specific Stress Responses. Cell Rep. 2020, 33, 108263. [Google Scholar] [CrossRef]
- Konig, R.; Chiang, C.-Y.; Tu, B.P.; Yan, S.F.; DeJesus, P.D.; Romero, A.; Bergauer, T.; Orth, A.; Krueger, U.; Zhou, Y.; et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 2007, 4, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.M.; Dowdle, W.E.; DeJesus, R.; Wang, Z.; Bergman, P.; Kobylarz, M.; Lindeman, A.; Xavier, R.J.; McAllister, G.; Nyfeler, B.; et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017, 20, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Cases, I.; Hentze, M.W.; Sanchez, M. SIREs: Searching for iron-responsive elements. Nucleic Acids Res. 2010, 38, W360–W367. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Li, H.; Zhao, H.; Hao, S.; Shang, L.; Wu, J.; Song, C.; Meyron-Holtz, E.G.; Qiao, T.; Li, K. Iron regulatory protein deficiency compromises mitochondrial function in murine embryonic fibroblasts. Sci. Rep. 2018, 8, 5118. [Google Scholar] [CrossRef]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 2015, 21, 311–323. [Google Scholar] [CrossRef]
- Galy, B.; Ferring-Appel, D.; Sauer, S.W.; Kaden, S.; Lyoumi, S.; Puy, H.; Kolker, S.; Grone, H.J.; Hentze, M.W. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010, 12, 194–201. [Google Scholar] [CrossRef]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar] [CrossRef]
- Stehling, O.; Mascarenhas, J.; Vashisht, A.A.; Sheftel, A.D.; Niggemeyer, B.; Rosser, R.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 2013, 18, 187–198. [Google Scholar] [CrossRef]
- Terzi, E.M.; Sviderskiy, V.O.; Alvarez, S.W.; Whiten, G.C.; Possemato, R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 2021, 7, eabg4302. [Google Scholar] [CrossRef]
- Wang, H.; Shi, H.; Rajan, M.; Canarie, E.R.; Hong, S.; Simoneschi, D.; Pagano, M.; Bush, M.F.; Stoll, S.; Leibold, E.A.; et al. FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster. Mol. Cell 2020, 78, 31–41.e35. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.D.; Crameri, J.J.; Muellner-Wong, L.; Frazier, A.E.; Palmer, C.S.; Formosa, L.E.; Hock, D.H.; Fujihara, K.M.; Stait, T.; Sharpe, A.J.; et al. Sideroflexin 4 is a complex I assembly factor that interacts with the MCIA complex and is required for the assembly of the ND2 module. Proc. Natl. Acad. Sci. USA 2022, 119, e2115566119. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Tesfay, L.; Winkler, C.R.; Torti, F.M.; Torti, S.V. Sideroflexin 4 affects Fe-S cluster biogenesis, iron metabolism, mitochondrial respiration and heme biosynthetic enzymes. Sci. Rep. 2019, 9, 19634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sun, L.; Hao, Y.; Suo, C.; Shen, S.; Wei, H.; Ma, W.; Zhang, P.; Wang, T.; Gu, X.; et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat. Cancer 2022, 3, 75–89. [Google Scholar] [CrossRef]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Investig. 2010, 120, 1749–1761. [Google Scholar] [CrossRef]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef]
- Bi, M.; Du, X.; Jiao, Q.; Liu, Z.; Jiang, H. alpha-Synuclein Regulates Iron Homeostasis via Preventing Parkin-Mediated DMT1 Ubiquitylation in Parkinson’s Disease Models. ACS Chem. Neurosci. 2020, 11, 1682–1691. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Aguirre, P.; Tapia, V.; Carrasco, C.M.; Mena, N.P.; Nunez, M.T. Cell death induced by mitochondrial complex I inhibition is mediated by Iron Regulatory Protein 1. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2202–2209. [Google Scholar] [CrossRef]
- Mena, N.P.; Bulteau, A.L.; Salazar, J.; Hirsch, E.C.; Nunez, M.T. Effect of mitochondrial complex I inhibition on Fe-S cluster protein activity. Biochem. Biophys. Res. Commun. 2011, 409, 241–246. [Google Scholar] [CrossRef]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef]
- Hara, Y.; Yanatori, I.; Tanaka, A.; Kishi, F.; Lemasters, J.J.; Nishina, S.; Sasaki, K.; Hino, K. Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep. 2020, 21, e50202. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, H.; Zhang, D.; Sehgal, S.A.; Zhang, D.; Wang, X.; Qin, Y.; Liu, L.; Chen, Q. Defective mitochondrial ISCs biogenesis switches on IRP1 to fine tune selective mitophagy. Redox Biol. 2020, 36, 101661. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Kuno, S.; Fujita, H.; Tanaka, Y.K.; Ogra, Y.; Iwai, K. Iron-induced NCOA4 condensation regulates ferritin fate and iron homeostasis. EMBO Rep. 2022, 23, e54278. [Google Scholar] [CrossRef]
- Fujimaki, M.; Furuya, N.; Saiki, S.; Amo, T.; Imamichi, Y.; Hattori, N. Iron Supply via NCOA4-Mediated Ferritin Degradation Maintains Mitochondrial Functions. Mol. Cell. Biol. 2019, 39, e00010-19. [Google Scholar] [CrossRef]
- Lee, H.E.; Jung, M.K.; Noh, S.G.; Choi, H.B.; Chae, S.H.; Lee, J.H.; Mun, J.Y. Iron Accumulation and Changes in Cellular Organelles in WDR45 Mutant Fibroblasts. Int. J. Mol. Sci. 2021, 22, 11650. [Google Scholar] [CrossRef]
- Aring, L.; Choi, E.K.; Kopera, H.; Lanigan, T.; Iwase, S.; Klionsky, D.J.; Seo, Y.A. A neurodegeneration gene, WDR45, links impaired ferritinophagy to iron accumulation. J. Neurochem. 2022, 160, 356–375. [Google Scholar] [CrossRef]
- Xiong, Q.; Li, X.; Li, W.; Chen, G.; Xiao, H.; Li, P.; Wu, C. WDR45 Mutation Impairs the Autophagic Degradation of Transferrin Receptor and Promotes Ferroptosis. Front. Mol. Biosci. 2021, 8, 645831. [Google Scholar] [CrossRef]
- Toulmay, A.; Whittle, F.B.; Yang, J.; Bai, X.; Diarra, J.; Banerjee, S.; Levine, T.P.; Golden, A.; Prinz, W.A. Vps13-like proteins provide phosphatidylethanolamine for GPI anchor synthesis in the ER. J. Cell Biol. 2022, 221, e202111095. [Google Scholar] [CrossRef]
- Nagae, M.; Hirata, T.; Morita-Matsumoto, K.; Theiler, R.; Fujita, M.; Kinoshita, T.; Yamaguchi, Y. 3D Structure and Interaction of p24beta and p24delta Golgi Dynamics Domains: Implication for p24 Complex Formation and Cargo Transport. J. Mol. Biol. 2016, 428, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Bonnon, C.; Wendeler, M.W.; Paccaud, J.-P.; Hauri, H.-P. Selective export of human GPI-anchored proteins from the endoplasmic reticulum. J. Cell Sci. 2010, 123, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Margraf, R.L.; Carey, J.C.; Zhou, H.; Newcomb, T.M.; Coonrod, E.; Durtschi, J.; Mallempati, K.; Kumanovics, A.; Katz, B.E.; et al. A novel germline PIGA mutation in Ferro-Cerebro-Cutaneous syndrome: A neurodegenerative X-linked epileptic encephalopathy with systemic iron-overload. Am. J. Med. Genet. A 2014, 164, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, L.; Marques, O.; Colucci, S.; Kunz, J.; Fabrowski, P.; Bast, T.; Altamura, S.; Hochsmann, B.; Schrezenmeier, H.; Langlotz, M.; et al. Constitutional PIGA mutations cause a novel subtype of hemochromatosis in patients with neurologic dysfunction. Blood 2022, 139, 1418–1422. [Google Scholar] [CrossRef]
- Flores-Torres, J.; Carver, J.D.; Sanchez-Valle, A. PIGA Mutations Can Mimic Neonatal Hemochromatosis. Pediatrics 2021, 147, e20200918. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Fazi, C.; Gerardi, G.; Levi, S.; Arosio, P.; Camaschella, C. Defective targeting of hemojuvelin to plasma membrane is a common pathogenetic mechanism in juvenile hemochromatosis. Blood 2007, 109, 4503–4510. [Google Scholar] [CrossRef]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef]
- Davids, M.; Menezes, M.; Guo, Y.; McLean, S.D.; Hakonarson, H.; Collins, F.; Worgan, L.; Billington, C.J., Jr.; Maric, I.; Littlejohn, R.O.; et al. Homozygous splice-variants in human ARV1 cause GPI-anchor synthesis deficiency. Mol. Genet. Metab. 2020, 130, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef]
- Kennard, M.L.; Richardson, D.R.; Gabathuler, R.; Ponka, P.; Jefferies, W.A. A novel iron uptake mechanism mediated by GPI-anchored human p97. EMBO J. 1995, 14, 4178–4186. [Google Scholar] [CrossRef]
- Akiyama, Y.; Oshima, K.; Kuhara, T.; Shin, K.; Abe, F.; Iwatsuki, K.; Nadano, D.; Matsuda, T. A lactoferrin-receptor, intelectin 1, affects uptake, sub-cellular localization and release of immunochemically detectable lactoferrin by intestinal epithelial Caco-2 cells. J. Biochem. 2013, 154, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.K.; Haldar, S.; Qian, J.; Beserra, A.; Suda, S.; Singh, A.; Hopfer, U.; Chen, S.G.; Garrick, M.D.; Turner, J.R.; et al. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic. Biol. Med. 2015, 84, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.V.; Sandri, C. The electromotor system of the electric eel investigated with horseradish peroxidase as a retrograde tracer. Brain Res. 1989, 488, 22–30. [Google Scholar] [CrossRef]
- Patel, B.N.; David, S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 1997, 272, 20185–20190. [Google Scholar] [CrossRef]
- Rothenberger, S.; Food, M.R.; Gabathuler, R.; Kennard, M.L.; Yamada, T.; Yasuhara, O.; McGeer, P.L.; Jefferies, W.A. Coincident expression and distribution of melanotransferrin and transferrin receptor in human brain capillary endothelium. Brain Res. 1996, 712, 117–121. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Nillesse, N.; Damier, P.; Spik, G.; Mouatt-Prigent, A.; Pierce, A.; Leveugle, B.; Kubis, N.; Hauw, J.J.; Agid, Y.; et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1995, 92, 9603–9607. [Google Scholar] [CrossRef]
- Liu, H.; Wu, H.; Zhu, N.; Xu, Z.; Wang, Y.; Qu, Y.; Wang, J. Lactoferrin protects against iron dysregulation, oxidative stress, and apoptosis in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson’s disease in mice. J. Neurochem. 2020, 152, 397–415. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Meng, F.; Fleming, B.A.; Jia, X.; Rousek, A.A.; Mulvey, M.A.; Ward, D.M. Lysosomal iron recycling in mouse macrophages is dependent upon both LcytB and Steap3 reductases. Blood Adv. 2022, 6, 1692–1707. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, A.; Sanuki, R.; Kakimoto, K.; Kojo, S.; Taketani, S. Involvement of 101F6, a homologue of cytochrome b561, in the reduction of ferric ions. J. Biochem. 2007, 142, 699–705. [Google Scholar] [CrossRef] [PubMed]
- El Behery, M.; Fujimura, M.; Kimura, T.; Tsubaki, M. Direct measurements of ferric reductase activity of human 101F6 and its enhancement upon reconstitution into phospholipid bilayer nanodisc. Biochem. Biophys. Rep. 2020, 21, 100730. [Google Scholar] [CrossRef] [PubMed]
- Recuenco, M.C.; Rahman, M.M.; Takeuchi, F.; Kobayashi, K.; Tsubaki, M. Electron transfer reactions of candidate tumor suppressor 101F6 protein, a cytochrome b561 homologue, with ascorbate and monodehydroascorbate radical. Biochemistry 2013, 52, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife 2019, 8, e51031. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef]
- Siow, W.X.; Kabiri, Y.; Tang, R.; Chao, Y.-K.; Plesch, E.; Eberhagen, C.; Flenkenthaler, F.; Frohlich, T.; Bracher, F.; Grimm, C.; et al. Lysosomal TRPML1 regulates mitochondrial function in hepatocellular carcinoma cells. J. Cell Sci. 2022, 135, jcs259455. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Inden, M.; Tsuboi, H.; Niwa, M.; Uchida, Y.; Naka, Y.; Hozumi, I.; Nagasawa, H. A Golgi-targeting fluorescent probe for labile Fe(ii) to reveal an abnormal cellular iron distribution induced by dysfunction of VPS35. Chem. Sci. 2019, 10, 1514–1521. [Google Scholar] [CrossRef]
- Zhao, S.-B.; Dean, N.; Gao, X.-D.; Fujita, M. MON2 Guides Wntless Transport to the Golgi through Recycling Endosomes. Cell Struct. Funct. 2020, 45, 77–92. [Google Scholar] [CrossRef]
- Chen, C.; Garcia-Santos, D.; Ishikawa, Y.; Seguin, A.; Li, L.; Fegan, K.H.; Hildick-Smith, G.J.; Shah, D.I.; Cooney, J.D.; Chen, W.; et al. Snx3 regulates recycling of the transferrin receptor and iron assimilation. Cell Metab. 2013, 17, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Mayank, A.K.; Pandey, V.; Vashisht, A.A.; Barshop, W.D.; Rayatpisheh, S.; Sharma, T.; Haque, T.; Powers, D.N.; Wohlschlegel, J.A. An Oxygen-Dependent Interaction between FBXL5 and the CIA-Targeting Complex Regulates Iron Homeostasis. Mol. Cell 2019, 75, 382–393. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Alberghini, A.; Recalcati, S.; Tacchini, L.; Santambrogio, P.; Campanella, A.; Cairo, G. Loss of the von Hippel Lindau tumor suppressor disrupts iron homeostasis in renal carcinoma cells. J. Biol. Chem. 2005, 280, 30120–30128. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bórquez, D.A.; Castro, F.; Núñez, M.T.; Urrutia, P.J. New Players in Neuronal Iron Homeostasis: Insights from CRISPRi Studies. Antioxidants 2022, 11, 1807. https://doi.org/10.3390/antiox11091807
Bórquez DA, Castro F, Núñez MT, Urrutia PJ. New Players in Neuronal Iron Homeostasis: Insights from CRISPRi Studies. Antioxidants. 2022; 11(9):1807. https://doi.org/10.3390/antiox11091807
Chicago/Turabian StyleBórquez, Daniel A., Francisco Castro, Marco T. Núñez, and Pamela J. Urrutia. 2022. "New Players in Neuronal Iron Homeostasis: Insights from CRISPRi Studies" Antioxidants 11, no. 9: 1807. https://doi.org/10.3390/antiox11091807
APA StyleBórquez, D. A., Castro, F., Núñez, M. T., & Urrutia, P. J. (2022). New Players in Neuronal Iron Homeostasis: Insights from CRISPRi Studies. Antioxidants, 11(9), 1807. https://doi.org/10.3390/antiox11091807