

Facile Detection of Light-Controlled Radical Scavengers from Natural Products Using In Situ UV-LED NMR Spectroscopy

 , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. In Situ UV-LED NMR Spectroscopy
2.2. NMR Data Processing
2.3. Preparation of the Subfractions from Licorice Root Extract
2.4. Tyrosinase Inhibition Activity Assay
2.5. Statistical Analysis
3. Results and Discussion
3.1. Novel NMR Acquisition and Processing Method Tailored to Detect Photoswitchable Compounds
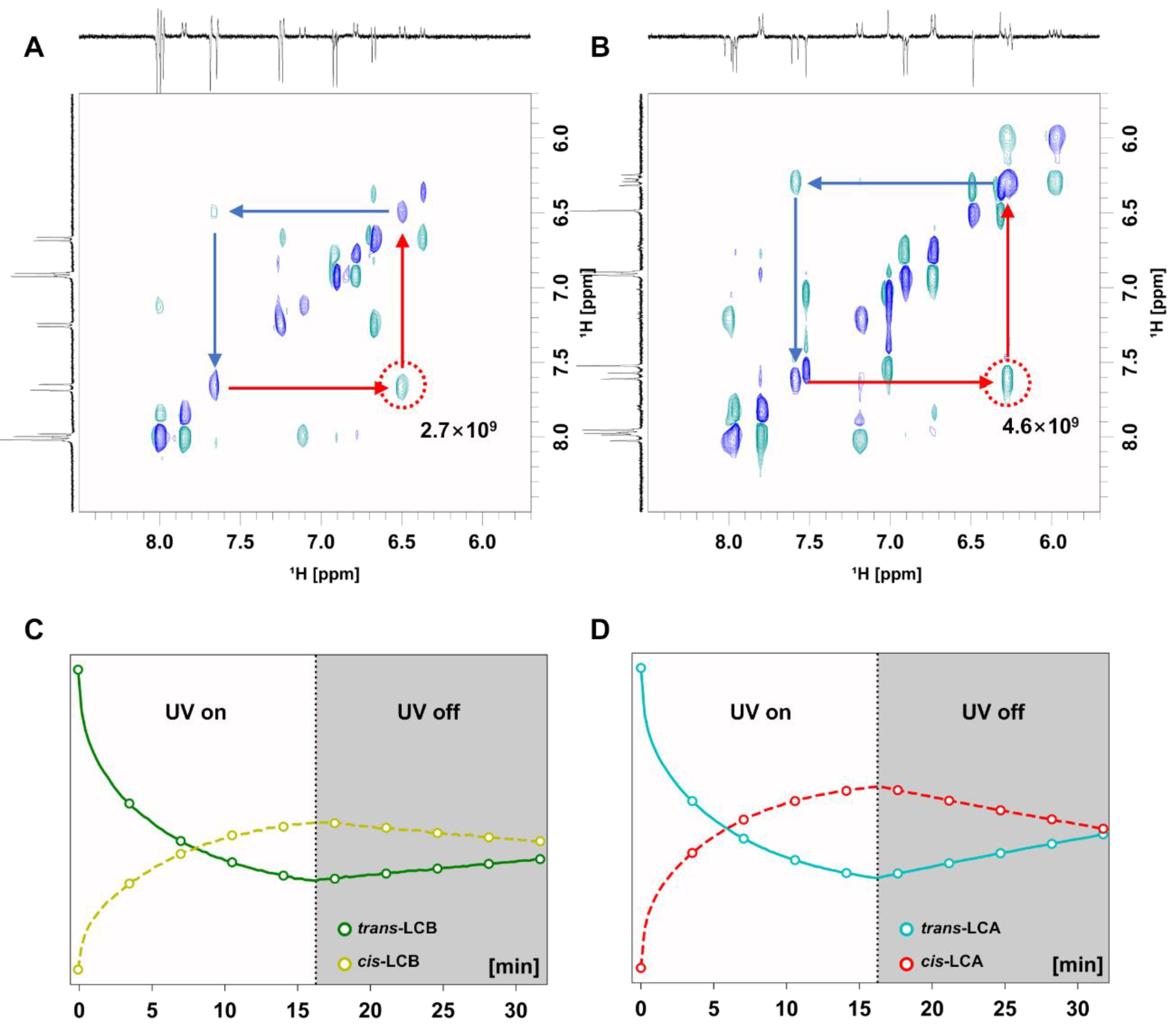
3.2. Evaluation of Photoisomerization by Kinetic Analysis and 2D Photo NMR Spectroscopy
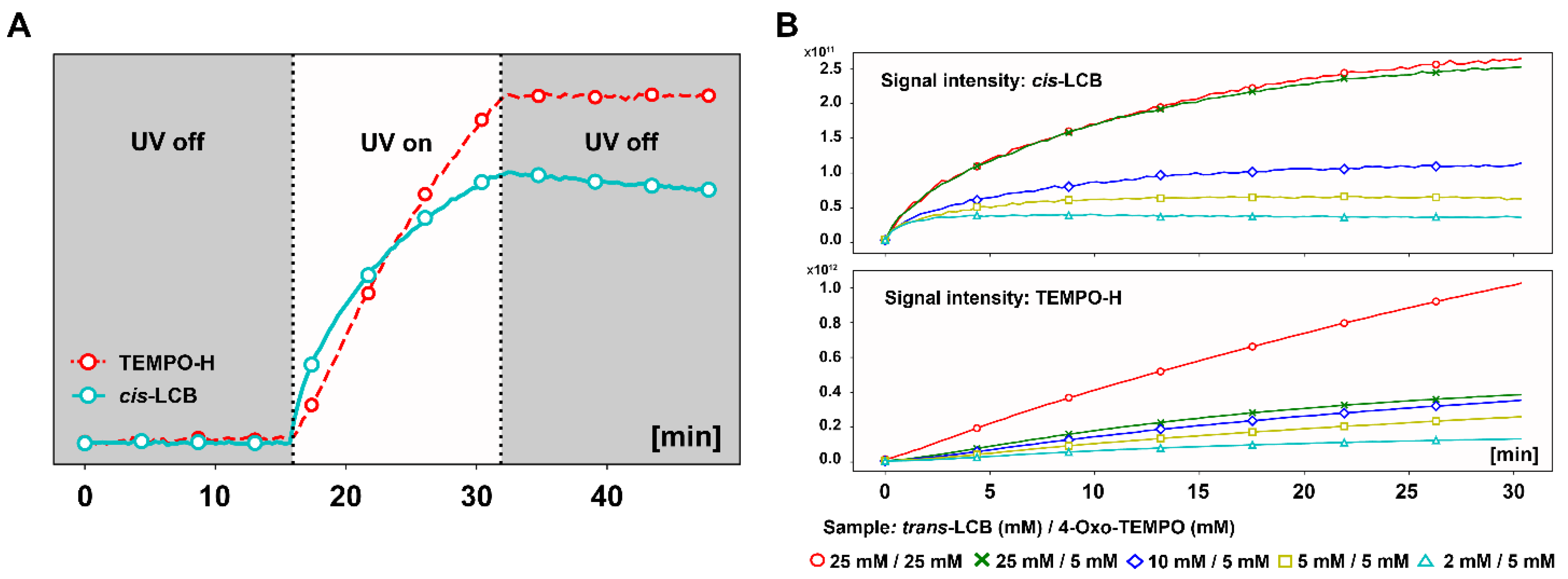
3.3. In Situ NMR Monitoring of the Radical Scavenging Activity of Licochalcones under UV Irradiation
3.4. Correlation between the Radical Scavenging Activity and Photoisomerization of Licochalcones
3.5. Tyrosinase Inhibition of Licochalcones by UV Irradiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barreiro, E.J. What Is Hidden in the Biodiversity? The Role of Natural Products and Medicinal Chemistry in the Drug Discovery Process. An. Acad. Bras. Ciênc. 2019, 91, e20190306. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S. The Role of Natural Product Chemistry in Drug Discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Y.; Yang, D.-P.; Tang, G.-Y. Multipotent Antioxidants: From Screening to Design. Drug Discov. Today 2006, 11, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Abourashed, E.A. Bioavailability of Plant-Derived Antioxidants. Antioxidants 2013, 2, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication Strategies in Natural Product Research: How Many Tools and Methodologies behind the Same Concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Zani, C.L.; Carroll, A.R. Database for Rapid Dereplication of Known Natural Products Using Data from Ms and Fast NMR Experiments. J. Nat. Prod. 2017, 80, 1758–1766. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Monteiro, A.F.; Bertonha, A.F.; Bernardi, D.I.; Gubiani, J.R.; Slivinski, J.; Michaliski, L.F.; Tonon, L.A.; Venancio, V.A.; Freire, V.F. Approaches for the Isolation and Identification of Hydrophilic, Light-Sensitive, Volatile and Minor Natural Products. Nat. Prod. Rep. 2019, 36, 981–1004. [Google Scholar] [CrossRef]
- Detoni, C.B.; Souto, G.D.; da Silva, A.L.M.; Pohlmann, A.R.; Guterres, S.S. Photostability and Skin Penetration of Different E-Resveratrol-Loaded Supramolecular Structures. Photochem. Photobiol. 2012, 88, 913–921. [Google Scholar] [CrossRef]
- Guan, W.; Zhang, W.; Tang, L.; Wang, Y.; Cui, H. Fabrication of Novel Avermectin Nanoemulsion Using a Polyurethane Emulsifier with Cleavable Disulfide Bonds. J. Agric. Food. Chem. 2017, 66, 6569–6577. [Google Scholar] [CrossRef]
- Bailey, C.S.; Zarins-Tutt, J.S.; Agbo, M.; Gao, H.; Diego-Taboada, A.; Gan, M.; Hamed, R.B.; Abraham, E.R.; Mackenzie, G.; Evans, P.A. A Natural Solution to Photoprotection and Isolation of the Potent Polyene Antibiotic, Marinomycin A. Chem. Sci. 2019, 10, 7549–7553. [Google Scholar] [CrossRef]
- Kreutzer, M.; Kage, H.; Nett, M. Cupriachelin, a Photoreactive Siderophore from the Freshwater Bacterium Cupriavidus necator H16. Planta Med. 2012, 78, PI42. [Google Scholar] [CrossRef]
- Barbeau, K.; Zhang, G.; Live, D.H.; Butler, A. Petrobactin, a Photoreactive Siderophore Produced by the Oil-Degrading Marine Bacterium Marinobacter hydrocarbonoclasticus. J. Am. Chem. Soc. 2002, 124, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Biji, M.; Radhakrishnan, K.V.; Lankalapalli, R.S. Tandem Photoisomerization and Transannular Cyclizations of Zerumbone Epoxide: A Model for Diversity-Oriented Synthesis Using Abundant Natural Products. Org. Lett. 2021, 23, 5871–5875. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Renslow, R.S.; Makola, M.M.; Webb, I.K.; Deng, L.; Thomas, D.G.; Govind, N.; Ibrahim, Y.M.; Kabanda, M.M.; Dubery, I.A. Structural Elucidation of Cis/Trans Dicaffeoylquinic Acid Photoisomerization Using Ion Mobility Spectrometry-Mass Spectrometry. J. Phys. Chem. Lett. 2017, 8, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Ko, K.; Kim, S.-H.; Lee, J.K.; Park, J.-S.; Park, K.; Kim, M.R.; Kang, K.; Oh, D.-C.; Kim, S.Y. Tropolone-Bearing Sesquiterpenes from Juniperus chinensis: Structures, Photochemistry and Bioactivity. J. Nat. Prod. 2021, 84, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Kim, S.-H.; Park, S.; Han, H.S.; Lee, J.K.; Cha, J.W.; Hwang, S.; Choi, K.Y.; Song, Y.-J.; Nam, S.-J.; et al. Discovery and Photoisomerization of New Pyrrolosesquiterpenoids Glaciapyrroles D and E, from Deep-Sea Sediment Streptomyces sp. Mar. Drugs 2022, 20, 281. [Google Scholar] [CrossRef]
- Jang, Y.H.; Koo, G.-B.; Kim, J.-Y.; Kim, Y.-S.; Kim, Y.C. Prolonged Activation of Erk Contributes to the Photorejuvenation Effect in Photodynamic Therapy in Human Dermal Fibroblasts. J. Investig. Dermatol. 2013, 133, 2265–2275. [Google Scholar] [CrossRef]
- Ristow, M. Unraveling the Truth About Antioxidants: Mitohormesis Explains Ros-Induced Health Benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- Shanker, N.; Dilek, O.; Mukherjee, K.; McGee, D.W.; Bane, S.L. Aurones: Small Molecule Visible Range Fluorescent Probes Suitable for Biomacromolecules. J. Fluoresc. 2011, 21, 2173–2184. [Google Scholar] [CrossRef]
- Rodriguez, R.A.; Lahoz, I.R.; Faza, O.N.; Cid, M.M.; Lopez, C.S. Theoretical and Experimental Exploration of the Photochemistry of Resveratrol: Beyond the Simple Double Bond Isomerization. Org. Biomol. Chem. 2012, 10, 9175–9182. [Google Scholar] [CrossRef]
- Kim, M.C.; Cullum, R.; Machado, H.; Smith, A.J.; Yang, I.; Rodvold, J.J.; Fenical, W. Photopiperazines a-D, Photosensitive Interconverting Diketopiperazines with Significant and Selective Activity against U87 Glioblastoma Cells, from a Rare, Marine-Derived Actinomycete of the Family Streptomycetaceae. J. Nat. Prod. 2019, 82, 2262–2267. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, R.; Barnes, C.A.; Trampel, R.L.; Wallace, S.J.; Kee, T.W.; Petrich, J.W. Photoinduced Trans-to-Cis Isomerization of Cyclocurcumin. J. Phys. Chem. B 2011, 115, 10707–10714. [Google Scholar] [CrossRef]
- Mishra, K.; Ojha, H.; Chaudhury, N.K. Estimation of Antiradical Properties of Antioxidants Using Dpph Center Dot Assay: A Critical Review and Results. Food Chem. 2012, 130, 1036–1043. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant Activity Applying an Improved Abts Radical Cation Decolorization Assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Polovka, M.; Brezova, V.; Stasko, A. Antioxidant Properties of Tea Investigated by Epr Spectroscopy. Biophys. Chem. 2003, 106, 39–56. [Google Scholar] [CrossRef]
- Banach, M.; Wiloch, M.; Zawada, K.; Cyplik, W.; Kujawski, W. Evaluation of Antioxidant and Anti-Inflammatory Activity of Anthocyanin-Rich Water-Soluble Aronia Dry Extracts. Molecules 2020, 25, 4055. [Google Scholar] [CrossRef]
- Wolff, C.; Kind, J.; Schenderlein, H.; Bartling, H.; Feldmeier, C.; Gschwind, R.M.; Biesalski, M.; Thiele, C.M. Studies of a Photochromic Model System Using NMR with Ex-Situ and in-Situ Irradiation Devices. Magn. Reson. Chem. 2016, 54, 485–491. [Google Scholar] [CrossRef]
- Prochazkova, E.; Cechova, L.; Kind, J.; Janeba, Z.; Thiele, C.M.; Dracinsky, M. Photoswitchable Intramolecular Hydrogen Bonds in 5-Phenylazopyrimidines Revealed by in situ Irradiation NMR Spectroscopy. Chem. Eur. J. 2018, 24, 492–498. [Google Scholar] [CrossRef]
- Ji, Y.N.; Bottecchia, C.; Levesque, F.; Narsimhan, K.; Lehnherr, D.; McMullen, J.P.; Dalby, S.M.; Xiao, K.J.; Reibarkh, M. Benzylic Photobromination for the Synthesis of Belzutifan: Elucidation of Reaction Mechanisms Using in situ Led-NMR. J. Org. Chem. 2022, 87, 2055–2062. [Google Scholar] [CrossRef]
- Bramham, J.E.; Golovanov, A.P. Sample Illumination Device Facilitates in situ Light-Coupled NMR Spectroscopy without Fibre Optics. Commun. Chem. 2022, 5, 90. [Google Scholar] [CrossRef]
- Toncheva-Moncheva, N.; Dangalov, M.; Vassilev, N.G.; Novakov, C.P. Thiol–ene coupling reaction achievement and monitoring by “in situ” UV irradiation NMR spectroscopy. RSC Adv. 2020, 10, 25214–25222. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. Nmrpipe: A Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Helmus, J.J.; Jaroniec, C.P. Nmrglue: An Open Source Python Package for the Analysis of Multidimensional NMR Data. J. Biomol. NMR 2013, 55, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.E.; Tassoti, S.; Glanzer, S.; Stadler, E.; Herges, R.; Gescheidt, G.; Zangger, K. Monitoring Fast Chemical Processes by Reaction-Interrupted Excitation Transfer (Extra) NMR Spectroscopy. Chem. Commun. 2019, 55, 12575–12578. [Google Scholar] [CrossRef] [PubMed]
- Seegerer, A.; Nitschke, P.; Gschwind, R.M. Combined in Situ Illumination-NMR-UV/Vis Spectroscopy: A New Mechanistic Tool in Photochemistry. Angew. Chem. Int. Ed. 2018, 57, 7493–7497. [Google Scholar] [CrossRef]
- Feldmeier, C.; Bartling, H.; Riedle, E.; Gschwind, R.M. Led Based NMR Illumination Device for Mechanistic Studies on Photochemical Reactions—Versatile and Simple, yet Surprisingly Powerful. J. Magn. Reson. 2013, 232, 39–44. [Google Scholar] [CrossRef]
- Ernst, R.R.; Bodenhausen, G.; Wokaun, A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Baishya, B.; Verma, A. Elimination of Zero-Quantum Artifacts and Sensitivity Enhancement in Perfect Echo Based 2d Noesy. J. Magn. Reson. 2015, 252, 41–48. [Google Scholar] [CrossRef]
- Stadler, E.; Tassoti, S.; Lentes, P.; Herges, R.; Glasnov, T.; Zangger, K.; Gescheidt, G. In Situ Observation of Photoswitching by NMR Spectroscopy: A Photochemical Analogue to the Exchange Spectroscopy Experiment. Anal. Chem. 2019, 91, 11367–11373. [Google Scholar] [CrossRef]
- Miyaji, A.; Kohno, M.; Inoue, Y.; Baba, T. Singlet Oxygen Generation during the Oxidation of L-Tyrosine and L-Dopa with Mushroom Tyrosinase. Biochem. Biophys. Res. Commun. 2016, 471, 450–453. [Google Scholar] [CrossRef]
- Satterlee, J.D. Fundamental Concepts of NMR in Paramagnetic Systems. Part II: Relaxation Effects. Concept. Magn. Reson. 1990, 2, 119–129. [Google Scholar] [CrossRef]
- Marano, S.; Minnelli, C.; Ripani, L.; Marcaccio, M.; Laudadio, E.; Mobbili, G.; Amici, A.; Armeni, T.; Stipa, P. Insights into the Antioxidant Mechanism of Newly Synthesized Benzoxazinic Nitrones: In Vitro and in Silico Studies with Dpph Model Radical. Antioxidants 2021, 10, 1224. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and Molecular Mechanisms of Antioxidants: Experimental Approaches and Model Systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Staig, S.J.; Weaver, J.D. Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc. 2014, 136, 5275–5278. [Google Scholar] [CrossRef]
- Schultz, T.; Quenneville, J.; Levine, B.; Toniolo, A.; Martinez, T.J.; Lochbrunner, S.; Schmitt, M.; Shaffer, J.P.; Zgierski, M.Z.; Stolow, A. Mechanism and Dynamics of Azobenzene Photoisomerization. J. Am. Chem. Soc. 2003, 125, 8098–8099. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Chen, X.Z.; Vudhya Gowrisankar, Y.; Yen, H.R.; Chuang, J.Y.; Yang, H.L. The Skin-Whitening Effects of Ectoine via the Suppression of Alpha-Msh-Stimulated Melanogenesis and the Activation of Antioxidant Nrf2 Pathways in Uva-Irradiated Keratinocytes. Antioxidants 2020, 9, 63. [Google Scholar] [CrossRef]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef]
- Espin, J.C.; Varon, R.; Fenoll, L.G.; Gilabert, M.A.; Garcia-Ruiz, P.A.; Tudela, J.; Garcia-Canovas, F. Kinetic Characterization of the Substrate Specificity and Mechanism of Mushroom Tyrosinase. Eur. J. Biochem. 2000, 267, 1270–1279. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, I.; Park, G.; Choi, Y.; Jo, S.-W.; Kwon, H.C.; Park, J.-S.; Cha, J.W. Facile Detection of Light-Controlled Radical Scavengers from Natural Products Using In Situ UV-LED NMR Spectroscopy. Antioxidants 2022, 11, 2206. https://doi.org/10.3390/antiox11112206
Park I, Park G, Choi Y, Jo S-W, Kwon HC, Park J-S, Cha JW. Facile Detection of Light-Controlled Radical Scavengers from Natural Products Using In Situ UV-LED NMR Spectroscopy. Antioxidants. 2022; 11(11):2206. https://doi.org/10.3390/antiox11112206
Chicago/Turabian StylePark, InWha, Goeun Park, Yoojin Choi, Seung-Woo Jo, Hak Cheol Kwon, Jin-Soo Park, and Jin Wook Cha. 2022. "Facile Detection of Light-Controlled Radical Scavengers from Natural Products Using In Situ UV-LED NMR Spectroscopy" Antioxidants 11, no. 11: 2206. https://doi.org/10.3390/antiox11112206
APA StylePark, I., Park, G., Choi, Y., Jo, S.-W., Kwon, H. C., Park, J.-S., & Cha, J. W. (2022). Facile Detection of Light-Controlled Radical Scavengers from Natural Products Using In Situ UV-LED NMR Spectroscopy. Antioxidants, 11(11), 2206. https://doi.org/10.3390/antiox11112206