
Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation


 ,
,
Abstract
:1. Pathophysiology of Diabetic Retinopathy
2. Role of Müller Cells in Retinal Physiology
3. Müller Cells in DR
3.1. Oxidative Stress
3.2. Inflammation
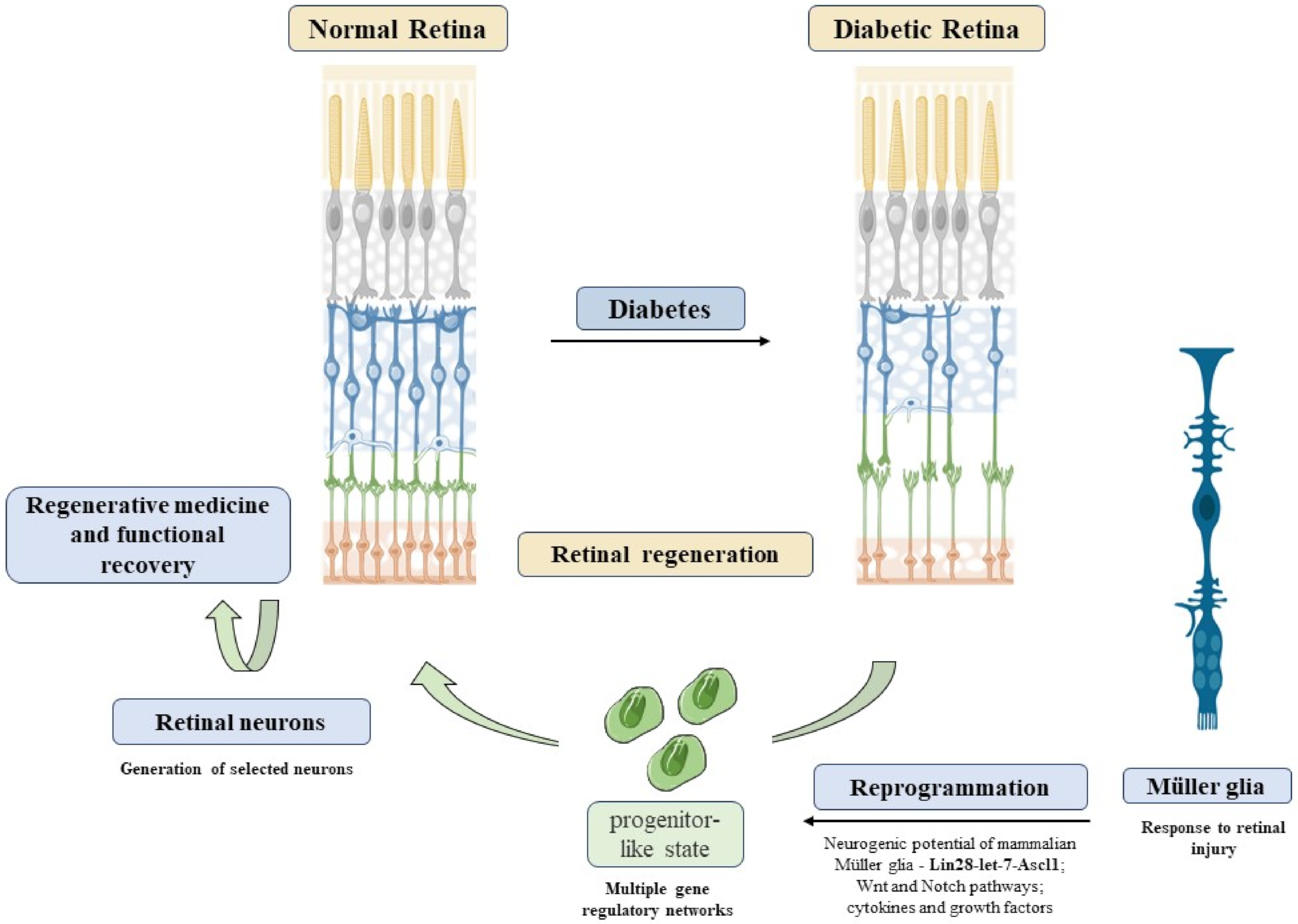
4. Future Directions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global Estimates of Diabetes Prevalence for 2017 and Projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Prevention of Diabetes Mellitus. Report of a WHO Study Group; WHO: Geneva, Switzerland, 1994; Volume 844.
- Pereira-Figueiredo, D.; Brito, R.; Araújo, D.S.M.; Nascimento, A.A.; Lyra, E.S.B.; Cheibub, A.M.S.S.; Pereira Netto, A.D.; Ventura, A.L.M.; Paes-de-Carvalho, R.C.K.; Calaza, K.C. Caffeine Exposure Ameliorates Acute Ischemic Cell Death in Avian Developing Retina. Purinergic Signal. 2020, 16, 41–59. [Google Scholar] [CrossRef]
- Lin, A.D.; Lee, A.Y.; Zhang, Q.; Rezaei, K.A.; Kinyoun, J.; Wang, R.K.; Lee, C.S. Association Between OCT-based Microangiography Perfusion Indices and Diabetic Retinopathy Severity. Br. J. Ophthalmol. 2018, 101, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Gong, C.; Chen, X.; Zhou, H.; Yan, J.; Hong, W. Associations between Vascular Endothelial Growth Factor Gene Polymorphisms and Different Types of Diabetic Retinopathy Susceptibility: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2021, 2021, 7059139. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Ripps, H. Neurovascular Interaction and the Pathophysiology of Diabetic Retinopathy. Exp. Diabetes Res. 2011, 2011, 693426. [Google Scholar] [CrossRef]
- Tarr, J.M.; Kaul, K.; Chopra, M.; Kohner, E.M.; Chibber, R. Pathophysiology of Diabetic Retinopathy. ISRN Ophthalmol. 2013, 2013, 343560. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.J. A New View of Diabetic Retinopathy: A Neurodegenerative Disease of the Eye. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2003, 27, 283–290. [Google Scholar] [CrossRef]
- Barber, A.J.; Gardner, T.W.; Abcouwer, S.F. The Significance of Vascular and Neural Apoptosis to the Pathology of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1156–1163. [Google Scholar] [CrossRef]
- Jonas, J.B.; Schneider, U.; Naumann, G.O. Count and Density of Human Retinal Photoreceptors. Graefe’s Arch. Clin. Exp. Ophthalmol. = Albr. von Graefes Arch. fur Klin. und Exp. Ophthalmol. 1992, 230, 505–510. [Google Scholar] [CrossRef]
- Curcio, C.A.; Sloan, K.R.; Kalina, R.E.; Hendrickson, A.E. Human Photoreceptor Topography. J. Comp. Neurol. 1990, 292, 497–523. [Google Scholar] [CrossRef]
- Watson, A.B. A Formula for Human Retinal Ganglion Cell Receptive Field Density as a Function of Visual Field Location. J. Vis. 2014, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong-Riley, M.T.T. Energy Metabolism of the Visual System. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Campos, V.S.; Calaza, K.C.; Adesse, D. Implications of TORCH Diseases in Retinal Development—Special Focus on Congenital Toxoplasmosis. Front. Cell. Infect. Microbiol. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Cheung, C.M.G.; Larsen, M.; Sharma, S.; Simó, R. Diabetic Retinopathy. Nat. Rev. Dis. Prim. 2016, 17, 16012. [Google Scholar] [CrossRef]
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015, 582060. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural Apoptosis in the Retina during Experimental and Human Diabetes. Early Onset and Effect of Insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, E.; Hernández, C.; Miralles, A.; Huguet, P.; Farrés, J.; Simó, R. Lower Somatostatin Expression Is an Early Event in Diabetic Retinopathy and Is Associated with Retinal Neurodegeneration. Diabetes Care 2007, 30, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.M.; Roon, P.; Van Ells, T.K.; Ganapathy, V.; Smith, S.B. Death of Retinal Neurons in Streptozotocin-Induced Diabetic Mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3330–3336. [Google Scholar] [CrossRef] [Green Version]
- Olivares, A.M.; Althoff, K.; Chen, G.F.; Wu, S.; Morrisson, M.A.; DeAngelis, M.M.; Haider, N. Animal Models of Diabetic Retinopathy. Curr. Diab. Rep. 2017, 17, 93. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Bearse, M.A.J.; Schneck, M.E.; Barez, S.; Jacobsen, C.H.; Adams, A.J. Multifocal Electroretinogram Delays Predict Sites of Subsequent Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 948–954. [Google Scholar] [CrossRef]
- Juen, S.; Kieselbach, G.F. Electrophysiological Changes in Juvenile Diabetics without Retinopathy. Arch. Ophthalmol. 1990, 108, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Shirao, Y.; Kawasaki, K. Electrical Responses from Diabetic Retina. Prog. Retin. Eye Res. 1998, 17, 59–76. [Google Scholar] [CrossRef]
- Gastinger, M.J.; Singh, R.S.J.; Barber, A.J. Loss of Cholinergic and Dopaminergic Amacrine Cells in Streptozotocin- Diabetic Rat and Ins2Akita-Diabetic Mouse Retinas. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3143–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roufail, E.; Soulis, T.; Boel, E.; Cooper, M.E.; Rees, S. Depletion of Nitric Oxide Synthase-Containing Neurons in the Diabetic Retina: Reversal by Aminoguanidine. Diabetologia 1998, 41, 1419–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, M.; Tanaka, T.; Nawa, H.; Usui, T.; Fukuchi, T.; Ikeda, K.; Abe, H.; Takei, N. Early Retinal Neuropathy of Streptozotocin-Induced. Am. Diabetes Assoc. 2004, 53, 1–8. [Google Scholar]
- Carpineto, P.; Toto, L.; Aloia, R.; Ciciarelli, V.; Borrelli, E.; Vitacolonna, E.; Nicola, M.D.; Antonio, L.D.; Mastropasqua, R. Neuroretinal Alterations in the Early Stages of Diabetic Retinopathy in Patients with Type 2 Diabetes Mellitus. Eye 2016, 30, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.S.K.; Chiang, P.P.C.; Tan, G.; Cheung, C.M.G.; Cheng, C.Y.; Cheung, C.Y.; Wong, T.Y.; Lamoureux, E.L.; Ikram, M.K. Retinal Ganglion Cell Neuronal Damage in Diabetes and Diabetic Retinopathy. Clin. Exp. Ophthalmol. 2016, 44, 243–250. [Google Scholar] [CrossRef]
- Sohn, E.H.; Van Dijk, H.W.; Jiao, C.; Kok, P.H.B.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; Van Velthoven, M.E.J.; et al. Retinal Neurodegeneration May Precede Microvascular Changes Characteristic of Diabetic Retinopathy in Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [Green Version]
- Vujosevic, S.; Muraca, A.; Gatti, V.; Masoero, L.; Brambilla, M.; Cannillo, B.; Villani, E.; Nucci, P.; De Cillà, S. Peripapillary Microvascular and Neural Changes in Diabetes Mellitus: An OCT-Angiography Study. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5074–5081. [Google Scholar] [CrossRef] [Green Version]
- Markan, A.; Agarwal, A.; Arora, A.; Bazgain, K.; Rana, V.; Gupta, V. Novel Imaging Biomarkers in Diabetic Retinopathy and Diabetic Macular Edema. Ther. Adv. Ophthalmol. 2020, 12, 2515841420950513. [Google Scholar] [CrossRef]
- Mendonça, H.R.; Carvalho, J.N.A.; Abreu, C.A.; Mariano de Souza Aguiar dos Santos, D.; Carvalho, J.R.; Marques, S.A.; da Costa Calaza, K.; Martinez, A.M.B. Lack of Galectin-3 Attenuates Neuroinflammation and Protects the Retina and Optic Nerve of Diabetic Mice. Brain Res. 2018, 1700, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Araujo, A.P.B.; Matias, I.; Garcia, M.N.; Barros-Aragão, F.G.Q.; de Melo Reis, R.A.; Foguel, D.; Braga, C.; Figueiredo, C.P.; Romão, L.; et al. Astrocyte Glutamate Transporters Are Increased in an Early Sporadic Model of Synucleinopathy. Neurochem. Int. 2020, 138, 104758. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Matias, I.; Siqueira, M.; Stipursky, J.; Gomes, F.C.A. Astrocytes and the TGF-Β1 Pathway in the Healthy and Diseased Brain: A Double-Edged Sword. Mol. Neurobiol. 2019, 56, 4653–4679. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Bérgamo Araujo, A.P.; Melo, H.M.; Seixas da Silva, G.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef] [Green Version]
- Amaral, R.F.; Geraldo, L.H.M.; Einicker-Lamas, M.; E Spohr, T.C.L. de S.; Mendes, F.; Lima, F.R.S. Microglial Lysophosphatidic Acid Promotes Glioblastoma Proliferation and Migration via LPA(1) Receptor. J. Neurochem. 2021, 156, 499–512. [Google Scholar] [CrossRef]
- Moraes, C.A.; Santos, G.; de Sampaio e Spohr, T.C.L.; D’Avila, J.C.; Lima, F.R.S.; Benjamim, C.F.; Bozza, F.A.; Gomes, F.C.A. Activated Microglia-Induced Deficits in Excitatory Synapses Through IL-1β: Implications for Cognitive Impairment in Sepsis. Mol. Neurobiol. 2015, 52, 653–663. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller Cells in the Healthy and Diseased Retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Hamon, A.; Roger, J.E.; Yang, X.-J.; Perron, M. Müller Glial Cell-Dependent Regeneration of the Neural Retina: An Overview across Vertebrate Model Systems. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2016, 245, 727–738. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Glia of the Human Retina. Glia 2020, 68, 768–796. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-Neuron Interactions in the Mammalian Retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Kefalov, V.J. The Cone-Specific Visual Cycle. Prog. Retin. Eye Res. 2011, 30, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Morshedian, A.; Kaylor, J.J.; Ng, S.Y.; Tsan, A.; Frederiksen, R.; Xu, T.; Yuan, L.; Sampath, A.P.; Radu, R.A.; Fain, G.L.; et al. Light-Driven Regeneration of Cone Visual Pigments through a Mechanism Involving RGR Opsin in Müller Glial Cells. Neuron 2019, 102, 1172–1183.e5. [Google Scholar] [CrossRef] [PubMed]
- Franze, K.; Grosche, J.; Skatchkov, S.N.; Schinkinger, S.; Foja, C.; Schild, D.; Uckermann, O.; Travis, K.; Reichenbach, A.; Guck, J. Müller Cells Are Living Optical Fibers in the Vertebrate Retina. Proc. Natl. Acad. Sci. USA 2007, 104, 8287–8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, A.; Syrbe, S.; Görner, K.; Kacza, J.; Francke, M.; Wiedemann, P.; Reichenbach, A. The Primate Fovea: Structure, Function and Development. Prog. Retin. Eye Res. 2018, 66, 49–84. [Google Scholar] [CrossRef] [PubMed]
- Khmelinskii, I.; Makarov, V. Intermediate Filaments in the Retinal Müller Cells as Natural Light Energy Guides. J. Photochem. Photobiol. B Biol. 2019, 200, 111641. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Zueva, L.; Golubeva, T.; Korneeva, E.; Khmelinskii, I.; Inyushin, M. Errata: Quantum Mechanism of Light Transmission by the Intermediate Filaments in Some Specialized Optically Transparent Cells. Neurophotonics 2017, 4, 19801. [Google Scholar] [CrossRef] [Green Version]
- Lindenau, W.; Kuhrt, H.; Ulbricht, E.; Körner, K.; Bringmann, A.; Reichenbach, A. Cone-to-Müller Cell Ratio in the Mammalian Retina: A Survey of Seven Mammals with Different Lifestyle. Exp. Eye Res. 2019, 181, 38–48. [Google Scholar] [CrossRef]
- Omri, S.; Omri, B.; Savoldelli, M.; Jonet, L.; Thillaye-Goldenberg, B.; Thuret, G.; Gain, P.; Jeanny, J.C.; Crisanti, P.; Behar-Cohen, F. The Outer Limiting Membrane (OLM) Revisited: Clinical Implications. Clin. Ophthalmol. 2010, 4, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Daruich, A.; Matet, A.; Moulin, A.; Kowalczuk, L.; Nicolas, M.; Sellam, A.; Rothschild, P.R.; Omri, S.; Gélizé, E.; Jonet, L.; et al. Mechanisms of Macular Edema: Beyond the Surface. Prog. Retin. Eye Res. 2018, 63, 20–68. [Google Scholar] [CrossRef]
- Bejarano-Escobar, R.; Sánchez-Calderón, H.; Otero-Arenas, J.; Martín-Partido, G.; Francisco-Morcillo, J. Müller Glia and Phagocytosis of Cell Debris in Retinal Tissue. J. Anat. 2017, 231, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Sakami, S.; Imanishi, Y.; Palczewski, K. Müller Glia Phagocytose Dead Photoreceptor Cells in a Mouse Model of Retinal Degenerative Disease. FASEB J. 2019, 33, 3680–3692. [Google Scholar] [CrossRef] [PubMed]
- Balaratnasingam, C.; Chae, B.; Remmer, M.H.; Gomez, E.; Suzuki, M.; Engelbert, M.; Spaide, R.F. The Spatial Profile of Macular Pigments Is Related to the Topological Characteristics of the Foveal Avascular Zone. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7859–7865. [Google Scholar] [CrossRef] [Green Version]
- Candiello, J.; Balasubramani, M.; Schreiber, E.M.; Cole, G.J.; Mayer, U.; Halfter, W.; Lin, H. Biomechanical Properties of Native Basement Membranes. FEBS J. 2007, 274, 2897–2908. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Jo, A.O.; Frye, A.M.; Vazquez-Chona, F.; MaCaulay, N.; Thoreson, W.B.; Križaj, D. Swelling and Eicosanoid Metabolites Differentially Gate TRPV4 Channels in Retinal Neurons and Glia. J. Neurosci. 2014, 34, 15689–15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisignano, M.; Park, C.K.; Angioni, C.; Zhang, D.D.; von Hehn, C.; Cobos, E.J.; Ghasemlou, N.; Xu, Z.Z.; Kumaran, V.; Lu, R.; et al. 5,6-EET Is Released upon Neuronal Activity and Induces Mechanical Pain Hypersensitivity via TRPA1 on Central Afferent Terminals. J. Neurosci. 2012, 32, 6364–6372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza Monteiro de Araújo, D.; De Logu, F.; Adembri, C.; Rizzo, S.; Janal, M.N.; Landini, L.; Magi, A.; Mattei, G.; Cini, N.; Pandolfo, P.; et al. TRPA1 Mediates Damage of the Retina Induced by Ischemia and Reperfusion in Mice. Cell Death Dis. 2020, 11, 633. [Google Scholar] [CrossRef]
- Uckermann, O.; Wolf, A.; Kutzera, F.; Kalisch, F.; Beck-Sickinger, A.G.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Glutamate Release by Neurons Evokes a Purinergic Inhibitory Mechanism of Osmotic Glial Cell Swelling in the Rat Retina: Activation by Neuropeptide Y. J. Neurosci. Res. 2006, 83, 538–550. [Google Scholar] [CrossRef]
- Pannicke, T.; Wurm, A.; Iandiev, I.; Hollborn, M.; Linnertz, R.; Binder, D.K.; Kohen, L.; Wiedemann, P.; Steinhäuser, C.; Reichenbach, A.; et al. Deletion of Aquaporin-4 Renders Retinal Glial Cells More Susceptible to Osmotic Stress. J. Neurosci. Res. 2010, 88, 2877–2888. [Google Scholar] [CrossRef]
- Moseley, H.; Foulds, W.S.; Allan, D.; Kyle, P.M. Routes of Clearance of Radioactive Water from the Rabbit Vitreous. Br. J. Ophthalmol. 1984, 68, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Karwoski, C.J.; Lu, H.-K.; Newman, E.A. Spatial Buffering of Light-Evoked Potassium Increases by Retinal Müller (Glial) Cells. Science (80-) 1989, 244, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Tout, S.; Chan-Ling, T.; Holländer, H.; Stone, J. The Role of Müller Cells in the Formation of the Blood-Retinal Barrier. Neuroscience 1993, 55, 291–301. [Google Scholar] [CrossRef]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.R.; Coorey, N.J.; Killingsworth, M.; Sherman, L.S.; et al. Conditional Müller Cell Ablation Causes Independent Neuronal and Vascular Pathologies in a Novel Transgenic Model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef] [PubMed]
- Metea, M.R.; Newman, E.A. Glial Cells Dilate and Constrict Blood Vessels: A Mechanism of Neurovascular Coupling. J. Neurosci. 2006, 26, 2862–2870. [Google Scholar] [CrossRef]
- Newman, E.A. Functional Hyperemia and Mechanisms of Neurovascular Coupling in the Retinal Vasculature. J. Cereb. Blood Flow Metab. 2013, 33, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.A. Glial Cell Regulation of Neuronal Activity and Blood Flow in the Retina by Release of Gliotransmitters. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 1–9. [Google Scholar] [CrossRef]
- Amann, B.; Kleinwort, K.J.H.; Hirmer, S.; Sekundo, W.; Kremmer, E.; Hauck, S.M.; Deeg, C.A. Expression and Distribution Pattern of Aquaporin 4, 5 and 11 in Retinas of 15 Different Species. Int. J. Mol. Sci. 2016, 17, 1145. [Google Scholar] [CrossRef] [Green Version]
- Vujosevic, S.; Midena, E. Retinal Layers Changes in Human Preclinical and Early Clinical Diabetic Retinopathy Support Early Retinal Neuronal and Müller Cells Alterations. J. Diabetes Res. 2013, 2013, 905058. [Google Scholar] [CrossRef]
- Orduña Ríos, M.; Noguez Imm, R.; Hernández Godínez, N.M.; Bautista Cortes, A.M.; López Escalante, D.D.; Liedtke, W.; Martínez Torres, A.; Concha, L.; Thébault, S. TRPV4 Inhibition Prevents Increased Water Diffusion and Blood-Retina Barrier Breakdown in the Retina of Streptozotocin-Induced Diabetic Mice. PLoS ONE 2019, 14, e0212158. [Google Scholar] [CrossRef] [Green Version]
- Arredondo Zamarripa, D.; Noguez Imm, R.; Bautista Cortés, A.M.; Vázquez Ruíz, O.; Bernardini, M.; Fiorio Pla, A.; Gkika, D.; Prevarskaya, N.; López-Casillas, F.; Liedtke, W.; et al. Dual Contribution of TRPV4 Antagonism in the Regulatory Effect of Vasoinhibins on Blood-Retinal Barrier Permeability: Diabetic Milieu Makes a Difference. Sci. Rep. 2017, 7, 13094. [Google Scholar] [CrossRef]
- Li, H.; Chen, D.; Sun, W.; Chen, J.; Luo, C.; Xu, H.; Ma, J.H.; Tang, S. Katp Opener Attenuates Diabetic-Induced Müller Gliosis and Inflammation by Modulating Kir6.1 in Microglia. Investig. Ophthalmol. Vis. Sci. 2021, 62, 3. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Dukic-Stefanovic, S.; Pannicke, T.; Ulbricht, E.; Reichenbach, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Expression of Aquaporins in the Retina of Diabetic Rats. Curr. Eye Res. 2011, 36, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Pannicke, T.; Iandiev, I.; Wurm, A.; Uckermann, O.; Vom Hagen, F.; Reichenbach, A.; Wiedemann, P.; Hammes, H.P.; Bringmann, A. Diabetes Alters Osmotic Swelling Characteristics and Membrane Conductance of Glial Cells in Rat Retina. Diabetes 2006, 55, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Oosuka, S.; Kida, T.; Oku, H.; Horie, T.; Morishita, S.; Fukumoto, M.; Sato, T.; Ikeda, T. Effects of an Aquaporin 4 Inhibitor, TGN-020, on Murine Diabetic Retina. Int. J. Mol. Sci. 2020, 21, 2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, B.; Sun, J.-H.; Xiang, F.-F.; Liu, L.; Li, W.-J. Aquaporin 4 Knockdown Exacerbates Streptozotocin-Induced Diabetic Retinopathy through Aggravating Inflammatory Response. Exp. Eye Res. 2012, 98, 37–43. [Google Scholar] [CrossRef]
- Picconi, F.; Parravano, M.; Sciarretta, F.; Fulci, C.; Nali, M.; Frontoni, S.; Varano, M.; Caccuri, A.M. Activation of Retinal Müller Cells in Response to Glucose Variability. Endocrine 2019, 65, 542–549. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, G.; Ling, Q.; Da, C. Expression of Aquaporin 4 and Kir4.1 in Diabetic Rat Retina: Treatment with Minocycline. J. Int. Med. Res. 2011, 39, 464–479. [Google Scholar] [CrossRef]
- Kida, T.; Oku, H.; Horie, T.; Fukumoto, M.; Okuda, Y.; Morishita, S.; Ikeda, T. Implication of VEGF and Aquaporin 4 Mediating Müller Cell Swelling to Diabetic Retinal Edema. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1149–1157. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New Functions of Müller Cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Fletcher, E.; Phipps, J.; Ward, M.; Puthussery, T.; Wilkinson-Berka, J. Neuronal and Glial Cell Abnormality as Predictors of Progression of Diabetic Retinopathy. Curr. Pharm. Des. 2007, 13, 2699–2712. [Google Scholar] [CrossRef]
- Curtis, T.M.; Hamilton, R.; Yong, P.H.; McVicar, C.M.; Berner, A.; Pringle, R.; Uchida, K.; Nagai, R.; Brockbank, S.; Stitt, A.W. Müller Glial Dysfunction during Diabetic Retinopathy in Rats Is Linked to Accumulation of Advanced Glycation End-Products and Advanced Lipoxidation End-Products. Diabetologia 2011, 54, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavira-Suárez, E.; Sandoval, A.; Felix, R.; Lamas, M. Expression and High Glucose-Mediated Regulation of K+ Channel Interacting Protein 3 (KChIP3) and KV4 Channels in Retinal Müller Glial Cells. Biochem. Biophys. Res. Commun. 2011, 404, 678–683. [Google Scholar] [CrossRef] [PubMed]
- McDowell, R.E.; Barabas, P.; Augustine, J.; Chevallier, O.; McCarron, P.; Chen, M.; McGeown, J.G.; Curtis, T.M. Müller Glial Dysfunction during Diabetic Retinopathy in Rats Is Reduced by the Acrolein-Scavenging Drug, 2-Hydrazino-4,6-Dimethylpyrimidine. Diabetologia 2018, 61, 2654–2667. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M. Abnormalities in Glutamate Metabolism and Excitotoxicity in the Retinal Diseases. Scientifica 2013, 2013, 528940. [Google Scholar] [CrossRef] [Green Version]
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial Reactivity and Impaired Glutamate Metabolism in Short-Term Experimental Diabetic Retinopathy. Penn State Retina Research Group. Diabetes 1998, 47, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.W.; Liu, Q.H.; Wang, J.L. Blocking IL-17A Alleviates Diabetic Retinopathy in Rodents. Cell. Physiol. Biochem. 2017, 41, 960–972. [Google Scholar] [CrossRef]
- Ma, M.; Zhao, S.; Zhang, J.; Sun, T.; Fan, Y.; Zheng, Z. High Glucose-Induced TRPC6 Channel Activation Decreases Glutamate Uptake in Rat Retinal Müller Cells. Front. Pharmacol. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Gu, L.; Xu, H.; Wang, F.; Xu, G.; Sinha, D.; Wang, J.; Xu, J.Y.; Tian, H.; Gao, F.; Li, W.; et al. Erythropoietin Exerts a Neuroprotective Function against Glutamate Neurotoxicity in Experimental Diabetic Retina. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8208–8222. [Google Scholar] [CrossRef] [Green Version]
- Solà-Adell, C.; Bogdanov, P.; Hernández, C.; Sampedro, J.; Valeri, M.; Garcia-Ramirez, M.; Pasquali, C.; Simó, R. Calcium Dobesilate Prevents Neurodegeneration and Vascular Leakage in Experimental Diabetes. Curr. Eye Res. 2017, 42, 1273–1286. [Google Scholar] [CrossRef]
- Ambati, J.; Chalam, K.V.; Chawla, D.K.; D’Angio, C.T.; Guillet, E.G.; Rose, S.J.; Vanderlinde, R.E.; Ambati, B.K. Elevated Gamma-Aminobutyric Acid, Glutamate, and Vascular Endothelial Growth Factor Levels in the Vitreous of Patients with Proliferative Diabetic Retinopathy. Arch. Ophthalmol. 1997, 115, 1161–1166. [Google Scholar] [CrossRef]
- Deng, J.; Wu, D.Z.; Gao, R. Detection of Glutamate and Gamma-Aminobutyric Acid in Vitreous of Patients with Proliferative Diabetic Retinopathy. Yan ke xue bao 2000, 16, 199–202. [Google Scholar]
- Li, Q.; Puro, D.G. Diabetes-Induced Dysfunction of the Glutamate Transporter in Retinal Müller Cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3109–3116. [Google Scholar]
- Gowda, K.; Zinnantif, W.J.; LaNoue, K.F. The Influence of Diabetes on Glutamate Metabolism in Retinas. J. Neurochem. 2011, 117, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Miya-Coreixas, V.S.; Maggesissi Santos, R.; Carpi Santos, R.; Gardino, P.F.; Calaza, K. Regulation of GABA Content by Glucose in the Chick Retina. Exp. Eye Res. 2013, 115, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Carpi-Santos, R.; Maggesissi, R.S.; von Seehausen, M.P.; Calaza, K.C. Retinal Exposure to High Glucose Condition Modifies the GABAergic System: Regulation by Nitric Oxide. Exp. Eye Res. 2017, 162, 116–125. [Google Scholar] [CrossRef]
- Ishikawa, A.; Ishiguro, S.I.; Tamai, M. Changes in GABA Metabolism in Streptozotocin-Induced Diabetic Rat Retinas. Curr. Eye Res. 1996, 15, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Ruzafa, N.; Pereiro, X.; Lepper, M.F.; Hauck, S.M.; Vecino, E. A Proteomics Approach to Identify Candidate Proteins Secreted by Müller Glia That Protect Ganglion Cells in the Retina. Proteomics 2018, 18, 1700321. [Google Scholar] [CrossRef]
- Eastlake, K.; Wang, W.; Jayaram, H.; Murray-Dunning, C.; Carr, A.J.F.; Ramsden, C.M.; Vugler, A.; Gore, K.; Clemo, N.; Stewart, M.; et al. Phenotypic and Functional Characterization of Müller Glia Isolated from Induced Pluripotent Stem Cell-Derived Retinal Organoids: Improvement of Retinal Ganglion Cell Function upon Transplantation. Stem Cells Transl. Med. 2019, 8, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Matteucci, A.; Gaddini, L.; Villa, M.; Varano, M.; Parravano, M.; Monteleone, V.; Cavallo, F.; Leo, L.; Mallozzi, C.; Malchiodi-Albedi, F.; et al. Neuroprotection by Rat Müller Glia against High Glucose-Induced Neurodegeneration through a Mechanism Involving ERK1/2 Activation. Exp. Eye Res. 2014, 125, 20–29. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, Z.; Liu, C.H.; Gong, Y.; Cakir, B.; Liegl, R.; Sun, Y.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Fibroblast Growth Factor 21 Protects Photoreceptor Function in Type 1 Diabetic Mice. Diabetes 2018, 67, 974–985. [Google Scholar] [CrossRef] [Green Version]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients with Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5594–5603. [Google Scholar] [CrossRef] [PubMed]
- Coorey, N.J.; Shen, W.; Chung, S.H.; Zhu, L.; Gillies, M.C. The Role of Glia in Retinal Vascular Disease. Clin. Exp. Optom. 2012, 95, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Nakagawa, K.; Khalil, A.; Ishibashi, T.; Inomata, H.; Sueishi, K. The Relation between Expression of Vascular Endothelial Growth Factor and Breakdown of the Blood-Retinal Barrier in Diabetic Rat Retinas. Lab. Investig. 1996, 74, 819–825. [Google Scholar] [PubMed]
- Joussen, A.M.; Murata, T.; Tsujikawa, A.; Kirchhof, B.; Bursell, S.E.; Adamis, A.P. Leukocyte-Mediated Endothelial Cell Injury and Death in the Diabetic Retina. Am. J. Pathol. 2001, 158, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of Leukostasis and Vascular Leakage in Streptozotocin-Induced Diabetic Retinopathy via Intercellular Adhesion Molecule-1 Inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.-Z.; Le, Y.-Z. Significance of Outer Blood-Retina Barrier Breakdown in Diabetes and Ischemia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2160–2164. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.-Z. Müller Cell-Derived VEGF Is Essential for Diabetes-Induced Retinal Inflammation and Vascular Leakage. Diabetes 2010, 59, 2297–2305. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Busoy, J.M.; Zaman, B.A.A.; Tan, Q.S.W.; Tan, G.S.W.; Barathi, V.A.; Cheung, N.; Wei, J.J.Y.; Hunziker, W.; Hong, W.; et al. A Novel Model of Persistent Retinal Neovascularization for the Development of Sustained Anti-VEGF Therapies. Exp. Eye Res. 2018, 174, 98–106. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a Neurotransmitter in the Healthy Brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [Green Version]
- Yafai, Y.; Iandiev, I.; Lange, J.; Unterlauft, J.D.; Wiedemann, P.; Bringmann, A.; Reichenbach, A.; Eichler, W. Müller Glial Cells Inhibit Proliferation of Retinal Endothelial Cells via TGF-Β2 and Smad Signaling. Glia 2014, 62, 1476–1485. [Google Scholar] [CrossRef]
- Mao, X.-B.; Cheng, Y.-H.; Peng, K.-S.; You, Z.-P. Sirtuin (Sirt) 3 Overexpression Prevents Retinopathy in Streptozotocin-Induced Diabetic Rats. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e920883. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Reh, T.A. Müller Glia Are a Potential Source of Neural Regeneration in the Postnatal Chicken Retina. Nat. Neurosci. 2001, 4, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Mendel, T.A.; Clabough, E.B.D.; Kao, D.S.; Demidova-Rice, T.N.; Durham, J.T.; Zotter, B.C.; Seaman, S.A.; Cronk, S.M.; Rakoczy, E.P.; Katz, A.J.; et al. Pericytes Derived from Adipose-Derived Stem Cells Protect against Retinal Vasculopathy. PLoS ONE 2013, 8, e65691. [Google Scholar] [CrossRef]
- Ezquer, M.; Urzua, C.A.; Montecino, S.; Leal, K.; Conget, P.; Ezquer, F. Intravitreal Administration of Multipotent Mesenchymal Stromal Cells Triggers a Cytoprotective Microenvironment in the Retina of Diabetic Mice. Stem Cell Res. Ther. 2016, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieth, E.; Gardner, T.W.; Barber, A.J.; Antonetti, D.A. Retinal Neurodegeneration: Early Pathology in Diabetes. Clin. Exp. Ophthalmol. 2000, 28, 3–8. [Google Scholar] [CrossRef]
- Mylonas, G.; Sacu, S.; Deák, G.; Dunavoelgyi, R.; Buehl, W.; Georgopoulos, M.; Schmidt-Erfurth, U. Macular Edema Following Cataract Surgery in Eyes with Previous 23-Gauge Vitrectomy and Peeling of the Internal Limiting Membrane. Am. J. Ophthalmol. 2013, 155, 253–259.e2. [Google Scholar] [CrossRef]
- Uji, A.; Murakami, T.; Unoki, N.; Ogino, K.; Horii, T.; Yoshitake, S.; Dodo, Y.; Yoshimura, N. Parallelism for Quantitative Image Analysis of Photoreceptor-Retinal Pigment Epithelium Complex Alterations in Diabetic Macular Edema. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3361–3367. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.-K.; Lee, E.-J.; Kim, Y.-H.; Kim, D.Y.; Oh, H.; Kim, S.-I.; Kang, Y.-H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 2018, 10, 1046. [Google Scholar] [CrossRef] [Green Version]
- Patrick, A.T.; He, W.; Madu, J.; Sripathi, S.R.; Choi, S.; Lee, K.; Samson, F.P.; Powell, F.L.; Bartoli, M.; Jee, D.; et al. Mechanistic Dissection of Diabetic Retinopathy Using the Protein-Metabolite Interactome. J. Diabetes Metab. Disord. 2020, 19, 829–848. [Google Scholar] [CrossRef]
- Tonade, D.; Kern, T.S. Photoreceptor Cells and RPE Contribute to the Development of Diabetic Retinopathy. Prog. Retin. Eye Res. 2021, 83, 100919. [Google Scholar] [CrossRef]
- Park, S.H.; Park, J.W.; Park, S.J.; Kim, K.Y.; Chung, J.W.; Chun, M.H.; Oh, S.J. Apoptotic Death of Photoreceptors in the Streptozotocin-Induced Diabetic Rat Retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor Cells Are Major Contributors to Diabetes-Induced Oxidative Stress and Local Inflammation in the Retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadfi, K.; Atlasz, T.; Kiss, P.; Reglodi, D.; Szabo, A.; Kovacs, K.; Szalontai, B.; Setalo, G.; Banki, E.; Csanaky, K.; et al. Protective Effects of the Neuropeptide PACAP in Diabetic Retinopathy. Cell Tissue Res. 2012, 348, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Énzsöly, A.; Szabó, A.; Kántor, O.; Dávid, C.; Szalay, P.; Szabó, K.; Szél, Á.; Németh, J.; Lukáts, Á. Pathologic Alterations of the Outer Retina in Streptozotocin-Induced Diabetes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3686–3699. [Google Scholar] [CrossRef] [Green Version]
- Bavinger, J.C.; Dunbar, G.E.; Stem, M.S.; Blachley, T.S.; Kwark, L.; Farsiu, S.; Jackson, G.R.; Gardner, T.W. The Effects of Diabetic Retinopathy and Pan-Retinal Photocoagulation on Photoreceptor Cell Function as Assessed by Dark Adaptometry. Investig. Ophthalmol. Vis. Sci. 2016, 57, 208–217. [Google Scholar] [CrossRef]
- Tavares Ferreira, J.; Alves, M.; Dias-Santos, A.; Costa, L.; Santos, B.O.; Cunha, J.P.; Papoila, A.L.; Abegão Pinto, L. Retinal Neurodegeneration in Diabetic Patients without Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6455–6460. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.J.; Le, Y.Z. Müller Glia Are a Major Cellular Source of Survival Signals for Retinal Neurons in Diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Nesper, P.L.; Scarinci, F.; Fawzi, A.A. Adaptive Optics Reveals Photoreceptor Abnormalities in Diabetic Macular Ischemia. PLoS ONE 2017, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lima, V.C.; Rosen, R.B.; Maia, M.; Prata, T.S.; Dorairaj, S.; Farah, M.E.; Sallum, J. Macular Pigment Optical Density Measured by Dual-Wavelength Autofluorescence Imaging in Diabetic and Nondiabetic Patients: A Comparative Study. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5840–5845. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, Lactate, and Shuttling of Metabolites in Vertebrate Retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Poitry-Yamate, C.L.; Poitry, S.; Tsacopoulos, M. Lactate Released by Müller Glial Cells Is Metabolized by Photoreceptors from Mammalian Retina. J. Neurosci. 1995, 15, 5179–5191. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.B.; Chao, J.R. It’s Never Too Late to Save a Photoreceptor. J. Clin. Invest. 2015, 125, 3424–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, B.S.; Arnold, M.J.; Brassell, M.A.; Puro, D.G. Energy Metabolism in Human Retinal Müller Cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3183–3190. [Google Scholar] [PubMed]
- Salceda, R.; Vilchis, C.; Coffe, V.; Hernández-Muñoz, R. Changes in the Redox State in the Retina and Brain during the Onset of Diabetes in Rats. Neurochem. Res. 1998, 23, 893–897. [Google Scholar] [CrossRef]
- Santiago, A.R.; Garrido, M.J.; Cristóvão, A.J.; Duarte, J.M.N.; Carvalho, R.A.; Ambrósio, A.F. Evaluation of the Impact of Diabetes on Retinal Metabolites by NMR Spectroscopy. Curr. Eye Res. 2010, 35, 992–1001. [Google Scholar] [CrossRef]
- Ramírez-Pérez, G.; Sánchez-Chávez, G.; Salceda, R. Mitochondrial Bound Hexokinase Type I in Normal and Streptozotocin Diabetic Rat Retina. Mitochondrion 2020, 52, 212–217. [Google Scholar] [CrossRef]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Müller Cell Metabolic Signatures: Evolutionary Conservation and Disruption in Disease. Trends Endocrinol. Metab. 2020, 31, 320–329. [Google Scholar] [CrossRef]
- Eastlake, K.; Banerjee, P.J.; Angbohang, A.; Charteris, D.G.; Khaw, P.T.; Limb, G.A. Müller Glia as an Important Source of Cytokines and Inflammatory Factors Present in the Gliotic Retina during Proliferative Vitreoretinopathy. Glia 2016, 64, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A Journey into the Retina: Müller Glia Commanding Survival and Death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef]
- Eymann, J.; Di-Poï, N. Glia-Mediated Regenerative Response Following Acute Excitotoxic Damage in the Postnatal Squamate Retina. Front. Cell Dev. Biol. 2020, 8, 406. [Google Scholar] [CrossRef]
- Schitine, C.S.; Mendez-Flores, O.G.; Santos, L.E.; Ornelas, I.; Calaza, K.C.; Pérez-Toledo, K.; López-Bayghen, E.; Ortega, A.; Gardino, P.F.; De Mello, F.G.; et al. Functional Plasticity of GAT-3 in Avian Müller Cells Is Regulated by Neurons via a Glutamatergic Input. Neurochem. Int. 2015, 82, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Ahmad, I. Let-7 MicroRNA Regulates Neurogliogenesis in the Mammalian Retina through Hmga2. Dev. Biol. 2016, 410, 70–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, J.; Wilken, M.S.; Ueki, Y.; Cox, K.E.; Sullivan, J.M.; Taylor, R.J.; Levine, E.M.; Reh, T.A. ASCL1 Reprograms Mouse Muller Glia into Neurogenic Retinal Progenitors. Development 2013, 140, 2619–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Zhu, J.; Duan, Y.; Li, G.; Cai, H.; Zheng, L.; Qian, H.; Zhang, C.; Jin, Z.; Fu, X.-D.; et al. Visual Function Restoration in Genetically Blind Mice via Endogenous Cellular Reprogramming. bioRxiv 2020. [Google Scholar] [CrossRef]
- Roska, B.; Sahel, J.-A. Restoring Vision. Nature 2018, 557, 359–367. [Google Scholar] [CrossRef]
- Stower, H. Restoring Sight with Native Cell Reprogramming. Nat. Med. 2018, 24, 1303. [Google Scholar] [CrossRef]
- Yao, K.; Qiu, S.; Wang, Y.V.; Park, S.J.H.; Mohns, E.J.; Mehta, B.; Liu, X.; Chang, B.; Zenisek, D.; Crair, M.C.; et al. Restoration of Vision after de Novo Genesis of Rod Photoreceptors in Mammalian Retinas. Nature 2018, 560, 484–488. [Google Scholar] [CrossRef]
- Konar, G.J.; Ferguson, C.; Flickinger, Z.; Kent, M.R.; Patton, J.G. MiRNAs and Müller Glia Reprogramming During Retina Regeneration. Front. Cell Dev. Biol. 2020, 8, 632632. [Google Scholar] [CrossRef]
- Xia, X.; Teotia, P.; Patel, H.; Van Hook, M.J.; Ahmad, I. Chemical Induction of Neurogenic Properties in Mammalian Müller Glia. Stem Cells 2021, 39, 1081–1090. [Google Scholar] [CrossRef]
- de Melo Reis, R.A.; Ventura, A.L.M.; Schitine, C.S.; de Mello, M.C.F.; de Mello, F.G. Müller Glia as an Active Compartment Modulating Nervous Activity in the Vertebrate Retina: Neurotransmitters and Trophic Factors. Neurochem. Res. 2008, 33, 1466–1474. [Google Scholar] [CrossRef]
- Devoldere, J.; Peynshaert, K.; De Smedt, S.C.; Remaut, K. Müller Cells as a Target for Retinal Therapy. Drug Discov. Today 2019, 24, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Schmalen, A.; Lorenz, L.; Grosche, A.; Pauly, D.; Deeg, C.A.; Hauck, S.M. Proteomic Phenotyping of Stimulated Müller Cells Uncovers Profound Pro-In Fl Ammatory Signaling and Antigen-Presenting Capacity. Front. Pharmacol. 2021, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza Salas, L.F.; García-Venzor, A.; Beltramone, N.; Capurro, C.; Toiber, D.; Silberman, D.M. Metabolic Imbalance Effect on Retinal Müller Glial Cells Reprogramming Capacity: Involvement of Histone Deacetylase SIRT6. Front. Genet. 2021, 12, 769723. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, M.; Hitchcock, P.F. Inflammation Regulates the Multi-Step Process of Retinal Regeneration in Zebrafish. Cells 2021, 10, 783. [Google Scholar] [CrossRef]
- Palazzo, I.; Deistler, K.; Hoang, T.V.; Blackshaw, S.; Fischer, A.J. NF-ΚB Signaling Regulates the Formation of Proliferating Müller Glia-Derived Progenitor Cells in the Avian Retina. Development 2020, 147, dev183418. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, H.; Yu, S.; Zhou, C.; Zhang, X.; Li, N.; Zhang, S.; Song, K.; Lu, Y.; Liu, D.; et al. Inflammation-Induced Mammalian Target of Rapamycin Signaling Is Essential for Retina Regeneration. Glia 2020, 68, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Zelinka, C.P.; Volkov, L.; Goodman, Z.A.; Todd, L.; Palazzo, I.; Bishop, W.A.; Fischer, A.J. MTor Signaling Is Required for the Formation of Proliferating Müller Glia-Derived Progenitor Cells in the Chick Retina. Development 2016, 143, 1859–1873. [Google Scholar] [CrossRef] [Green Version]
- Todd, L.; Finkbeiner, C.; Wong, C.K.; Hooper, M.J.; Reh, T.A. Microglia Suppress Ascl1-Induced Retinal Regeneration in Mice. Cell Rep. 2020, 33, 108507. [Google Scholar] [CrossRef]
- Shen, W.; Li, S.; Chung, S.H.; Gillies, M.C. Retinal Vascular Changes after Glial Disruption in Rats. J. Neurosci. Res. 2010, 88, 1485–1499. [Google Scholar] [CrossRef]
- Garcia-Ramírez, M.; Hernández, C.; Villarroel, M.; Canals, F.; Alonso, M.A.; Fortuny, R.; Masmiquel, L.; Navarro, A.; García-Arumí, J.; Simó, R. Interphotoreceptor Retinoid-Binding Protein (IRBP) Is Downregulated at Early Stages of Diabetic Retinopathy. Diabetologia 2009, 52, 2633–2641. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, H.; Carpi-Santos, R.; Da Costa Calaza, K.; Blanco Martinez, A. Neuroinflammation and Oxidative Stress Act in Concert to Promote Neurodegeneration in the Diabetic Retina and Optic Nerve: Galectin-3 Participation. Neural Regen. Res. 2020, 15, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Rübsam, A.; Parikh, S.; Fort, P.E. Role of Inflammation in Diabetic Retinopathy. Int. J. Mol. Sci. 2018, 19, 942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalam, K.V.; Grover, S.; Sambhav, K.; Balaiya, S.; Murthy, R.K. Aqueous Interleukin-6 Levels Are Superior to Vascular Endothelial Growth Factor in Predicting Therapeutic Response to Bevacizumab in Age-Related Macular Degeneration. J. Ophthalmol. 2014, 2014, 502174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.; Li, L.; Zhu, L.; Guo, Y.; Du, S.; Zhang, Y.; Wang, Z.; Zhang, Y.; Zhu, M. Geniposide Attenuates Hyperglycemia-Induced Oxidative Stress and Inflammation by Activating the Nrf2 Signaling Pathway in Experimental Diabetic Retinopathy. Oxid. Med. Cell. Longev. 2021, 2021, 9247947. [Google Scholar] [CrossRef]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-Induced pro-Inflammatory Responses by Retinal Müller Glia Are Regulated by the Receptor for Advanced Glycation End-Products (RAGE). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert-Garay, J.S.; Riesgo-Escovar, J.; Salceda, R. High Glucose Concentrations Induce Oxidative Stress by Inhibiting Nrf2 Expression in Rat Müller Retinal Cells in Vitro. Sci. Rep. 2022, 12, 1261. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; De, J.B.; Wilkinson-berka, J.L. Nrf2 Activation Is a Potential Therapeutic Approach to Attenuate Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, H.; Chen, X. Protective Effects of Sulforaphane on Diabetic Retinopathy: Activation of the Nrf2 Pathway and Inhibition of NLRP3 Inflammasome Formation. Exp. Anim. 2019, 68, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Nonarath, H.J.; Hall, A.E.; SenthilKumar, G.; Abroe, B.; Eells, J.T.; Liedhegner, E.S. 670nm Photobiomodulation Modulates Bioenergetics and Oxidative Stress, in Rat Müller Cells Challenged with High Glucose. PLoS ONE 2021, 16, e0260968. [Google Scholar] [CrossRef]
- Walker, R.J.; Steinle, J.J. Role of Beta-Adrenergic Receptors in Inflammatory Marker Expression in Müller Cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5276–5281. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.-R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 Plays a Protective Role in Diabetic Retinopathy in Mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, X.; Gao, L.; Hatala, D.A.; Smith, D.G.; Codispoti, M.C.; Gong, B.; Kern, T.S.; Zhang, J.-Z. Chronically Elevated Glucose-Induced Apoptosis Is Mediated by Inactivation of Akt in Cultured Müller Cells. Biochem. Biophys. Res. Commun. 2005, 326, 548–553. [Google Scholar] [CrossRef]
- Jiang, Y.; Pagadala, J.; Miller, D.; Steinle, J.J. Reduced Insulin Receptor Signaling in Retinal Müller Cells Cultured in High Glucose. Mol. Vis. 2013, 19, 804–811. [Google Scholar] [PubMed]
- Han, N.; Yu, L.; Song, Z.; Luo, L.; Wu, Y. Agmatine Protects Müller Cells from High-Concentration Glucose-Induced Cell Damage via N-Methyl-D-Aspartic Acid Receptor Inhibition. Mol. Med. Rep. 2015, 12, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Ao, H.; Li, H.; Zhao, X.; Liu, B.; Lu, L. TXNIP Positively Regulates the Autophagy and Apoptosis in the Rat Müller Cell of Diabetic Retinopathy. Life Sci. 2021, 267, 118988. [Google Scholar] [CrossRef]
- Duarte, D.A.; Papadimitriou, A.; Gilbert, R.E.; Thai, K.; Zhang, Y.; Rosales, M.A.B.; De Faria, J.B.L.; De Faria, J.M.L. Conditioned Medium from Early-Outgrowth Bone Marrow Cells Is Retinal Protective in Experimental Model of Diabetes. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Lee, I.; Hüttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP Links Innate Host Defense Mechanisms to Oxidative Stress and Inflammation in Retinal Muller Glia under Chronic Hyperglycemia: Implications for Diabetic Retinopathy. Exp. Diabetes Res. 2012, 2012, 438238. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.P.; Devi, T.S. Potential Combination Drug Therapy to Prevent Redox Stress and Mitophagy Dysregulation in Retinal Müller Cells under High Glucose Conditions: Implications for Diabetic Retinopathy. Diseases 2021, 9, 91. [Google Scholar] [CrossRef]
- Trueblood, K.E.; Mohr, S.; Dubyak, G.R. Purinergic Regulation of High-Glucose-Induced Caspase-1 Activation in the Rat Retinal Müller Cell Line RMC-1. Am. J. Physiol. Cell Physiol. 2011, 301, 1–22. [Google Scholar] [CrossRef]
- Hernández-Ramírez, E.; Sánchez-Chávez, G.; Estrella-Salazar, L.A.; Salceda, R. Nitrosative Stress in the Rat Retina at the Onset of Streptozotocin-Induced Diabetes. Cell. Physiol. Biochem. 2017, 42, 2353–2363. [Google Scholar] [CrossRef]
- Lieth, E.; LaNoue, K.F.; Antonetti, D.A.; Ratz, M. Diabetes Reduces Glutamate Oxidation and Glutamine Synthesis in the Retina. The Penn State Retina Research Group. Exp. Eye Res. 2000, 70, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Rungger-Brändle, E.; Dosso, A.A.; Leuenberger, P.M. Glial Reactivity, an Early Feature of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1971–1980. [Google Scholar]
- Syrbe, S.; Kuhrt, H.; Gärtner, U.; Habermann, G.; Wiedemann, P.; Bringmann, A.; Reichenbach, A. Müller Glial Cells of the Primate Foveola: An Electron Microscopical Study. Exp. Eye Res. 2018, 167, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Udaondo, P.; Adan, A.; Arias-Barquet, L.; Ascaso, F.J.; Cabrera-López, F.; Castro-Navarro, V.; Donate-López, J.; García-Layana, A.; Lavid, F.J.; Rodríguez-Maqueda, M.; et al. Challenges in Diabetic Macular Edema Management: An Expert Consensus Report. Clin. Ophthalmol. 2021, 15, 3183–3195. [Google Scholar] [CrossRef]
- Ashraf, M.; Souka, A.; Adelman, R. Predicting Outcomes to Anti-Vascular Endothelial Growth Factor (VEGF) Therapy in Diabetic Macular Oedema: A Review of the Literature. Br. J. Ophthalmol. 2016, 100, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Muftuoglu, I.K.; Mendoza, N.; Gaber, R.; Alam, M.; You, Q.; Freeman, W.R. Integrity of outer retinal layers after resolution of central involved diabetic macular edema. Retina 2017, 37, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Ramasamy, K.; Sivaprasad, S. Indicators of Visual Prognosis in Diabetic Macular Oedema. J. Pers. Med. 2021, 11, 449. [Google Scholar] [CrossRef]
- Choi, M.; Yun, C.; Oh, J.-H.; Kim, S.-W. Foveal Müller Cell Cone as a Prognostic OCT Biomarker for Initial Response to Anti-VEGF Treatment in Cystoid Diabetic Macular Edema. Retina 2021. [Google Scholar] [CrossRef]
- Pierce, E.A.; Avery, R.L.; Foley, E.D.; Aiello, L.P.; Smith, L.E. Vascular Endothelial Growth Factor/Vascular Permeability Factor Expression in a Mouse Model of Retinal Neovascularization. Proc. Natl. Acad. Sci. USA 1995, 92, 905–909. [Google Scholar] [CrossRef] [Green Version]
- Robbins, S.G.; Conaway, J.R.; Ford, B.L.; Roberto, K.A.; Penn, J.S. Detection of Vascular Endothelial Growth Factor (VEGF) Protein in Vascular and Non-Vascular Cells of the Normal and Oxygen-Injured Rat Retina. Growth Factors 1997, 14, 229–241. [Google Scholar] [CrossRef]
- Bai, Y.; Ma, J.; Guo, J.; Wang, J.; Zhu, M.; Chen, Y.; Le, Y.-Z. Müller Cell-Derived VEGF Is a Significant Contributor to Retinal Neovascularization. J. Pathol. 2009, 219, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Hauck, S.M.; von Toerne, C.; Ueffing, M. The Neuroprotective Potential of Retinal Müller Glial Cells. Adv. Exp. Med. Biol. 2014, 801, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.K.; Benyajati, S.; Le, Y.; Cheng, R.; Zhang, W.; Ma, J. Interruption of Wnt Signaling in Müller Cells Ameliorates Ischemia-Induced Retinal Neovascularization. PLoS ONE 2014, 9, e108454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, F.; Yan, J.; Ling, Y.; Liu, Z.; Fu, C.; Li, X.; Qin, Y. Effect of Shuangdan Mingmu Capsule, a Chinese Herbal Formula, on Oxidative Stress- Induced Apoptosis of Pericytes through PARP/GAPDH Pathway. BMC Complementary Med. Ther. 2021, 21, 118. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; An, Y.; He, X.; Zhang, D.; He, W. Protection of Kaempferol on Oxidative Stress-Induced Retinal Pigment Epithelial Cell Damage. Oxidative Med. Cell. Longev. 2018, 2018, 438238. [Google Scholar] [CrossRef]
- Haque, R.; Iuvone, P.M.; He, L.; Hur, E.H.; Su, K.; Choi, C.; Park, D.; Farrell, A.N.; Ngo, A.; Gokhale, S.; et al. Prorenin receptor (PRR)-mediated NADPH oxidase (Nox) signaling regulates VEGF synthesis under hyperglycemic condition in ARPE-19 cells. J. Recept. Signal Transduct. 2017, 37, 560–568. [Google Scholar] [CrossRef]
- Rossino, M.G.; Lulli, M.; Amato, R.; Cammalleri, M.; Monte, M.D.; Casini, G. Oxidative Stress Induces a VEGF Autocrine Loop in the Retina: Relevance for Diabetic Retinopathy. Cells 2020, 9, 1452. [Google Scholar] [CrossRef]
- Wang, J.; Shanmugam, A.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Sigma 1 Receptor Regulates the Oxidative Stress Response in Primary Retinal Müller Glial Cells via NRF2 Signaling and System Xc(-), the Na(+)-Independent Glutamate-Cystine Exchanger. Free Radic. Biol. Med. 2015, 86, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.; Chen, L. The Cells Involved in the Pathological Process of Diabetic Retinopathy. Biomed. Pharmacother. 2020, 132, 110818. [Google Scholar] [CrossRef]
- Chalke, S.D.; Kale, P.P. Combinational Approaches Targeting Neurodegeneration, Oxidative Stress, and Inflammation in the Treatment of Diabetic Retinopathy. Curr. Drug Targets 2021, 22, 1810–1824. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Laddha, A.P.; Kulkarni, Y.A. NADPH Oxidase: A Membrane-Bound Enzyme and Its Inhibitors in Diabetic Complications. Eur. J. Pharmacol. 2020, 881, 173206. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ai, S.; Chen, Z.; Li, C. Probucol Promotes High Glucose-Induced Proliferation and Inhibits Apoptosis by Reducing Reactive Oxygen Species Generation in Müller Cells. Int. Ophthalmol. 2019, 39, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Ho, Y.-J.; Chou, H.-C.; Liao, E.-C.; Tsai, Y.-T.; Wei, Y.-S.; Lin, L.-H.; Lin, M.-W.; Wang, Y.-S.; Ko, M.-L.; et al. The Role of Transforming Growth Factor-Beta in Retinal Ganglion Cells with Hyperglycemia and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 6482. [Google Scholar] [CrossRef]
- Chen, N.; Li, Y.; Huang, N.; Yao, J.; Luo, W.-F.; Jiang, Q. The Nrf2 Activator MIND4-17 Protects Retinal Ganglion Cells from High Glucose-Induced Oxidative Injury. J. Cell. Physiol. 2020, 235, 7204–7213. [Google Scholar] [CrossRef]
- Sun, W.; Yu, J.; Kang, Q. Upregulation of Heme Oxygenase-1 by Brahma-Related Gene 1 through Nrf2 Signaling Confers Protective Effect against High Glucose-Induced Oxidative Damage of Retinal Ganglion Cells. Eur. J. Pharmacol. 2020, 875, 173038. [Google Scholar] [CrossRef]
- Rezzola, S.; Guerra, J.; Chandran, A.M.K.; Loda, A.; Cancarini, A.; Sacristani, P.; Semeraro, F.; Presta, M. Vegf-independent Activation of Müller Cells by the Vitreous from Proliferative Diabetic Retinopathy Patients. Int. J. Mol. Sci. 2021, 22, 2179. [Google Scholar] [CrossRef]
- Hammes, H.-P. Diabetic Retinopathy: Hyperglycaemia, Oxidative Stress and Beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Gui, F.; You, Z.; Fu, S.; Wu, H.; Zhang, Y. Endothelial Dysfunction in Diabetic Retinopathy. Front. Endocrinol. 2020, 11, 591. [Google Scholar] [CrossRef]
- Trumpower, B.L. The Protonmotive Q Cycle. Energy Transduction by Coupling of Proton Translocation to Electron Transfer by the Cytochrome Bc1 Complex. J. Biol. Chem. 1990, 265, 11409–11412. [Google Scholar] [CrossRef]
- Duarte, D.A.; Rosales, M.A.B.; Papadimitriou, A.; Silva, K.C.; Amancio, V.H.O.; Mendonça, J.N.; Lopes, N.P.; Lopes de Faria, J.B.; Lopes de Faria, J.M. Polyphenol-Enriched Cocoa Protects the Diabetic Retina from Glial Reaction through the Sirtuin Pathway. J. Nutr. Biochem. 2015, 26, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Gupta, S.K.; Nag, T.C.; Srivastava, S.; Saxena, R.; Jha, K.A.; Srinivasan, B.P. Retinal Neuroprotective Effects of Quercetin in Streptozotocin-Induced Diabetic Rats. Exp. Eye Res. 2014, 125, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Al-Dosary, D.I.; Alhomida, A.S.; Ola, M.S. Protective Effects of Dietary Flavonoids in Diabetic Induced Retinal Neurodegeneration. Curr. Drug Targets 2017, 18, 1468–1476. [Google Scholar] [CrossRef]
- Silva, K.C.; Rosales, M.A.B.; Hamassaki, D.E.; Saito, K.C.; Faria, A.M.; Ribeiro, P.A.O.; de Faria, J.B.L.; de Faria, J.M.L. Green Tea Is Neuroprotective in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tao, J.; Jiang, M.; Yao, Y. Apocynin Ameliorates Diabetic Retinopathy in Rats: Involvement of TLR4/NF-ΚB Signaling Pathway. Int. Immunopharmacol. 2019, 73, 49–56. [Google Scholar] [CrossRef]
- Matos, A.L.; Bruno, D.F.; Ambr, F. The Benefits of Flavonoids in Diabetic Retinopathy. Nutrients 2020, 12, 3169. [Google Scholar] [CrossRef]
- Rossino, M.G.; Casini, G. Nutraceuticals for the Treatment of Diabetic Retinopathy. Nutrients 2019, 11, 771. [Google Scholar] [CrossRef] [Green Version]
- Domanico, D.; Fragiotta, S.; Cutini, A.; Carnevale, C.; Zompatori, L.; Vingolo, E.M. Circulating Levels of Reactive Oxygen Species in Patients with Nonproliferative Diabetic Retinopathy and the Influence of Antioxidant Supplementation: 6-Month Follow-Up. Indian J. Ophthalmol. 2015, 63, 9–14. [Google Scholar] [CrossRef]
- Garcia-Medina, J.J.; Rubio-Velazquez, E.; Foulquie-Moreno, E.; Casaroli-Marano, R.P.; Pinazo-Duran, M.D.; Zanon-Moreno, V.; Del-Rio-vellosillo, M. Update on the Effects of Antioxidants on Diabetic Retinopathy: In Vitro Experiments, Animal Studies and Clinical Trials. Antioxidants 2020, 9, 561. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Ardeshirlarijani, E.; Namazi, N.; Nikfar, S.; Jalili, R.B.; Larijani, B. Dietary Antioxidative Supplements and Diabetic Retinopathy; a Systematic Review. J. Diabetes Metab. Disord. 2019, 18, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Roig-Revert, M.J.; Lleó-Pérez, A.; Zanón-Moreno, V.; Vivar-Llopis, B.; Marín-Montiel, J.; Dolz-Marco, R.; Alonso-Muñoz, L.; Albert-Fort, M.; López-Gálvez, M.I.; Galarreta-Mira, D.; et al. Enhanced Oxidative Stress and Other Potential Biomarkers for Retinopathy in Type 2 Diabetics: Beneficial Effects of the Nutraceutic Supplements. Biomed Res. Int. 2015, 2015, 408180. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Medina, J.J.; Pinazo-Duran, M.D.; Garcia-Medina, M.; Zanon-Moreno, V.; Pons-Vazquez, S. A 5-Year Follow-up of Antioxidant Supplementation in Type 2 Diabetic Retinopathy. Eur. J. Ophthalmol. 2011, 21, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Mares, J. Lutein and Zeaxanthin Isomers in Eye Health and Disease. Annu. Rev. Nutr. 2016, 36, 571–602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-C.; Wu, C.-R.; Wang, Z.-L.; Wang, L.-Y.; Han, Y.; Sun, S.-L.; Li, Q.-S.; Ma, L. Effect of Lutein Supplementation on Visual Function in Nonproliferative Diabetic Retinopathy. Asia Pac. J. Clin. Nutr. 2017, 26, 406–411. [Google Scholar] [CrossRef]
- Ren, Y.-B.; Qi, Y.-X.; Su, X.-J.; Luan, H.-Q.; Sun, Q. Therapeutic Effect of Lutein Supplement on Non-Proliferative Diabetic Retinopathy: A Retrospective Study. Medicine 2019, 98, e15404. [Google Scholar] [CrossRef]
- de Sampaio e Spohr, T.C.L.; Stipursky, J.; Sasaki, A.C.; Barbosa, P.R.; Martins, V.; Benjamim, C.F.; Roque, N.F.; Costa, S.L.; Gomes, F.C.A. Effects of the Flavonoid Casticin from Brazilian Croton Betulaster in Cerebral Cortical Progenitors in Vitro: Direct and Indirect Action through Astrocytes. J. Neurosci. Res. 2010, 88, 530–541. [Google Scholar] [CrossRef]
- Nones, J.; de Sampaio Spohr, T.C.L.; Gomes, F.C.A. Effects of the Flavonoid Hesperidin in Cerebral Cortical Progenitors in Vitro: Indirect Action through Astrocytes. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2012, 30, 303–313. [Google Scholar] [CrossRef]
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia Increases Mitochondrial Superoxide in Retina and Retinal Cells. Free Radic. Biol. Med. 2003, 35, 1491–1499. [Google Scholar] [CrossRef]
- Frenzel, J.; Richter, J.; Eschrich, K. Pyruvate Protects Glucose-Deprived Müller Cells from Nitric Oxide-Induced Oxidative Stress by Radical Scavenging. Glia 2005, 52, 276–288. [Google Scholar] [CrossRef]
- Huster, D.; Reichenbach, A.; Reichelt, W. The Glutathione Content of Retinal Müller (Glial) Cells: Effect of Pathological Conditions. Neurochem. Int. 2000, 36, 461–469. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential Compartmentalization of Brain Ascorbate and Glutathione between Neurons and Glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Freitas, H.R.; Ferraz, G.; Ferreira, G.C.; Ribeiro-Resende, V.T.; Chiarini, L.B.; Nascimento, J.L.M.D.; Oliveira, K.R.H.M.; De Pereira, T.L.; Ferreira, L.G.B.; Kubrusly, R.C.; et al. Glutathione-Induced Calcium Shifts in Chick Retinal Glial Cells. PLoS ONE 2016, 11, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Carpi-Santos, R.; Ferreira, M.J.; Pereira Netto, A.D.; Giestal-de-Araujo, E.; Ventura, A.L.M.; Cossenza, M.; Calaza, K.C. Early Changes in System [Formula: See Text] and Glutathione in the Retina of Diabetic Rats. Exp. Eye Res. 2016, 146, 35–42. [Google Scholar] [CrossRef]
- Carpi-Santos, R.; Calaza, K.C. Alterations in System x(c)(-) Expression in the Retina of Type 1 Diabetic Rats and the Role of Nrf2. Mol. Neurobiol. 2018, 55, 7941–7948. [Google Scholar] [CrossRef]
- Kern, T.S.; Kowluru, R.A.; Engerman, R.L. Abnormalities of Retinal Metabolism in Diabetes or Galactosemia: ATPases and Glutathione. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2962–2967. [Google Scholar]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription Factor Nrf2-Mediated Antioxidant Defense System in the Development of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Bannai, S. Exchange of Cystine and Glutamate across Plasma Membrane of Human Fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [CrossRef]
- Martis, R.M.; Knight, L.J.; Donaldson, P.J.; Lim, J.C. Identification, Expression, and Roles of the Cystine/Glutamate Antiporter in Ocular Tissues. Oxid. Med. Cell. Longev. 2020, 2020, 4594606. [Google Scholar] [CrossRef]
- Kato, S.; Ishita, S.; Sugawara, K.; Mawatari, K. Cystine/Glutamate Antiporter Expression in Retinal Müller Glial Cells: Implications for DL-Alpha-Aminoadipate Toxicity. Neuroscience 1993, 57, 473–482. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef]
- Shanab, A.Y.; Nakazawa, T.; Ryu, M.; Tanaka, Y.; Himori, N.; Taguchi, K.; Yasuda, M.; Watanabe, R.; Takano, J.; Saido, T.; et al. Metabolic Stress Response Implicated in Diabetic Retinopathy: The Role of Calpain, and the Therapeutic Impact of Calpain Inhibitor. Neurobiol. Dis. 2012, 48, 556–567. [Google Scholar] [CrossRef]
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 Exacerbates the Visual Deficits and Optic Neuritis Elicited by Experimental Autoimmune Encephalomyelitis. Mol. Vis. 2016, 22, 1503–1513. [Google Scholar]
- Qin, X.; Li, N.; Zhang, M.; Lin, S.; Zhu, J.; Xiao, D.; Cui, W.; Zhang, T.; Lin, Y.; Cai, X. Tetrahedral Framework Nucleic Acids Prevent Retina Ischemia-Reperfusion Injury from Oxidative Stress via Activating the Akt/Nrf2 Pathway. Nanoscale 2019, 11, 20667–20675. [Google Scholar] [CrossRef]
- Wang, J.-J. Functions of Müller Cell-Derived Vascular Endothelial Growth Factor in Diabetic Retinopathy. World J. Diabetes 2015, 6, 726. [Google Scholar] [CrossRef]
- Tu, Y.; Zhu, M.; Wang, Z.; Wang, K.; Chen, L.; Liu, W.; Shi, Q.; Zhao, Q.; Sun, Y.; Wang, X.; et al. Melatonin Inhibits Müller Cell Activation and Pro-Inflammatory Cytokine Production via Upregulating the MEG3/MiR-204/Sirt1 Axis in Experimental Diabetic Retinopathy. J. Cell. Physiol. 2020, 235, 8724–8735. [Google Scholar] [CrossRef] [PubMed]
- Lopes de Faria, J.M.; Duarte, D.A.; Montemurro, C.; Papadimitriou, A.; Consonni, S.R.; Lopes de Faria, J.B. Defective Autophagy in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4356–4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Chen, X.; Chen, W.; Huang, S.; Peng, X.; Tian, L.; Wu, X.; Huang, Y. Curcumin Is a Potential Adjuvant to Alleviates Diabetic Retinal Injury via Reducing Oxidative Stress and Maintaining Nrf2 Pathway Homeostasis. Front. Pharmacol. 2021, 12, 796565. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The Stress Response Protein REDD1 Promotes Diabetes-Induced Oxidative Stress in the Retina by Keap1-Independent Nrf2 Degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 Cell Defence Pathway: Keap1-Dependent and -Independent Mechanisms of Regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, F.; Zhang, X.; Cheng, R.; Ma, J.X.; Yi, J.; Li, J. Fenofibrate Ameliorates Diabetic Retinopathy by Modulating Nrf2 Signaling and NLRP3 Inflammasome Activation. Mol. Cell. Biochem. 2018, 445, 105–115. [Google Scholar] [CrossRef]
- Mysona, B.; Dun, Y.; Duplantier, J.; Ganapathy, V.; Smith, S.B. Effects of Hyperglycemia and Oxidative Stress on the Glutamate Transporters GLAST and System Xc− in Mouse Retinal Müller Glial Cells. Cell Tissue Res. 2009, 335, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Yang, S.; Elliott, M.H.; Fu, D.; Wilson, K.; Zhang, J.; Du, M.; Chen, J.; Lyons, T. Oxidative and Endoplasmic Reticulum Stresses Mediate Apoptosis Induced by Modified LDL in Human Retinal Müller Cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4595–4604. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Sun, H.; Wu, L.; Guo, X.; Dou, H.; Tso, M.O.M.; Zhao, L.; Li, S. Baicalein Reduces Inflammatory Process in a Rodent Model of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Canning, P.; Glenn, J.V.; Hsu, D.K.; Liu, F.T.; Gardiner, T.A.; Stitt, A.W. Inhibition of Advanced Glycation and Absence of Galectin-3 Prevent Blood-Retinal Barrier Dysfunction during Short-Term Diabetes. Exp. Diabesity Res. 2007, 2007, 3169. [Google Scholar] [CrossRef]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.C.; Allen, D.M.; Cardona, A.E. The Role of Microglia in Diabetic Retinopathy. J. Ophthalmol. 2014, 2014, 705783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, B.M.Y.; Li, C. Diabetes and Hypertension: Is There a Common Metabolic Pathway? Curr. Atheroscler. Rep. 2012, 14, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujosevic, S.; Simó, R. Local and Systemic Inflammatory Biomarkers of Diabetic Retinopathy: An Integrative Approach. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO68–BIO75. [Google Scholar] [CrossRef] [PubMed]
- Stahel, M.; Becker, M.; Graf, N.; Michels, S. SYSTEMIC INTERLEUKIN 1β INHIBITION IN PROLIFERATIVE DIABETIC RETINOPATHY: A Prospective Open-Label Study Using Canakinumab. Retina 2016, 36, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.Y.; Sun, J.; Kawasaki, R.; Ruamviboonsuk, P.; Gupta, N.; Lansingh, V.C.; Maia, M.; Mathenge, W.; Moreker, S.; Muqit, M.M.K.; et al. Guidelines on Diabetic Eye Care: The International Council of Ophthalmology Recommendations for Screening, Follow-up, Referral, and Treatment Based on Resource Settings. Ophthalmology 2018, 125, 1608–1622. [Google Scholar] [CrossRef] [Green Version]
- Diniz, L.P.; Tortelli, V.; Garcia, M.N.; Araújo, A.P.B.; Melo, H.M.; da Silva, G.S.S.; De Felice, F.G.; Alves-Leon, S.V.; de Souza, J.M.; Romão, L.F.; et al. Astrocyte Transforming Growth Factor Beta 1 Promotes Inhibitory Synapse Formation via CaM Kinase II Signaling. Glia 2014, 62, 1917–1931. [Google Scholar] [CrossRef]
- Matias, I.; Diniz, L.P.; Buosi, A.; Neves, G.; Stipursky, J.; Gomes, F.C.A. Flavonoid Hesperidin Induces Synapse Formation and Improves Memory Performance through the Astrocytic TGF-Β1. Front. Aging Neurosci. 2017, 9, 184. [Google Scholar] [CrossRef] [Green Version]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Gerhardinger, C.; Costa, M.B.; Coulombe, M.C.; Toth, I.; Hoehn, T.; Grosu, P. Expression of Acute-Phase Response Proteins in Retinal Müller Cells in Diabetes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 349–357. [Google Scholar] [CrossRef]
- Coughlin, B.A.; Feenstra, D.J.; Mohr, S. Müller Cells and Diabetic Retinopathy. Vision Res. 2017, 139, 93–100. [Google Scholar] [CrossRef]
- Liu, X.; Ye, F.; Xiong, H.; Hu, D.-N.; Limb, G.A.; Xie, T.; Peng, L.; Zhang, P.; Wei, Y.; Zhang, W.; et al. IL-1β Induces IL-6 Production in Retinal Müller Cells Predominantly through the Activation of P38 MAPK/NF-ΚB Signaling Pathway. Exp. Cell Res. 2015, 331, 223–231. [Google Scholar] [CrossRef]
- Mohammad, G.; AlSharif, H.M.; Siddiquei, M.M.; Ahmad, A.; Alam, K.; El-Asrar, A.M.A. Rho-Associated Protein Kinase-1 Mediates the Regulation of Inflammatory Markers in Diabetic Retina and in Retinal Müller Cells. Ann. Clin. Lab. Sci. 2018, 48, 137–145. [Google Scholar] [PubMed]
- Sipos, F.; Firneisz, G.; Műzes, G. Therapeutic Aspects of C-MYC Signaling in Inflammatory and Cancerous Colonic Diseases. World J. Gastroenterol. 2016, 22, 7938–7950. [Google Scholar] [CrossRef] [PubMed]
- Naskar, R.; Thanos, S. Retinal Gene Profiling in a Hereditary Rodent Model of Elevated Intraocular Pressure. Mol. Vis. 2006, 12, 1199–1210. [Google Scholar] [PubMed]
- Zhang, J.; Chen, C.; Wu, L.; Wang, Q.; Chen, J.; Zhang, S.; Chen, Z. C-Myc Contributes to the Release of Müller Cells-Derived Proinflammatory Cytokines by Regulating LncRNA MIAT/XNIP Pathway. Int. J. Biochem. Cell Biol. 2019, 114, 105574. [Google Scholar] [CrossRef] [PubMed]
- Portillo, J.-A.C.; Lopez Corcino, Y.; Miao, Y.; Tang, J.; Sheibani, N.; Kern, T.S.; Dubyak, G.R.; Subauste, C.S. CD40 in Retinal Müller Cells Induces P2X7-Dependent Cytokine Expression in Macrophages/Microglia in Diabetic Mice and Development of Early Experimental Diabetic Retinopathy. Diabetes 2017, 66, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Faria, R.X.; Reis, R.A.M.; Ferreira, L.G.B.; Cezar-de-Mello, P.F.T.; Moraes, M.O. P2X7R Large Pore Is Partially Blocked by Pore Forming Proteins Antagonists in Astrocytes. J. Bioenerg. Biomembr. 2016, 48, 309–324. [Google Scholar] [CrossRef]
- Faria, R.X.; Freitas, H.R.; Reis, R.A.M. P2X7 Receptor Large Pore Signaling in Avian Müller Glial Cells. J. Bioenerg. Biomembr. 2017, 49, 215–229. [Google Scholar] [CrossRef]
- Subauste, C.S. The CD40-ATP-P2X (7) Receptor Pathway: Cell to Cell Cross-Talk to Promote Inflammation and Programmed Cell Death of Endothelial Cells. Front. Immunol. 2019, 10, 2958. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Purinergic Signaling in Retinal Degeneration and Regeneration. Neuropharmacology 2016, 104, 194–211. [Google Scholar] [CrossRef]
- Pavlou, S.; Augustine, J.; Cunning, R.; Harkin, K.; Stitt, A.W.; Xu, H.; Chen, M. Attenuating Diabetic Vascular and Neuronal Defects by Targeting P2rx7. Int. J. Mol. Sci. 2019, 20, 2101. [Google Scholar] [CrossRef] [Green Version]
- Leasher, J.L.; Bourne, R.R.A.; Flaxman, S.R.; Jonas, J.B.; Keeffe, J.; Naidoo, N.; Pesudovs, K.; Price, H.; White, R.A.; Wong, T.Y.; et al. Erratum. Global Estimates on the Number of People Blind or Visually Impaired by Diabetic Retinopathy: A Meta-Analysis From 1990-2010. Diabetes Care 2016;39:1643-1649. Diabetes Care 2016, 39, 2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantalone, K.M.; Hobbs, T.M.; Wells, B.J.; Kong, S.X.; Kattan, M.W.; Bouchard, J.; Yu, C.; Sakurada, B.; Milinovich, A.; Weng, W.; et al. Clinical Characteristics, Complications, Comorbidities and Treatment Patterns among Patients with Type 2 Diabetes Mellitus in a Large Integrated Health System. BMJ Open Diabetes Res. Care 2015, 3, e000093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic Retinopathy: Current Understanding, Mechanisms, and Treatment Strategies. JCI Insight 2017, 2, e93751. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Sundstrom, J.M.; Antonetti, D.A. Ocular Anti-VEGF Therapy for Diabetic Retinopathy: The Role of VEGF in the Pathogenesis of Diabetic Retinopathy. Diabetes Care 2014, 37, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Singh, R.P. The Role of Anti-Vascular Endothelial Growth Factor (Anti-VEGF) in the Management of Proliferative Diabetic Retinopathy. Drugs Context 2018, 7, 212532. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.-I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.-S.; Kwon, S.-H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 Astrocyte Conversion by Microglia Is Neuroprotective in Models of Parkinson’s Disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic Insights into Pathological Changes in the Diabetic Retina Implications for Targeting Diabetic Retinopathy. Am. J. Pathol. 2017, 187, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Jiang, J.X.; Li, A.-F.; Kim, D. Connexin Channel and Its Role in Diabetic Retinopathy. Prog. Retin. Eye Res. 2017, 61, 35–59. [Google Scholar] [CrossRef]
- Iandiev, I.; Tenckhoff, S.; Pannicke, T.; Biedermann, B.; Hollborn, M.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Differential Regulation of Kir4.1 and Kir2.1 Expression in the Ischemic Rat Retina. Neurosci. Lett. 2006, 396, 97–101. [Google Scholar] [CrossRef]
- Le, Y.-Z. VEGF Production and Signaling in Müller Glia Are Critical to Modulating Vascular Function and Neuronal Integrity in Diabetic Retinopathy and Hypoxic Retinal Vascular Diseases. Vision Res. 2017, 139, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Huang, X.; Chen, H.; Xu, H. Müller Glia-Mediated Retinal Regeneration. Mol. Neurobiol. 2021, 58, 2342–2361. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D. Müller Glial Cell Reprogramming and Retina Regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, R.; Fausett, B.V.; Goldman, D. Ascl1a Regulates Müller Glia Dedifferentiation and Retinal Regeneration through a Lin-28-Dependent, Let-7 MicroRNA Signalling Pathway. Nat. Cell Biol. 2015, 17, 532. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.; Wang, J.; Boyd, P.; Wang, F.; Santiago, C.; Jiang, L.; Yoo, S.; Lahne, M.; Todd, L.J.; Jia, M.; et al. Gene Regulatory Networks Controlling Vertebrate Retinal Regeneration. Science 2020, 370, eabb8598. [Google Scholar] [CrossRef]
- Lahne, M.; Nagashima, M.; Hyde, D.R.; Hitchcock, P.F. Reprogramming Müller Glia to Regenerate Retinal Neurons. Annu. Rev. Vis. Sci. 2020, 6, 171–193. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Role | Retinal Changes in Hyperglycemia/DM | Alteration of Müller in Hyperglycemia/DM |
|---|---|---|
| Regulation of water and retinal volume [55,58,59,60,61,68] | Increased thickness of INL and OPL in patients [69] Inhibition of TRPV4 associated with decrease in the water diffusion of diabetic mice retina [70] and prevention of BRB breakdown [71] Alteration in AQP4 expression and/or localization [50,72] Increase in AQP1 and decrease in AQP6 and AQP11 expression [73] | Increase in cellular volume of Müller cells [74,75] Increase in AQP4 expression [76,77,78] in Müller cell [75,79] Swelling of Müller cells and cyst formation [49,75] |
| Regulation of ionic balance [62,80] | Reduction in Kir4.1 [72] and potassium conductance [74,81] | Alteration in Kir4.1 and KV4 channels content and activity [40,74,78,82,83,84] |
| Control of glutamate neurotransmission [40] | Decrease in EAAT1 and glutamine synthetase expression [85,86,87,88] Reduced conversion of glutamate to glutamine and increase in retinal glutamate [88,89,90] Elevation in glutamate levels in retinal cell culture, animal diabetic retina [87], and PDR patients vitreous [91,92] | Decrease in EAAT1 function [93] and expression [88] Reduction in glutamate metabolism [94] |
| Control of GABA neurotransmission [41] | Increase in retinal GABA content [95,96] Enhancement of GABA levels in PDR patients vitreous [91,92] | Increase in GABA immunolabeling in Müller cells [97] |
| Survival of retinal neurons by providing neurotrophic factors and antioxidants [41,98,99] | ND | Neuroprotection [100,101] Increased protein content of neurotrophic factors, but also of inflammatory signals [102] |
| Blood–retinal barrier formation, maintenance and enhancement [63,103] | Increase in GFAP and reduction/redistribution in occludin expression in retinal vessels [104] Increase in adherent leukocytes in blood vessels [105,106] Increase in macromolecules leakage in outer BRB [107] | Decrease in occludin and ZO-1 expression by Müller cell-derived VEGF [108] Leakage resulting from Müller cell ablation [109] Protection against vascular alterations and angiogenesis [110,111] |
| Neurovascular coupling [65,67] | Alteration in neurovascular coupling [66] | ND |
| ILM formation [41,54] | ND | Alteration of ILM [112] Better prognostic after foveal sparing ILM peeling in idiopathic macular hole [88] |
| Neural regeneration [113] | Intravitreal injected adipose-derived stem cells differentiated into pericytes and integrate retinal vasculature [114] Intravitreal injected multipotent mesenchymal stromal cells prevented retinal ganglion cell loss [115] | ND |
| Contribution to the outer retinal barrier, forming OLM [40,49,50] | Alteration in occludin content and distribution in diabetic retinas [49,116] Alteration of OLM in DME [117,118] | ND |
| Participation in the visual cycle [42,43] | Alteration in visual cycle-related proteins [119,120] Involvement of visual cycle proteins with oxidative stress and vascular alterations in DR [121] | ND |
| Contribution to photoreceptors integrity [51] | Degeneration of photoreceptor cells [122,123] Alteration in morphology [124,125,126,127] and disorganization of inner and/or outer segments [128] Disruption of cone arrangement [129] | ND |
| Support high acuity vision in the foveola, by expressing macular pigments [45,48,130] | Decrease in macular pigment [130] | ND |
| Impact on retinal metabolism: glycogen storage, primary use, and possible donation, of lactate, sparing oxygen to neurons [131,132,133,134] | Alteration in glycogen and lactate contents [135,136,137] | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpi-Santos, R.; de Melo Reis, R.A.; Gomes, F.C.A.; Calaza, K.C. Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation. Antioxidants 2022, 11, 617. https://doi.org/10.3390/antiox11040617
Carpi-Santos R, de Melo Reis RA, Gomes FCA, Calaza KC. Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation. Antioxidants. 2022; 11(4):617. https://doi.org/10.3390/antiox11040617
Chicago/Turabian StyleCarpi-Santos, Raul, Ricardo A. de Melo Reis, Flávia Carvalho Alcantara Gomes, and Karin C. Calaza. 2022. "Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation" Antioxidants 11, no. 4: 617. https://doi.org/10.3390/antiox11040617
APA StyleCarpi-Santos, R., de Melo Reis, R. A., Gomes, F. C. A., & Calaza, K. C. (2022). Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation. Antioxidants, 11(4), 617. https://doi.org/10.3390/antiox11040617