
Combinatorial Effects of the Natural Products Arctigenin, Chlorogenic Acid, and Cinnamaldehyde Commit Oxidation Assassination on Breast Cancer Cells



Abstract
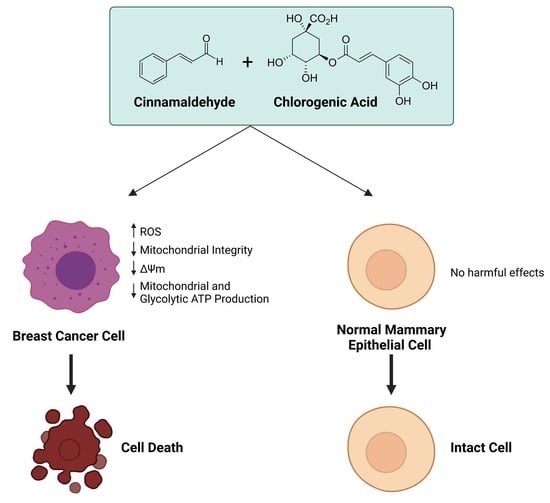
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cell Treatments
2.3. Cell Growth Curves
2.4. Cell Death Assay
2.5. 40× Fluorescence Microscopy and Phase–Contrast Imaging
2.6. 100× Fluorescence Microscopy Imaging
2.7. Mitochondrial Superoxide Production Assay
2.8. ATP Production Rate Assay
2.9. Statistical Analyses
3. Results
3.1. IC50 Concentrations of CA, CGA, and Arc Treatments of Breast Cancer Cells
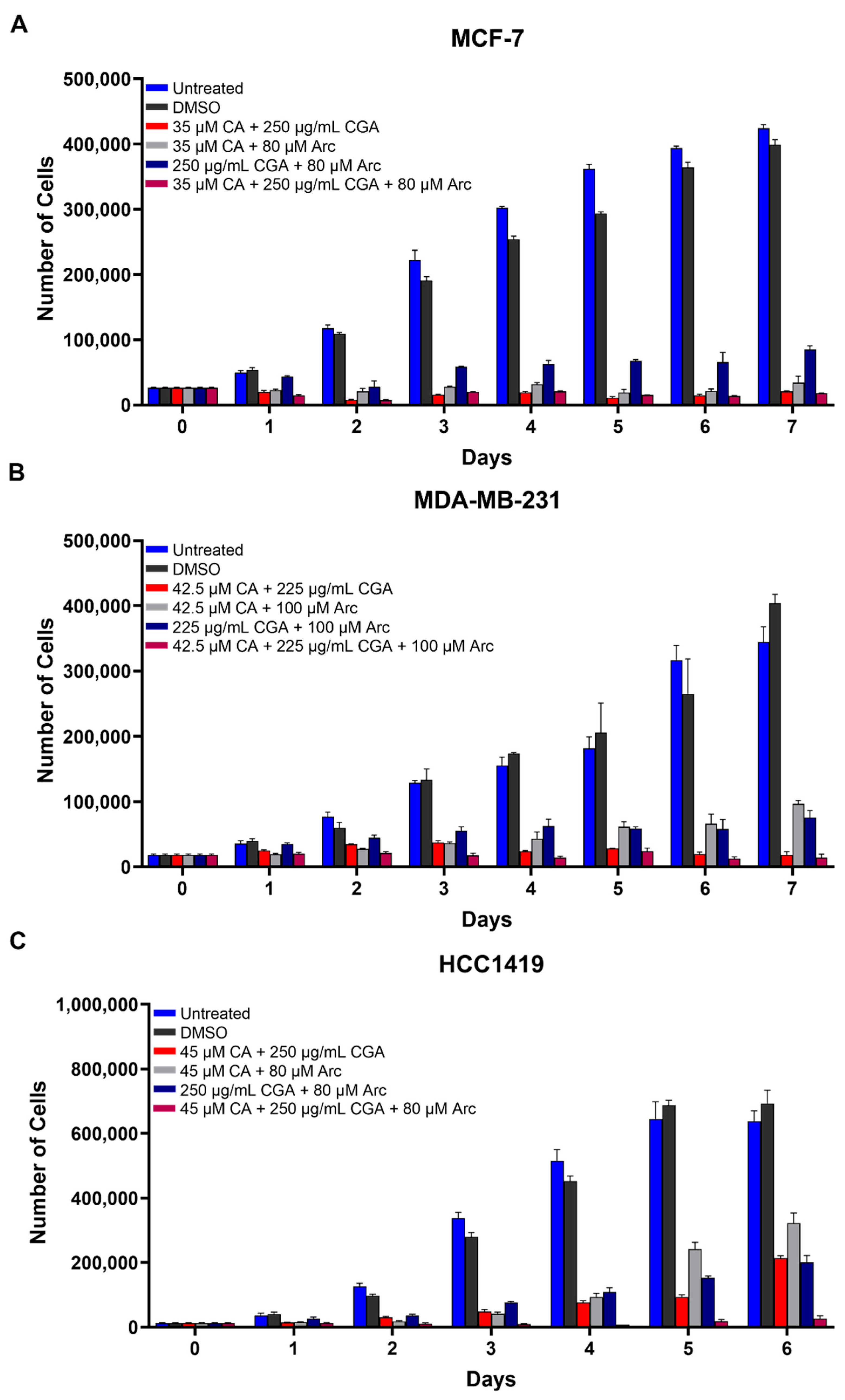
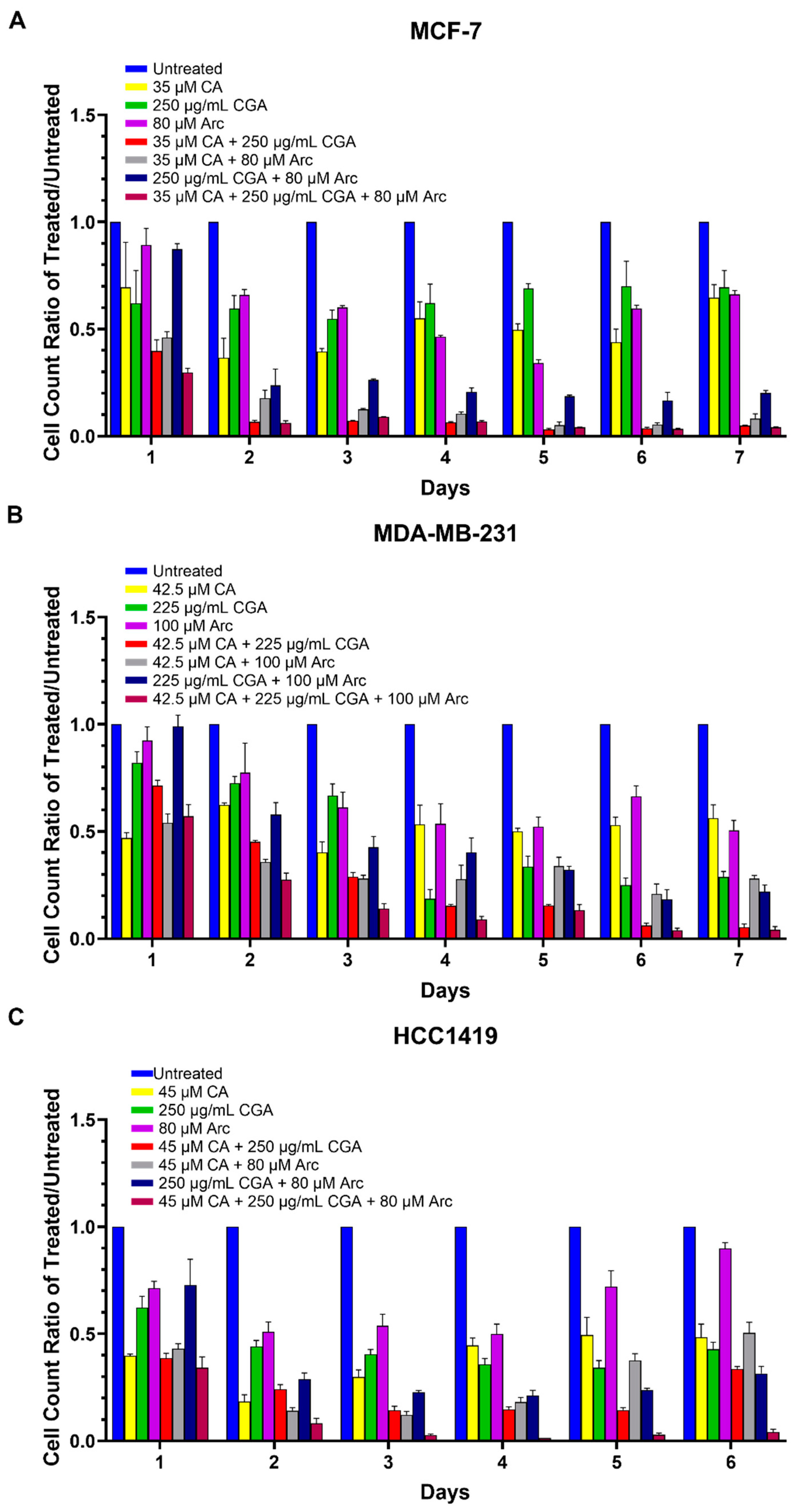
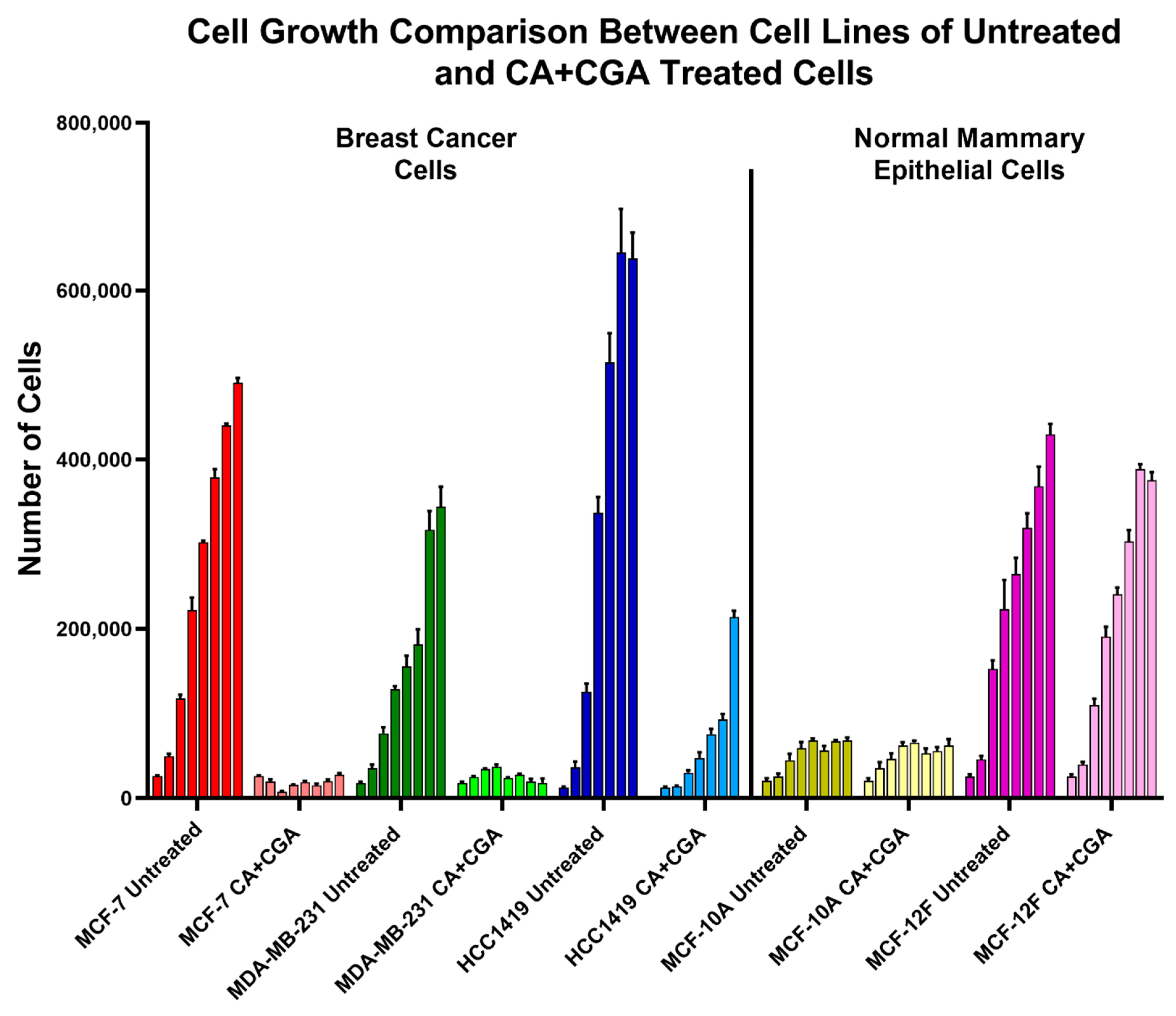
3.2. Combination Treatments of CA, CGA, and Arc Are Effective in Reducing Breast Cancer Cell Growth
3.3. Combination Treatments of CA, CGA, and Arc Are More Effective in Reducing Breast Cancer Cell Growth than Single Treatments
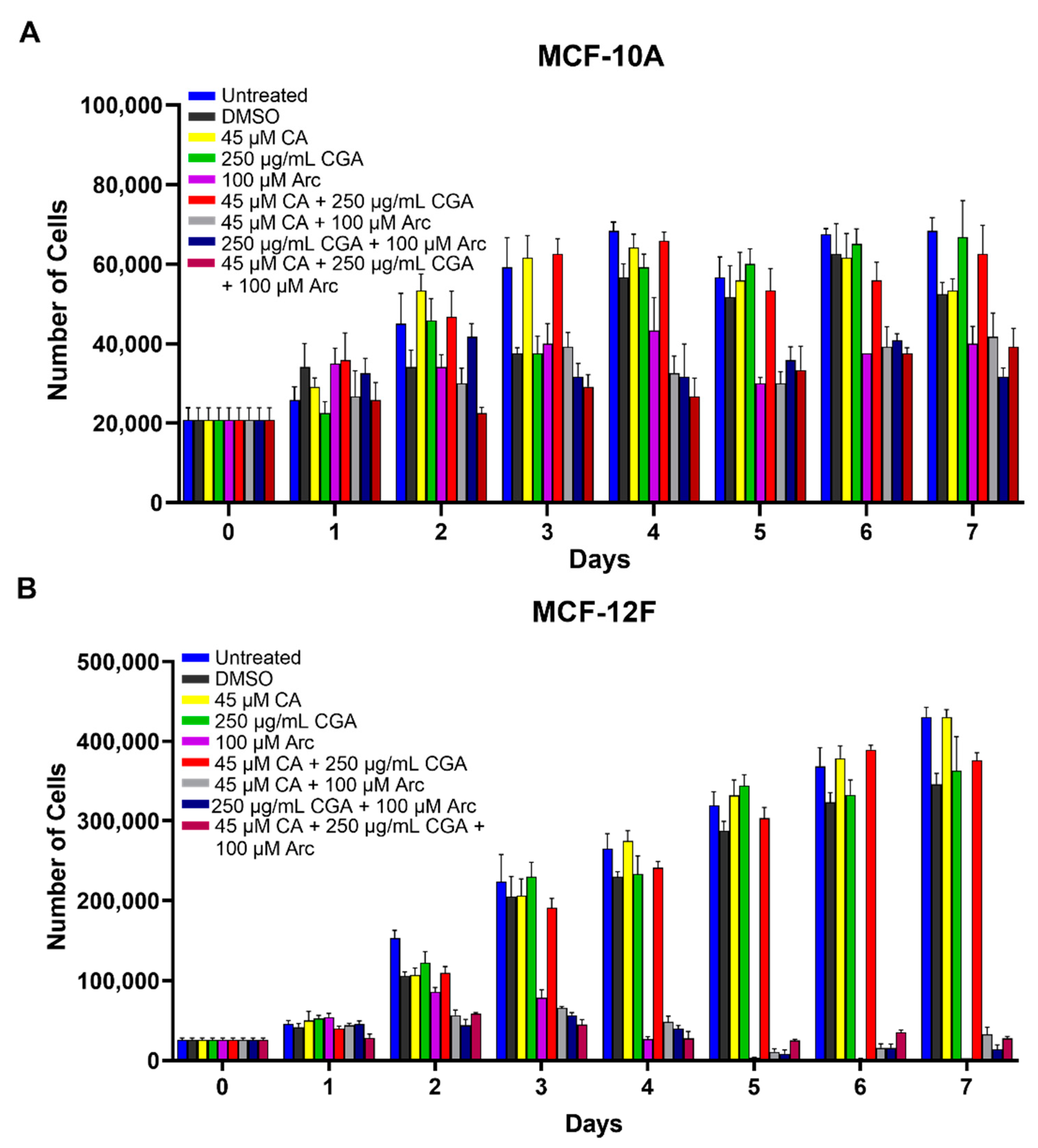
3.4. Single CA, CGA, and the Combination of CA + CGA Show No Harmful Effects While Arc Treated Groups, Alone or in Combination, Show Cytotoxic Effects in Normal Mammary Epithelial Cells
3.5. Effects of Single and Combination Treatments of CA, CGA, and Arc on Cell Death of Breast Cancer and Normal Mammary Epithelial Cells
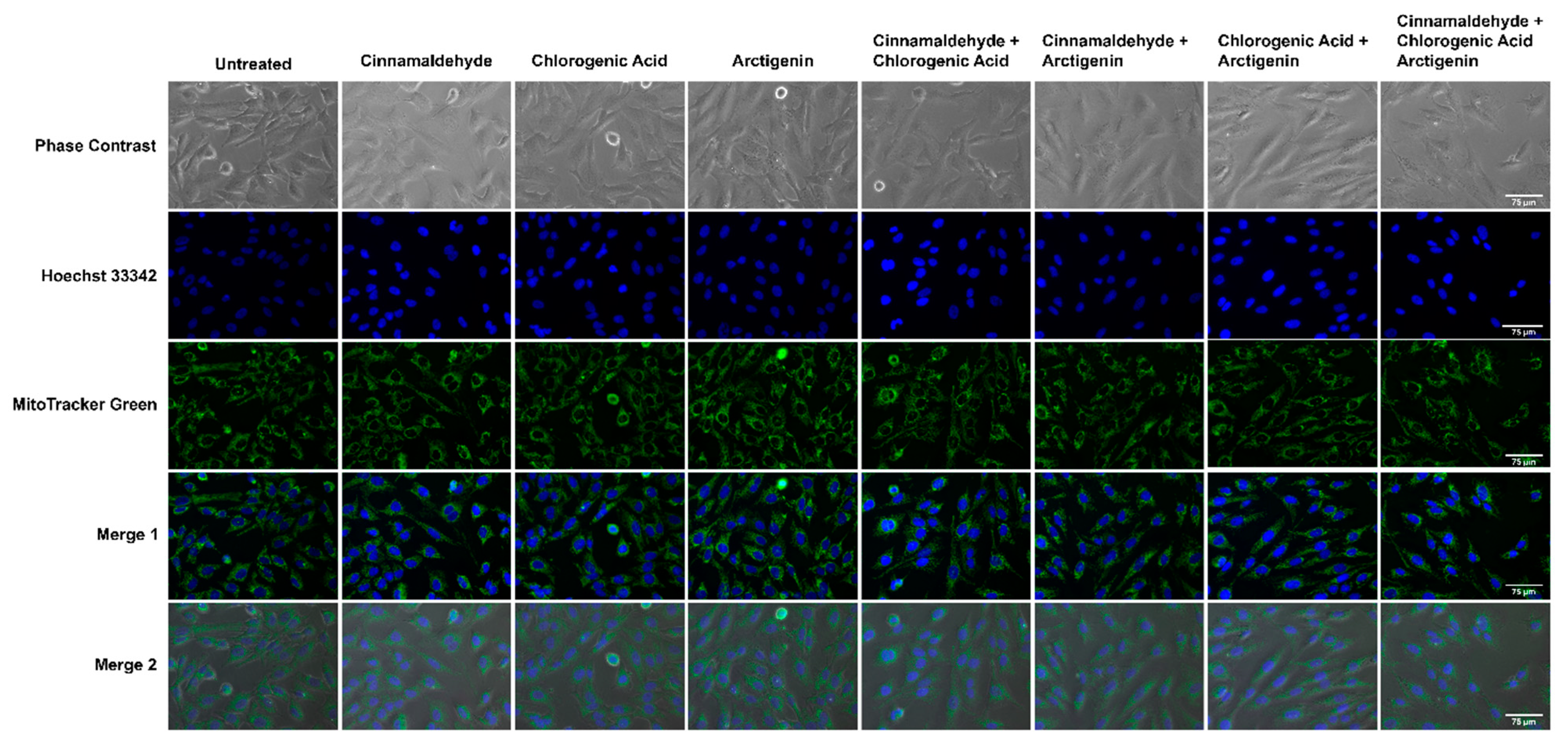
3.6. Effects of CA, CGA, and Arc, Alone and in Combination, on General Cell and Mitochondrial Morphology in Breast Cancer and Normal Mammary Epithelial Cells
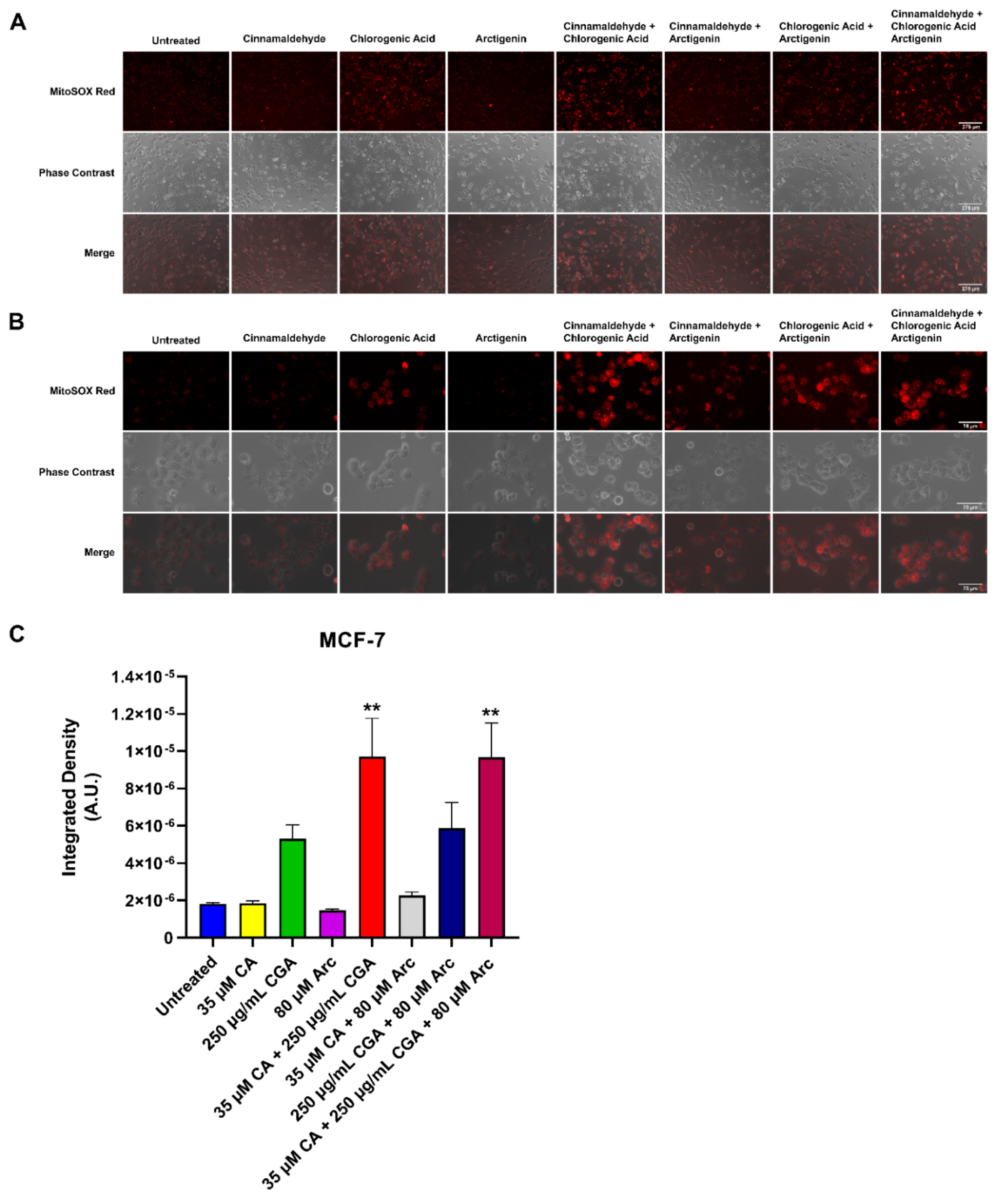
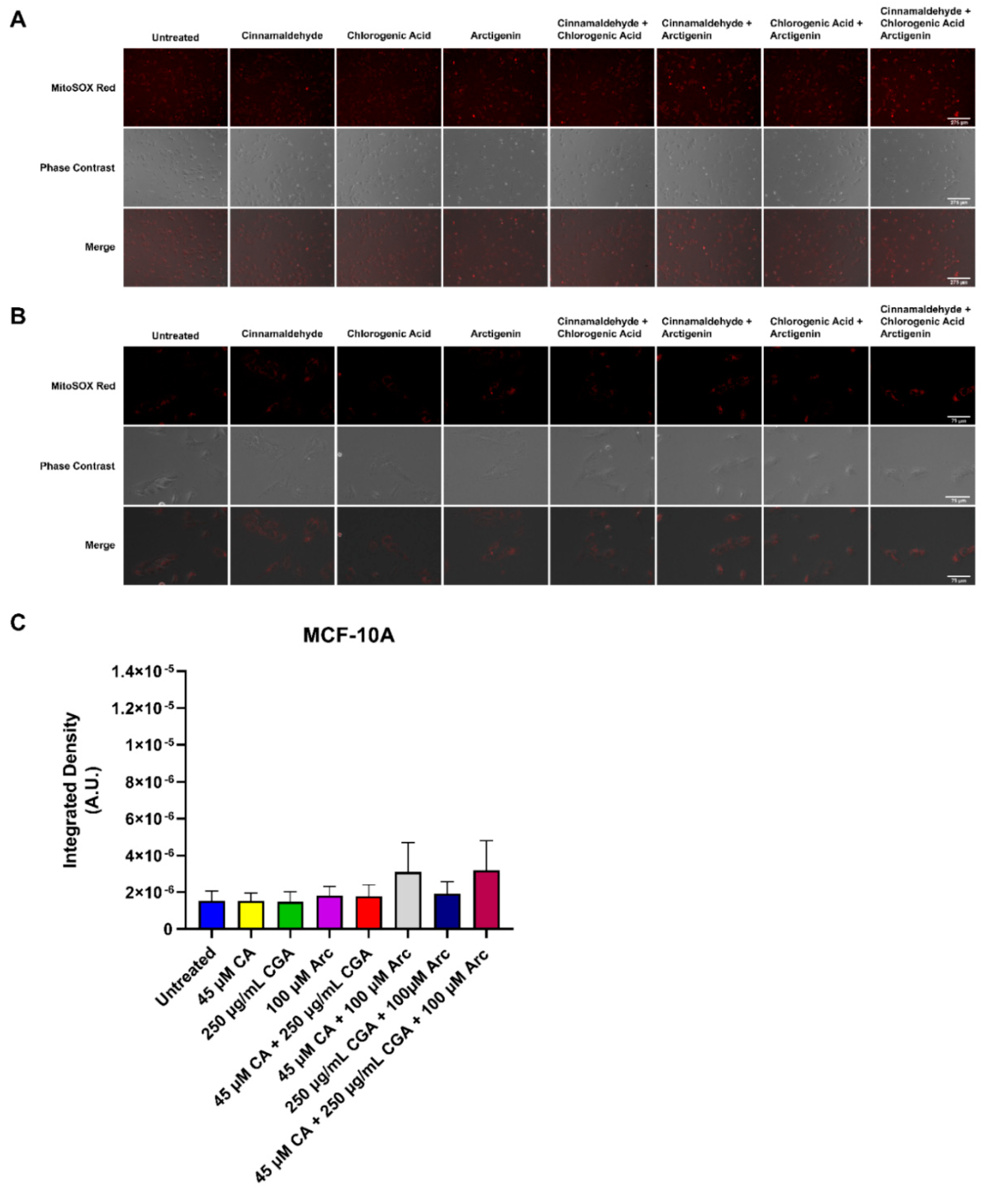
3.7. Effects of Single and Combination Treatments of CA, CGA, and Arc on Mitochondrial Superoxide Production in Breast Cancer and Normal Mammary Epithelial Cells
3.8. Effects of CA, CGA, Arc, CA + CGA, and CA + CGA + Arc Treatments on Cellular ATP Production by Glycolysis and Oxidative Phosphorylation in Breast Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial Function and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial Metabolism and Cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial Mutations in Cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive Oxygen Species in Normal and Tumor Stem Cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. Regulators of Mitochondrial Dynamics in Cancer. Curr. Opin. Cell Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W.G. Mitochondrial Apoptosis: Killing Cancer Using the Enemy Within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; van der Heiden, M.G. Altered Metabolite Levels in Cancer: Implications for Tumour Biology and Cancer Therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.; Grishin, N.V.; Brugarolas, J. Succination of Keap1 and Activation of Nrf2-Dependent Antioxidant Pathways in FH-Deficient Papillary Renal Cell Carcinoma Type 2. Cancer Cell 2011, 20, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting Mitochondria Metabolism for Cancer Therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef]
- Morandi, A.; Indraccolo, S. Linking Metabolic Reprogramming to Therapy Resistance in Cancer. Biochim. Et Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Machana, S.; Weerapreeyakul, N.; Barusrux, S. Anticancer Effect of the Extracts from Polyalthia Evecta against Human Hepatoma Cell Line (HepG2). Asian Pac. J. Trop. Biomed. 2012, 2, 368–374. [Google Scholar] [CrossRef]
- Wagner, H. Synergy Research: Approaching a New Generation of Phytopharmaceuticals. Fitoterapia 2011, 82, 34–37. [Google Scholar] [CrossRef]
- Ma, X.H.; Zheng, C.J.; Han, L.Y.; Xie, B.; Jia, J.; Cao, Z.W.; Li, Y.X.; Chen, Y.Z. Synergistic Therapeutic Actions of Herbal Ingredients and Their Mechanisms from Molecular Interaction and Network Perspectives. Drug Discov. Today 2009, 14, 579–588. [Google Scholar] [CrossRef]
- Rizeq, B.; Gupta, I.; Ilesanmi, J.; AlSafran, M.; Rahman, M.D.M.; Ouhtit, A. The Power of Phytochemicals Combination in Cancer Chemoprevention. J. Cancer 2020, 11, 4521–4533. [Google Scholar] [CrossRef]
- Kim, N.Y.; Trinh, N.T.; Ahn, S.G.; Kim, S.A. Cinnamaldehyde Protects against Oxidative Stress and Inhibits the TNF-α-Induced Inflammatory Response in Human Umbilical Vein Endothelial Cells. Int. J. Mol. Med. 2020, 46, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, P.; Wan, J.; Hou, J.; Zhou, P. Cinnamaldehyde Attenuates High Glucose-Induced Oxidative Stress and Endothelial Dysfunction as an Nrf2 Activator. Atherosclerosis 2016, 252, e145. [Google Scholar] [CrossRef]
- Gong, W.; Li, J.; Zhu, G.; Wang, Y.; Zheng, G.; Kan, Q. Chlorogenic Acid Relieved Oxidative Stress Injury in Retinal Ganglion Cells through IncRNA-TUG1/Nrf2. Cell Cycle 2019, 18, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Chen, W.; Gu, X.; Shan, R.; Zou, J.; Liu, G.; Shahid, M.; Gao, J.; Han, B. Cytoprotective Effect of Chlorogenic Acid against Hydrogen Peroxide-Induced Oxidative Stress in MC3T3-E1 Cells through PI3K/Akt-Mediated Nrf2/HO-1 Signaling Pathway. Oncotarget 2017, 8, 14680–14692. [Google Scholar] [CrossRef]
- Cha, J.W.; Piao, M.J.; Kim, K.C.; Yao, C.W.; Zheng, J.; Kim, S.M.; Hyun, C.L.; Ahn, Y.S.; Hyun, J.W. The Polyphenol Chlorogenic Acid Attenuates UVB-Mediated Oxidative Stress in Human HaCaT Keratinocytes. Biomol. Ther. 2014, 22, 136–142. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, L.; Ruan, Z.; Mi, S.; Jiang, M.; Li, X.; Wu, X.; Deng, Z.; Yin, Y. Chlorogenic Acid Ameliorates Intestinal Mitochondrial Injury by Increasing Antioxidant Effects and Activity of Respiratory Complexes. Biosci. Biotechnol. Biochem. 2016, 80, 962–971. [Google Scholar] [CrossRef]
- Liang, N.; Dupuis, J.H.; Yada, R.Y.; Kitts, D.D. Chlorogenic Acid Isomers Directly Interact with Keap 1-Nrf2 Signaling in Caco-2 Cells. Mol. Cell. Biochem. 2019, 457, 105–118. [Google Scholar] [CrossRef]
- Han, D.; Gu, X.; Gao, J.; Wang, Z.; Liu, G.; Barkema, H.W.; Han, B. Chlorogenic Acid Promotes the Nrf2/HO-1 Anti-Oxidative Pathway by Activating P21 Waf1/Cip1 to Resist Dexamethasone-Induced Apoptosis in Osteoblastic Cells. Free Radic. Biol. Med. 2019, 137, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Z.; Jiang, Z.K.; He, B.X.; Liu, X.B. Arctigenin Protects against Lipopolysaccharide-Induced Pulmonary Oxidative Stress and Inflammation in a Mouse Model via Suppression of MAPK, HO-1, and INOS Signaling. Inflammation 2015, 38, 1406–1414. [Google Scholar] [CrossRef]
- Yang, J.; Yin, H.S.; Cao, Y.J.; Jiang, Z.A.; Li, Y.J.; Song, M.C.; Wang, Y.F.; Wang, Z.H.; Yang, R.; Jiang, Y.F.; et al. Arctigenin Attenuates Ischemia/Reperfusion Induced Ventricular Arrhythmias by Decreasing Oxidative Stress in Rats. Cell. Physiol. Biochem. 2018, 49, 728–742. [Google Scholar] [CrossRef]
- Feng, T.; Cao, W.; Shen, W.; Zhang, L.; Gu, X.; Guo, Y.; Tsai, H.I.; Liu, X.; Li, J.; Zhang, J.; et al. Arctigenin Inhibits STAT3 and Exhibits Anticancer Potential in Human Triple-Negative Breast Cancer Therapy. Oncotarget 2017, 8, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.P.; Wang, G.; Sun, Q.M.; Wu, J.; Zou, X. Inhibitory Effect of Cinnamaldehyde on Invasion Capacities of Human Breast Cancer Cell Line MDA-MB-435S and Its Relation with Regulating the Expression of MiR-27a. Zhongguo Zhong Xi Yi Jie He Za Zhi Zhongguo Zhongxiyi Jiehe Zazhi = Chin. J. Integr. Tradit. West. Med./Zhongguo Zhong Xi Yi Jie He Xue Hui Zhongguo Zhong Yi Yan Jiu Yuan Zhu Ban 2014, 34, 964–969. [Google Scholar]
- Chiang, Y.F.; Chen, H.Y.; Huang, K.C.; Lin, P.H.; Hsia, S.M. Dietary Antioxidant Trans-Cinnamaldehyde Reduced Visfatin-Induced Breast Cancer Progression: In Vivo and in Vitro Study. Antioxidants 2019, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Wani, K.D.; Kadu, B.S.; Mansara, P.; Gupta, P.; Deore, A.V.; Chikate, R.C.; Poddar, P.; Dhole, S.D.; Kaul-Ghanekar, R. Synthesis, Characterization and in Vitro Study of Biocompatible Cinnamaldehyde Functionalized Magnetite Nanoparticles (CPGF Nps) for Hyperthermia and Drug Delivery Applications in Breast Cancer. PLoS ONE 2014, 9, e107315. [Google Scholar] [CrossRef] [PubMed]
- Rad, S.K.; Kanthimathi, M.S.; Malek, N.A.; Lee, G.S.; Looi, C.Y.; Wong, W.F. Cinnamomum Cassia Suppresses Caspase-9 through Stimulation of AKT1 in MCF-7 Cells but Not in MDA-MB-231 Cells. PLoS ONE 2015, 10, e0145216. [Google Scholar] [CrossRef]
- Najar, B.; Shortrede, J.E.; Pistelli, L.; Buhagiar, J. Chemical Composition and in Vitro Cytotoxic Screening of Sixteen Commercial Essential Oils on Five Cancer Cell Lines. Chem. Biodivers. 2020, 17, e1900478. [Google Scholar] [CrossRef]
- Kubatka, P.; Kello, M.; Kajo, K.; Samec, M.; Jasek, K.; Vybohova, D.; Uramova, S.; Liskova, A.; Sadlonova, V.; Koklesova, L.; et al. Chemopreventive and Therapeutic Efficacy of Cinnamomum Zeylanicum L. Bark in Experimental Breast Carcinoma: Mechanistic In Vivo and In Vitro Analyses. Molecules 2020, 25, 1399. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, K.; Nam, S.; Anderson, R.A.; Jove, R.; Wen, W. Novel Angiogenesis Inhibitory Activity in Cinnamon Extract Blocks VEGFR2 Kinase and Downstream Signaling. Carcinogenesis 2010, 31, 481–488. [Google Scholar] [CrossRef]
- Deka, S.J.; Gorai, S.; Manna, D.; Trivedi, V. Evidence of PKC Binding and Translocation to Explain the Anticancer Mechanism of Chlorogenic Acid in Breast Cancer Cells. Curr. Mol. Med. 2017, 17, 79–89. [Google Scholar] [CrossRef]
- Changizi, Z.; Moslehi, A.; Rohani, A.H.; Eidi, A. Chlorogenic Acid Induces 4T1 Breast Cancer Tumor’s Apoptosis via P53, Bax, Bcl-2, and Caspase-3 Signaling Pathways in BALB/c Mice. J. Biochem. Mol. Toxicol. 2021, 35, e22642. [Google Scholar] [CrossRef]
- Lee, W.J.; Zhu, B.T. Inhibition of DNA Methylation by Caffeic Acid and Chlorogenic Acid, Two Common Catechol-Containing Coffee Polyphenols. Carcinogenesis 2006, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Yue, C.H.; Pan, Y.R.; Chiu, Y.W.; Liu, J.Y.; Lin, K.I.; Lee, C.J. Isochlorogenic Acid C Reverses Epithelial–Mesenchymal Transition via down-Regulation of EGFR Pathway in MDA-MB-231 Cells. Anticancer Res. 2018, 38, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Liang, X.; Zhu, S.; Liu, C.; Wang, S.; Zhang, Q.; Zhao, J.; Song, L. Chlorogenic Acid Induces Apoptosis, Inhibits Metastasis and Improves Antitumor Immunity in Breast Cancer via the NF-ΚB Signaling Pathway. Oncol. Rep. 2021, 45, 717–727. [Google Scholar] [CrossRef]
- Hsieh, C.-J.; Kuo, P.-L.; Hsu, Y.-C.; Huang, Y.-F.; Tsai, E.-M.; Hsu, Y.-L. Arctigenin, A Dietary Phytoestrogen, Induces Apoptosis of Estrogen Receptor-Negative Breast Cancer Cells through the ROS/P38 MAPK Pathway and Epigenetic Regulation. Free Radic. Biol. Med. 2014, 67, 159–170. [Google Scholar] [CrossRef]
- Shi, H.; Zhao, L.; Guo, X.; Fang, R.; Zhang, H.; Dong, G.; Fu, J.; Yan, F.; Zhang, J.; Ning, Z.; et al. Arctigenin Attenuates Breast Cancer Progression through Decreasing GM-CSF/TSLP/STAT3/β-Catenin Signaling. Int. J. Mol. Sci. 2020, 21, 6357. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Imm, J.Y.; Lee, S.H. Β-Catenin Mediates Anti-Adipogenic and Anticancer Effects of Arctigenin in Preadipocytes and Breast Cancer Cells. J. Agric. Food Chem. 2017, 65, 2513–2520. [Google Scholar] [CrossRef]
- Zhu, L.; Shen, X.-B.; Yuan, P.-C.; Shao, T.-L.; Wang, G.-D.; Liu, X.-P. Arctigenin Inhibits Proliferation of ER-Positive Breast Cancer Cells through Cell Cycle Arrest Mediated by GSK3-Dependent Cyclin D1 Degradation. Life Sci. 2020, 256, 117983. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, T.; Chun, S.Y.; Lee, K.S.; Kim, S.; Nam, K.S. The Anti-Metastatic Effects of the Phytoestrogen Arctigenin on Human Breast Cancer Cell Lines Regardless of the Status of ER Expression. Int. J. Oncol. 2017, 50, 727–735. [Google Scholar] [CrossRef]
- Huang, Q.; Qin, S.; Yuan, X.; Zhang, L.; Ji, J.; Liu, X.; Ma, W.; Zhang, Y.; Liu, P.; Sun, Z.; et al. Arctigenin Inhibits Triple-Negative Breast Cancers by Targeting CIP2A to Reactivate Protein Phosphatase 2A. Oncol. Rep. 2017, 38, 598–606. [Google Scholar] [CrossRef]
- Lou, C.; Zhu, Z.; Zhao, Y.; Zhu, R.; Zhao, H. Arctigenin, a Lignan from Arctium Lappa L., Inhibits Metastasis of Human Breast Cancer Cells through the Downregulation of MMP-2/-9 and Heparanase in MDA-MB-231 Cells. Oncol. Rep. 2017, 37, 179–184. [Google Scholar] [CrossRef]
- Lee, M.; Lee, K.; Nam, K. Anti-metastatic Effects of Arctigenin Are Regulated by MAPK/AP-1 Signaling in 4T-1 Mouse Breast Cancer Cells. Mol. Med. Rep. 2020, 21, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, B.; Chung, S.; Wu, Y.; Henning, S.M.; Vadgama, J.V. Increased Chemopreventive Effect by Combining Arctigenin, Green Tea Polyphenol and Curcumin in Prostate and Breast Cancer Cells. RSC Adv. 2014, 4, 35242–35250. [Google Scholar] [CrossRef]
- Maxwell, T.; Lee, K.S.; Kim, S.; Nam, K.-S. Arctigenin Inhibits the Activation of the MTOR Pathway, Resulting in Autophagic Cell Death and Decreased ER Expression in ER-Positive Human Breast Cancer Cells. Int. J. Oncol. 2018, 52, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.-R.; Kim, H.S.; Lee, J.M.; Choi, J.H.; Kim, S.N.; Kim, D.K.; Kim, J.H.; Mun, S.H.; Kim, J.W.; Jeon, H.S. Arctigenin Suppresses Receptor Activator of Nuclear Factor ΚB Ligand (RANKL)-Mediated Osteoclast Differentiation in Bone Marrow-Derived Macrophages. Eur. J. Pharmacol. 2012, 682, 29–36. [Google Scholar] [CrossRef]
- Wu, S.J.; Ng, L.T. Antiproliferative Activity of Cinnamomum Cassia Constituents and Effects of Pifithrin-Alpha on Their Apoptotic Signaling Pathways in Hep G2 Cells. Evid. Based Complementary Altern. Med. 2011, 2011, 492148. [Google Scholar] [CrossRef]
- Li, J.; Teng, Y.; Liu, S.; Wang, Z.; Chen, Y.; Zhang, Y.; Xi, S.; Xu, S.; Wang, R.; Zou, X. Cinnamaldehyde Affects the Biological Behavior of Human Colorectal Cancer Cells and Induces Apoptosis via Inhibition of the PI3K/Akt Signaling Pathway. Oncol. Rep. 2016, 35, 1501–1510. [Google Scholar] [CrossRef]
- Wu, S.J.; Ng, L.T.; Lin, C.C. Cinnamaldehyde-Induced Apoptosis in Human PLC/PRF/5 Cells through Activation of the Proapoptotic Bcl-2 Family Proteins and MAPK Pathway. Life Sci. 2005, 77, 938–951. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-T.; Tai, C.-J.; Chang, S.-P.; Chen, J.-L.; Wu, S.-J.; Lin, C.-C. Cinnamaldehyde-Induced Apoptosis in Human Hepatoma PLC/PRF/5 Cells Involves the Mitochondrial Death Pathway and Is Sensitive to Inhibition by Cyclosporin A and z-VAD-Fmk. Anti-Cancer Agents Med. Chem. 2013, 13, 1565–1574. [Google Scholar] [CrossRef]
- Yang, J.S.; Liu, C.W.; Ma, Y.S.; Weng, S.W.; Tang, N.Y.; Wu, S.H.; Ji, B.C.; Ma, C.Y.; Ko, Y.C.; Funayama, S.; et al. Chlorogenic Acid Induces Apoptotic Cell Death in U937 Leukemia Cells through Caspase-and Mitochondria-Dependent Pathways. Vivo 2012, 26, 971–978. [Google Scholar] [PubMed]
- Rakshit, S.; Mandal, L.; Pal, B.C.; Bagchi, J.; Biswas, N.; Chaudhuri, J.; Chowdhury, A.A.; Manna, A.; Chaudhuri, U.; Konar, A.; et al. Involvement of ROS in Chlorogenic Acid-Induced Apoptosis of Bcr-Abl+ CML Cells. Biochem. Pharmacol. 2010, 80, 1662–1675. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Izawa, Y.; Onodera, D.; Tagami, M. Chlorogenic Acid Regulates Apoptosis and Stem Cell Marker-Related Gene Expression in A549 Human Lung Cancer Cells. Mol. Cell. Biochem. 2018, 441, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Xie, Z.; Rao, J.; Xu, G.; Huang, K.; Li, W.; Yin, Z. Chlorogenic Acid Inhibits Proliferation and Induces Apoptosis in A498 Human Kidney Cancer Cells via Inactivating PI3K/Akt/MTOR Signalling Pathway. J. Pharm. Pharmacol. 2019, 71, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Cao, S.; Zhou, H.; Hua, L.; Zhang, S.; Cao, J. Mechanism of Arctigenin-Induced Specific Cytotoxicity against Human Hepatocellular Carcinoma Cell Lines: Hep G2 and SMMC7721. PLoS ONE 2015, 10, e0125727. [Google Scholar] [CrossRef]
- Lu, Z.; Zhou, H.; Zhang, S.; Dai, W.; Zhang, Y.; Hong, L.; Chen, F.; Cao, J. Activation of Reactive Oxygen Species-Mediated Mitogen-Activated Protein Kinases Pathway Regulates Both Extrinsic and Intrinsic Apoptosis Induced by Arctigenin in Hep G2. J. Pharm. Pharmacol. 2020, 72, 29–43. [Google Scholar] [CrossRef]
- Gu, Y.; Qi, C.; Sun, X.; Ma, X.; Zhang, H.; Hu, L.; Yuan, J.; Yu, Q. Arctigenin Preferentially Induces Tumor Cell Death under Glucose Deprivation by Inhibiting Cellular Energy Metabolism. Biochem. Pharmacol. 2012, 84, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Shigemi, Z.; Hara, N.; Moriguchi, M.; Ikeda, M.; Watanabe, T.; Fujimuro, M. Arctigenin Induces the Apoptosis of Primary Effusion Lymphoma Cells under Conditions of Glucose Deprivation. Int. J. Oncol. 2018, 52, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Brecht, K.; Riebel, V.; Couttet, P.; Paech, F.; Wolf, A.; Chibout, S.D.; Pognan, F.; Krähenbühl, S.; Uteng, M. Mechanistic Insights into Selective Killing of OXPHOS-Dependent Cancer Cells by Arctigenin. Toxicol. Vitr. 2017, 40, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Nam, H.S.; Cho, M.K.; Lee, S.H. Arctigenin Induces Necroptosis through Mitochondrial Dysfunction with CCN1 Upregulation in Prostate Cancer Cells under Lactic Acidosis. Mol. Cell. Biochem. 2020, 467, 45–56. [Google Scholar] [CrossRef]
- Lee, Y.J.; Oh, J.E.; Lee, S.H. Arctigenin Shows Preferential Cytotoxicity to Acidity-Tolerant Prostate Carcinoma PC-3 cells through ROS-Mediated Mitochondrial Damage and the Inhibition of PI3K/Akt/MTOR Pathway. Biochem. Biophys. Res. Commun. 2018, 505, 1244–1250. [Google Scholar] [CrossRef]
- Hou, N.; Liu, N.; Han, J.; Yan, Y.; Li, J. Chlorogenic Acid Induces Reactive Oxygen Species Generation and Inhibits the Viability of Human Colon Cancer Cells. Anti-Cancer Drugs 2017, 28, 59–65. [Google Scholar] [CrossRef]
- Ka, H.; Park, H.J.; Jung, H.J.; Choi, J.W.; Cho, K.S.; Ha, J.; Lee, K.T. Cinnamaldehyde Induces Apoptosis by ROS-Mediated Mitochondrial Permeability Transition in Human Promyelocytic Leukemia HL-60 Cells. Cancer Lett. 2003, 196, 143–152. [Google Scholar] [CrossRef]
- Dornish, J.M.; Pettersen, E.O.; Oftebro, R. Synergistic Cell Inactivation of Human NHIK 3025 Cells by Cinnamaldehyde in Combination with Cis-Diamminedichloroplatinum(II). Cancer Res. 1988, 48, 938–942. [Google Scholar] [PubMed]
- Yu, C.; Liu, S.L.; Qi, M.H.; Zou, X. Cinnamaldehyde/ Chemotherapeutic Agents Interaction and Drug-Metabolizing Genes in Colorectal Cancer. Mol. Med. Rep. 2014, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.E.; Zhuang, Y.W.; Zhou, J.Y.; Liu, S.L.; Wang, R.P.; Shu, P. Cinnamaldehyde Enhances Apoptotic Effect of Oxaliplatin and Reverses Epithelial-Mesenchymal Transition and Stemnness in Hypoxic Colorectal Cancer Cells. Exp. Cell Res. 2019, 383, 111500. [Google Scholar] [CrossRef] [PubMed]
- Refolo, M.G.; Lippolis, C.; Carella, N.; Cavallini, A.; Messa, C.; D’Alessandro, R. Chlorogenic Acid Improves the Regorafenib Effects in Human Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 1518. [Google Scholar] [CrossRef]
- Yan, Y.; Li, J.; Han, J.; Hou, N.; Song, Y.; Dong, L. Chlorogenic Acid Enhances the Effects of 5-Fluorouracil in Human Hepatocellular Carcinoma Cells through the Inhibition of Extracellular Signal-Regulated Kinases. Anti-Cancer Drugs 2015, 26, 540–546. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Yao, Z.T.; Liang, G.K.; Chen, X.; Wu, H.H.; Jin, L.; Ding, L. Combination of Lapatinib with Chlorogenic Acid Inhibits Breast Cancer Metastasis by Suppressing Macrophage M2 Polarization. Zhejiang Da Xue Xue Bao. Yi Xue Ban J. Zhejiang Univ. Med. Sci. 2015, 44, 493–499. [Google Scholar]
- Yao, X.; Zhu, F.; Zhao, Z.; Liu, C.; Luo, L.; Yin, Z. Arctigenin Enhances Chemosensitivity of Cancer Cells to Cisplatin through Inhibition of the STAT3 Signaling Pathway. J. Cell. Biochem. 2011, 112, 2837–2849. [Google Scholar] [CrossRef]
- Wang, H.Q.; Jin, J.J.; Wang, J. Arctigenin Enhances Chemosensitivity to Cisplatin in Human Nonsmall Lung Cancer H460 Cells through Downregulation of Survivin Expression. J. Biochem. Mol. Toxicol. 2014, 28, 39–45. [Google Scholar] [CrossRef]
- Wang, Y.; Lina, L.; Xu, L.; Yang, Z.; Qian, Z.; Zhou, J.; Suoni, L. Arctigenin Enhances the Sensitivity of Cisplatin Resistant Colorectal Cancer Cell by Activating Autophagy. Biochem. Biophys. Res. Commun. 2019, 520, 20–26. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, M.G.; Kwon, Y.S.; Nam, K.S. Arctigenin Enhances the Cytotoxic Effect of Doxorubicin in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2997. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Geng, S.; Du, Z.; Yao, J.; Zheng, Y.; Li, Z.; Zhang, Z.; Li, J.; Duan, Y.; Du, G. Berberine and Cinnamaldehyde Together Prevent Lung Carcinogenesis. Oncotarget 2017, 8, 76385–76397. [Google Scholar] [CrossRef] [PubMed]
- Ekbatan, S.S.; Li, X.Q.; Ghorbani, M.; Azadi, B.; Kubow, S. Chlorogenic Acid and Its Microbial Metabolites Exert Anti-Proliferative Effects, S-Phase Cell-Cycle Arrest and Apoptosis in Human Colon Cancer Caco-2 Cells. Int. J. Mol. Sci. 2018, 19, 723. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Cuiping, C.; Pu, R.; Weihua, Y. Study on the Anticancer Effect of an Astragaloside- And Chlorogenic Acid-Containing Herbal Medicine (RLT-03) in Breast Cancer. Evid. Based Complementary Altern. Med. 2020, 2020, 1515081. [Google Scholar] [CrossRef]
- Wang, P.; Phan, T.; Gordon, D.; Chung, S.; Henning, S.M.; Vadgama, J.V. Arctigenin in Combination with Quercetin Synergistically Enhances the Antiproliferative Effect in Prostate Cancer Cells. Mol. Nutr. Food Res. 2015, 59, 250–261. [Google Scholar] [CrossRef]
- Lv, C.; Yuan, X.; Zeng, H.W.; Liu, R.H.; Zhang, W.D. Protective Effect of Cinnamaldehyde against Glutamate-Induced Oxidative Stress and Apoptosis in PC12 Cells. Eur. J. Pharmacol. 2017, 815, 487–494. [Google Scholar] [CrossRef]
- Jiang, Y.; Kusama, K.; Satoh, K.; Takayama, F.; Watanabe, S.; Sakagami, H. Induction of Cytotoxicity by Chlorogenic Acid in Human Oral Tumor Cell Lines. Phytomedicine 2000, 7, 483–491. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Liu, J.; Xu, M.; Tong, X.; Wang, J. Chlorogenic Acid Prevents Isoproterenol-Induced DNA Damage in Vascular Smooth Muscle Cells. Mol. Med. Rep. 2016, 14, 4063–4068. [Google Scholar] [CrossRef]
- Huang, S.; Wang, L.L.; Xue, N.N.; Li, C.; Guo, H.H.; Ren, T.K.; Zhan, Y.; Li, W.B.; Zhang, J.; Chen, X.G.; et al. Chlorogenic Acid Effectively Treats Cancers through Induction of Cancer Cell Differentiation. Theranostics 2019, 9, 6745–6763. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Calderón-Montaño, J.M.; Orta, M.L.; Pastor, N.; Pérez-Guerrero, C.; Austin, C.; Mateos, S.; López-Lázaro, M. The Coffee Constituent Chlorogenic Acid Induces Cellular DNA Damage and Formation of Topoisomerase I- and II-DNA Complexes in Cells. J. Agric. Food Chem. 2012, 60, 7384–7391. [Google Scholar] [CrossRef]
- Han, Y.H.; Kee, J.Y.; Kim, D.S.; Mun, J.G.; Jeong, M.Y.; Park, S.H.; Choi, B.M.; Park, S.J.; Kim, H.J.; Um, J.Y.; et al. Arctigenin Inhibits Lung Metastasis of Colorectal Cancer by Regulating Cell Viability and Metastatic Phenotypes. Molecules 2016, 21, 1135. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Solorzano, W.; Diaz, T.; Magyar, C.E.; Henning, S.M.; Vadgama, J.V. Arctigenin Inhibits Prostate Tumor Cell Growth In Vitro and In Vivo. Clin. Nutr. Exp. 2017, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Susanti, S.; Iwasaki, H.; Itokazu, Y.; Nago, M.; Taira, N.; Saitoh, S.; Oku, H. Tumor Specific Cytotoxicity of Arctigenin Isolated from Herbal Plant Arctium Lappa L. J. Nat. Med. 2012, 66, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Susanti, S.; Iwasaki, H.; Inafuku, M.; Taira, N.; Oku, H. Mechanism of Arctigenin-Mediated Specific Cytotoxicity against Human Lung Adenocarcinoma Cell Lines. Phytomedicine 2013, 21, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular Profiling of Breast Cancer Cell Lines Defines Relevant Tumor Models and Provides a Resource for Cancer Gene Discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Van Genderen, H.; Kenis, H.; Lux, P.; Ungeth, L.; Maassen, C.; Deckers, N.; Narula, J.; Hofstra, L.; Reutelingsperger, C. In Vitro Measurement of Cell Death with the Annexin A5 Affinity Assay. Nat. Protoc. 2006, 1, 363–367. [Google Scholar] [CrossRef]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The Mitochondrial Membrane Potential (Δψm) in Apoptosis; an Update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Chazotte, B. Labeling Mitochondria with Mitotracker Dyes. Cold Spring Harb. Protoc. 2011, 6, 990–992. [Google Scholar] [CrossRef]
- Henics, T.; Wheatley, D.N. Cytoplasmic Vacuolation, Adaptation and Cell Death: A View on New Perspectives and Features. Biol. Cell 1999, 91, 485–498. [Google Scholar] [CrossRef]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic Vacuolization in Cell Death and Survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; di Donato, M.; Pezone, A.; di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and Modulation of Apoptosis by Oxidative Stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Li, L.; Ishdorj, G.; Gibson, S.B. Reactive Oxygen Species Regulation of Autophagy in Cancer: Implications for Cancer Treatment. Free Radic. Biol. Med. 2012, 53, 1399–1410. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Kotamraju, S.; Chitambar, C.R.; Kalivendi, S.V.; Joseph, J.; Kalyanaraman, B. Transferrin Receptor-Dependent Iron Uptake Is Responsible for Doxorubicin-Mediated Apoptosis in Endothelial Cells. Role of Oxidant-Induced Iron Signaling in Apoptosis. J. Biol. Chem. 2002, 277, 17179–17187. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Berndtsson, M.; Hägg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute Apoptosis by Cisplatin Requires Induction of Reactive Oxygen Species but Is Not Associated with Damage to Nuclear DNA. Int. J. Cancer 2007, 120, 175–180. [Google Scholar] [CrossRef]
- Cappetta, D.; de Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxidative Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Santos, N.A.G.; Catão, C.S.; Martins, N.M.; Curti, C.; Bianchi, M.L.P.; Santos, A.C. Cisplatin-Induced Nephrotoxicity Is Associated with Oxidative Stress, Redox State Unbalance, Impairment of Energetic Metabolism and Apoptosis in Rat Kidney Mitochondria. Arch. Toxicol. 2007, 81, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Focaccetti, C.; Bruno, A.; Magnani, E.; Bartolini, D.; Principi, E.; Dallaglio, K.; Bucci, E.O.; Finzi, G.; Sessa, F.; Noonan, D.M.; et al. Effects of 5-Fluorouracil on Morphology, Cell Cycle, Proliferation, Apoptosis, Autophagy and Ros Production in Endothelial Cells and Cardiomyocytes. PLoS ONE 2015, 10, e0115686. [Google Scholar] [CrossRef]
- American Cancer Society How Chemotherapy Drugs Work. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/chemotherapy/how-chemotherapy-drugs-work.html (accessed on 29 January 2020).
- American Cancer Society Chemotherapy Side Effects. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/chemotherapy/chemotherapy-side-effects.html (accessed on 30 January 2020).
- American Cancer Society Chemotherapy for Breast Cancer. Available online: https://www.cancer.org/cancer/breast-cancer/treatment/chemotherapy-for-breast-cancer.html (accessed on 3 April 2020).
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Gaur, R.L.; Abdraboh, M.; Ireland, S.K.; Rao, P.N.; Raj, S.G.; Al-Riyami, H.; Shanmuganathan, S.; Gupta, I.; Murthy, S.N.; et al. Simultaneous Inhibition of Cell-Cycle, Proliferation, Survival, Metastatic Pathways and Induction of Apoptosis in Breast Cancer Cells by a Phytochemical Super-Cocktail: Genes That Underpin Its Mode of Action. J. Cancer 2014, 4, 703–715. [Google Scholar] [CrossRef]
- Raj, M.H.G.; Abd Elmageed, Z.Y.; Zhou, J.; Gaur, R.L.; Nguyen, L.; Azam, G.A.; Braley, P.; Rao, P.N.; Fathi, I.M.; Ouhtit, A. Synergistic Action of Dietary Phyto-Antioxidants on Survival and Proliferation of Ovarian Cancer Cells. Gynecol. Oncol. 2008, 110, 432–438. [Google Scholar] [CrossRef]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is There a Role for Mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Tokarchuk, A.V.; Panteleeva, A.A.; Mulkidjanian, A.Y.; Skulachev, V.P.; Chernyak, B.V. Induction of Autophagy by Depolarization of Mitochondria. Autophagy 2018, 14, 921–924. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide Is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple Cell Death Modalities and Their Key Features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane Blebbing during Apoptosis Results from Caspase-Mediated Activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef]
- Sperandio, S.; Poksay, K.S.; Schilling, B.; Crippen, D.; Gibson, B.W.; Bredesen, D.E. Identification of New Modulators and Protein Alterations in Non-Apoptotic Programmed Cell Death. J. Cell. Biochem. 2010, 111, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.; Swain, P.M.; Kam, Y.; Rogers, G.; Dranka, B.P. Abstract 3487: Bioenergetic Profiling of Cancer Cell Lines: Quantifying the Impact of Glycolysis on Cell Proliferation. Cancer Res. 2018, 78, 3487. [Google Scholar] [CrossRef]
- Owens, K.M.; Aykin-Burns, N.; Dayal, D.; Coleman, M.C.; Domann, F.E.; Spitz, D.R. Genomic Instability Induced by Mutant Succinate Dehydrogenase Subunit D (SDHD) Is Mediated by O2-• and H2O2. Free Radic. Biol. Med. 2012, 52, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Dreher, D.; Junod, A.F. Role of Oxygen Free Radicals in Cancer Development. Eur. J. Cancer 1996, 32, 30–38. [Google Scholar] [CrossRef]
- Itsumi, M.; Inoue, S.; Elia, A.J.; Murakami, K.; Sasaki, M.; Lind, E.F.; Brenner, D.; Harris, I.S.; Chio, I.I.C.; Afzal, S.; et al. Idh1 Protects Murine Hepatocytes from Endotoxin-Induced Oxidative Stress by Regulating the Intracellular NADP +/NADPH Ratio. Cell Death Differ. 2015, 22, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, Y.; Aryal, B.; Chehab, L.; Rao, V.A. Atg7- and Keap1-Dependent Autophagy Protects Breast Cancer Cell Lines against Mitoquinone-Induced Oxidative Stress. Oncotarget 2014, 5, 1526–1537. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, J.; Xu, L.; Cao, Y.; Xu, F.; Yan, L.; Nie, M.; Yuan, N.; Zhang, S.; Zhao, R.; et al. Autophagy Maintains Ubiquitination-Proteasomal Degradation of Sirt3 to Limit Oxidative Stress in K562 Leukemia Cells. Oncotarget 2016, 7, 35692–35702. [Google Scholar] [CrossRef][Green Version]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell Division in Normal and Transformed Cells: The Possible Role of Superoxide and Hydrogen Peroxide. Med. Hypotheses 1981, 7, 21–42. [Google Scholar] [CrossRef]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-Modulated Therapeutic Approaches in Cancer Treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Storz, P. KRas, ROS and the Initiation of Pancreatic Cancer. Small GTPases 2017, 8, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Gal, K.L.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants Can Increase Melanoma Metastasis in Mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Cancer: Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Li, Q.C.; Liang, Y.; Tian, Y.; Hu, G.R. Arctigenin Induces Apoptosis in Colon Cancer Cells through ROS/P38MAPK Pathway. J. Buon 2016, 21, 87–94. [Google Scholar] [PubMed]
- Han, L.; Mei, J.; Ma, J.; Wang, F.; Gu, Z.; Li, J.; Zhang, Z.; Zeng, Y.; Lou, X.; Yao, X.; et al. Cinnamaldehyde Induces Endogenous Apoptosis of the Prostate Cancer-Associated Fibroblasts via Interfering the Glutathione-Associated Mitochondria Function. Med. Oncol. 2020, 37, 91. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Villeneuve, N.F.; Lamore, S.D.; Bause, A.S.; Jiang, T.; Zhang, D.D. The Cinnamon-Derived Dietary Factor Cinnamic Aldehyde Activates the Nrf2-Dependent Antioxidant Response in Human Epithelial Colon Cells. Molecules 2010, 15, 3338–3355. [Google Scholar] [CrossRef]
- Long, M.; Tao, S.; De La Vega, M.R.; Jiang, T.; Wen, Q.; Park, S.L.; Zhang, D.D.; Wondrak, G.T. Nrf2-Dependent Suppression of Azoxymethane/Dextran Sulfate Sodium-Induced Colon Carcinogenesis by the Cinnamon-Derived Dietary Factor Cinnamaldehyde. Cancer Prev. Res. 2015, 8, 444–454. [Google Scholar] [CrossRef]
- Chew, E.H.; Nagle, A.A.; Zhang, Y.; Scarmagnani, S.; Palaniappan, P.; Bradshaw, T.D.; Holmgren, A.; Westwell, A.D. Cinnamaldehydes Inhibit Thioredoxin Reductase and Induce Nrf2: Potential Candidates for Cancer Therapy and Chemoprevention. Free Radic. Biol. Med. 2010, 48, 98–111. [Google Scholar] [CrossRef]
- Huang, T.C.; Chung, Y.L.; Wu, M.L.; Chuang, S.M. Cinnamaldehyde Enhances Nrf2 Nuclear Translocation to Upregulate Phase II Detoxifying Enzyme Expression in HepG2 Cells. J. Agric. Food Chem. 2011, 59, 5164–5171. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.Y.; Choi, J.S.; Kim, J.E.; Jeong, J.W. Cinnamic Aldehyde Suppresses Hypoxia-Induced Angiogenesis via Inhibition of Hypoxia-Inducible Factor-1α Expression during Tumor Progression. Biochem. Pharmacol. 2015, 98, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.; Jana, S.; Sarkar, A.; Mandal, D.P.; Bhattacharjee, S. The Inhibition of Hypoxia-Induced Angiogenesis and Metastasis by Cinnamaldehyde Is Mediated by Decreasing HIF-1α Protein Synthesis via PI3K/Akt Pathway. BioFactors 2019, 45, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lu, Y.; Yang, G.; Wu, J. Research on Tumorigenicity of Cinnamaldehyde in Melanoma Cell Lines and Its Mechanism. Tumor Biol. 2014, 35, 5717–5722. [Google Scholar] [CrossRef]
- Lee, M.S.; Lee, S.O.; Kim, K.R.; Lee, H.J. Sphingosine Kinase-1 Involves the Inhibitory Action of HIF-1α by Chlorogenic Acid in Hypoxic DU145 Cells. Int. J. Mol. Sci. 2017, 18, 325. [Google Scholar] [CrossRef]
- Park, J.J.; Hwang, S.J.; Park, J.H.; Lee, H.J. Chlorogenic Acid Inhibits Hypoxia-Induced Angiogenesis via down-Regulation of the HIF-1α/AKT Pathway. Cell. Oncol. 2015, 38, 111–118. [Google Scholar] [CrossRef]
- Chiu, D.K.-C.; Tse, A.P.-W.; Law, C.-T.; Xu, I.M.-J.; Lee, D.; Chen, M.; Lai, R.K.-H.; Yuen, V.W.-H.; Cheu, J.W.-S.; Ho, D.W.-H.; et al. Hypoxia Regulates the Mitochondrial Activity of Hepatocellular Carcinoma Cells through HIF/HEY1/PINK1 Pathway. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factor 1: Regulator of Mitochondrial Metabolism and Mediator of Ischemic Preconditioning. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | FSC Voltage | SSC Voltage | BL1 Voltage | YL1 Voltage |
|---|---|---|---|---|
| MCF-7 | 150 | 300 | 185 | 290 |
| MDA-MB-231 | 150 | 300 | 190 | 270 |
| HCC1419 | 160 | 290 | 210 | 285 |
| MCF-10A | 150 | 300 | 210 | 300 |
| MCF-12F | 150 | 300 | 220 | 300 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuster, C.; Wolpert, N.; Moustaid-Moussa, N.; Gollahon, L.S. Combinatorial Effects of the Natural Products Arctigenin, Chlorogenic Acid, and Cinnamaldehyde Commit Oxidation Assassination on Breast Cancer Cells. Antioxidants 2022, 11, 591. https://doi.org/10.3390/antiox11030591
Schuster C, Wolpert N, Moustaid-Moussa N, Gollahon LS. Combinatorial Effects of the Natural Products Arctigenin, Chlorogenic Acid, and Cinnamaldehyde Commit Oxidation Assassination on Breast Cancer Cells. Antioxidants. 2022; 11(3):591. https://doi.org/10.3390/antiox11030591
Chicago/Turabian StyleSchuster, Caroline, Nicholas Wolpert, Naima Moustaid-Moussa, and Lauren S. Gollahon. 2022. "Combinatorial Effects of the Natural Products Arctigenin, Chlorogenic Acid, and Cinnamaldehyde Commit Oxidation Assassination on Breast Cancer Cells" Antioxidants 11, no. 3: 591. https://doi.org/10.3390/antiox11030591
APA StyleSchuster, C., Wolpert, N., Moustaid-Moussa, N., & Gollahon, L. S. (2022). Combinatorial Effects of the Natural Products Arctigenin, Chlorogenic Acid, and Cinnamaldehyde Commit Oxidation Assassination on Breast Cancer Cells. Antioxidants, 11(3), 591. https://doi.org/10.3390/antiox11030591