
Indolium 1 Exerts Activity against Vemurafenib-Resistant Melanoma In Vivo

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
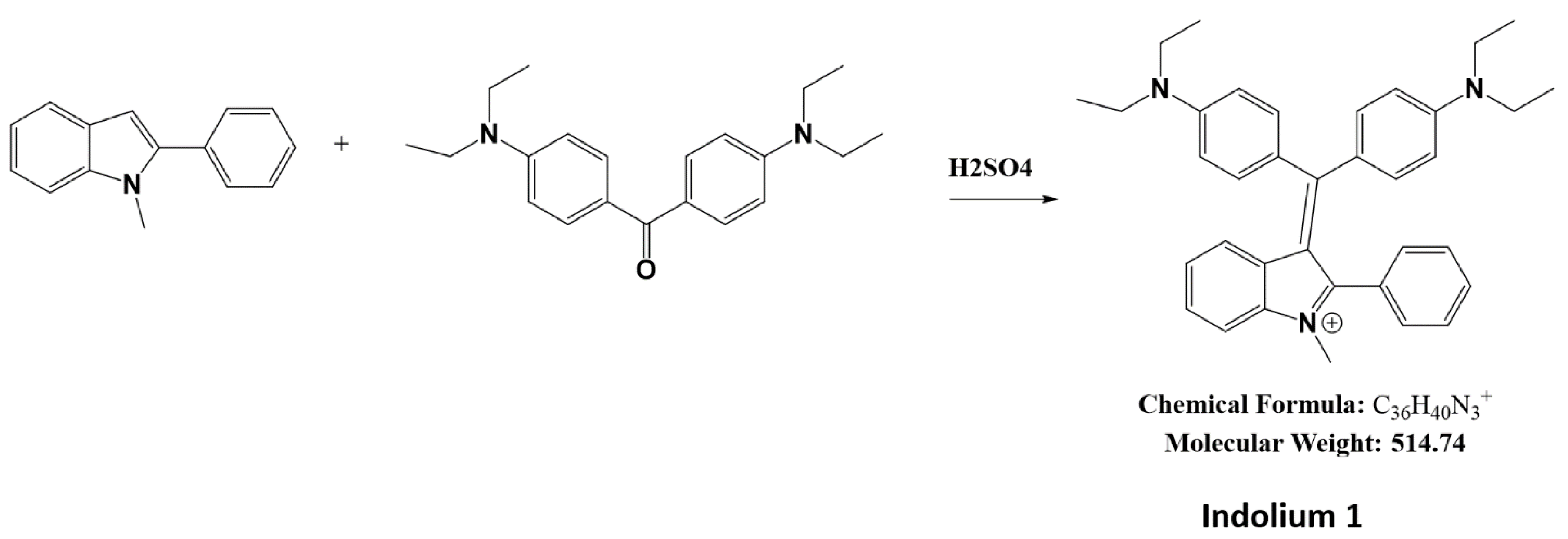
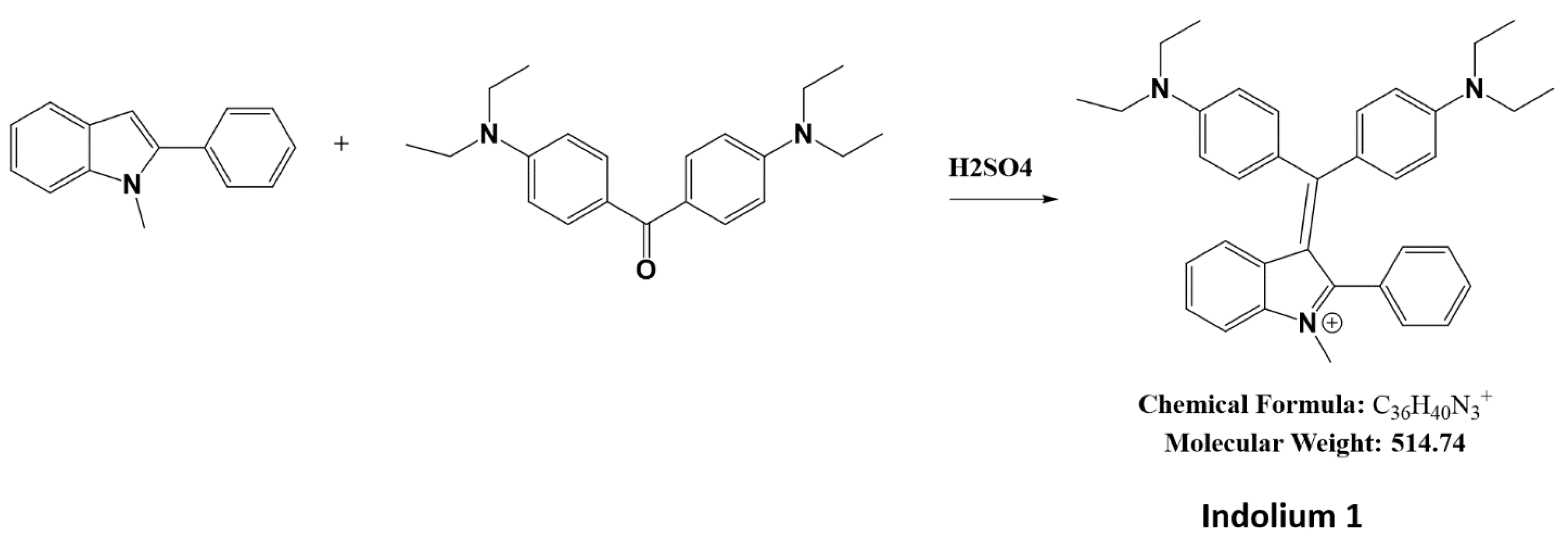
2.1. Synthesis of Indolium 1
2.2. Cell Culture
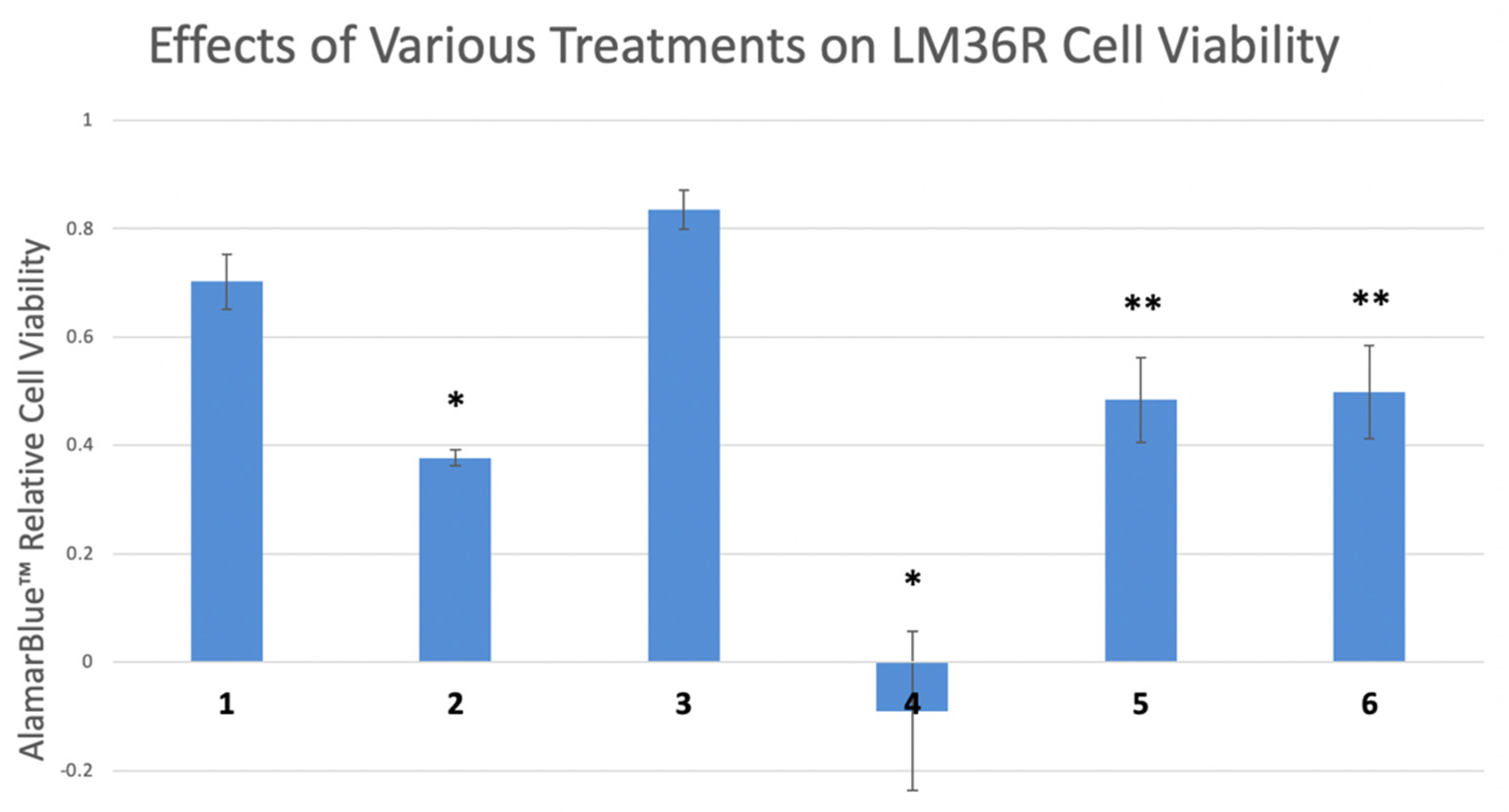
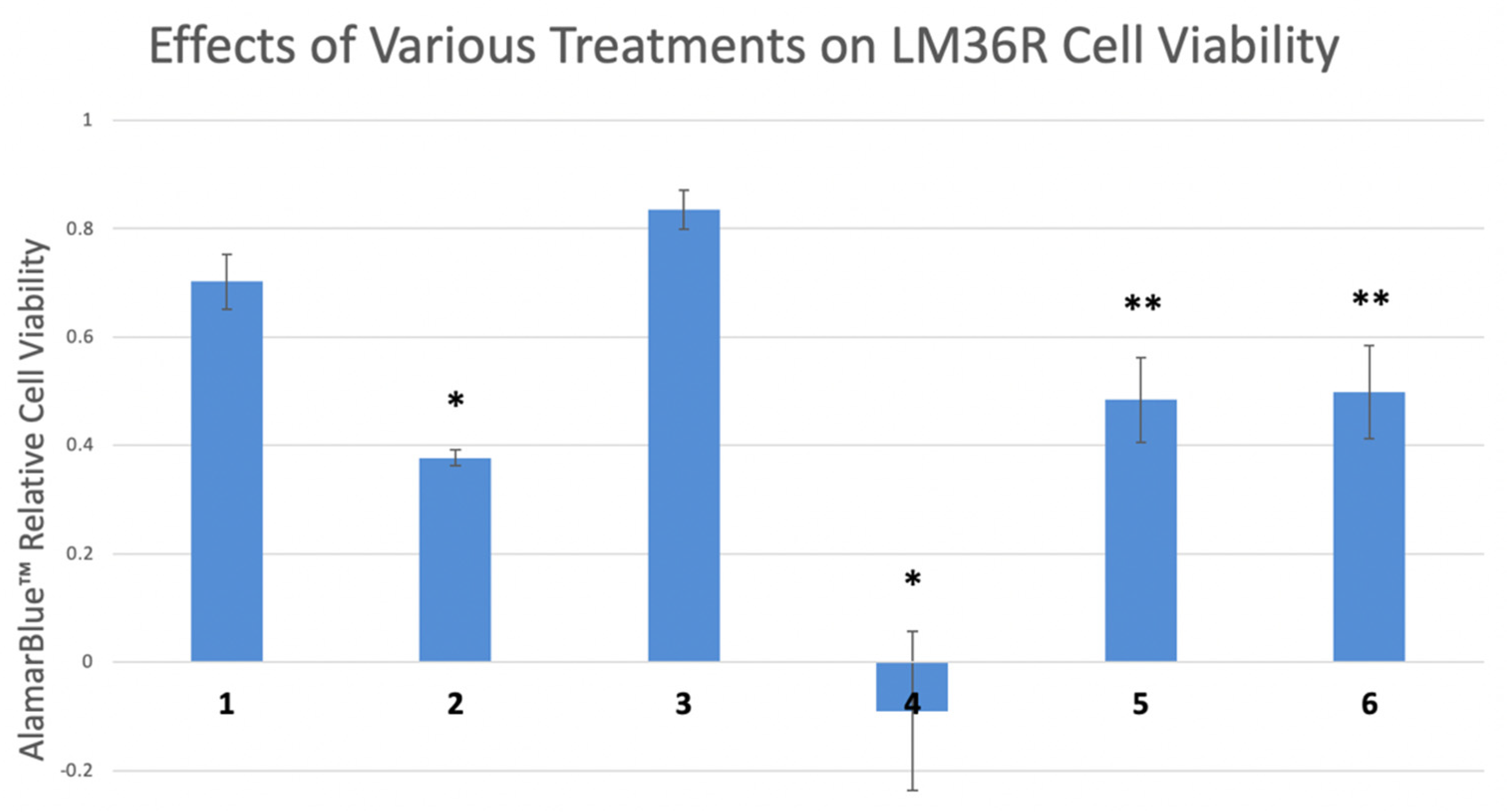
2.3. Proliferation Assay
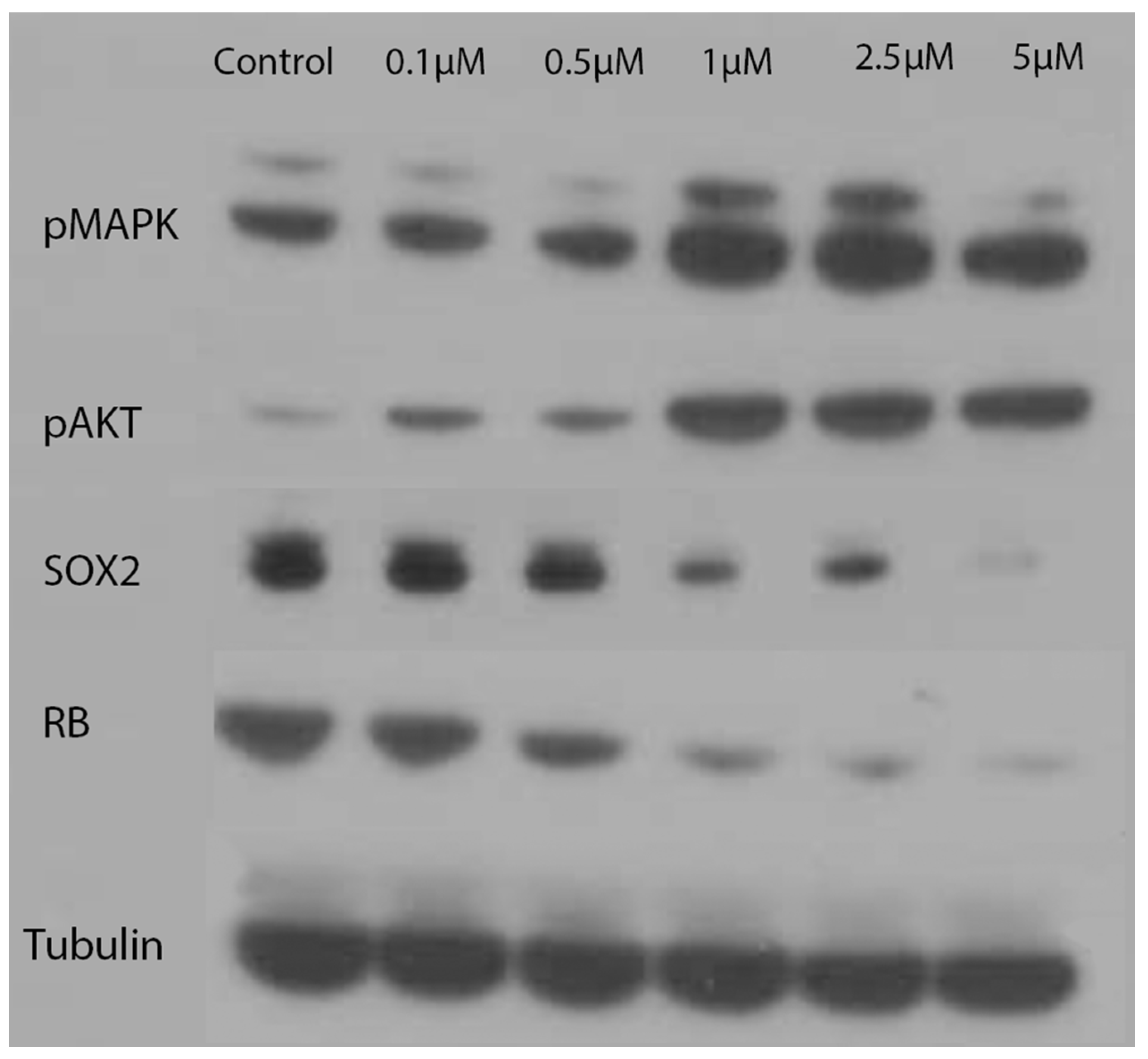
2.4. Western Blotting
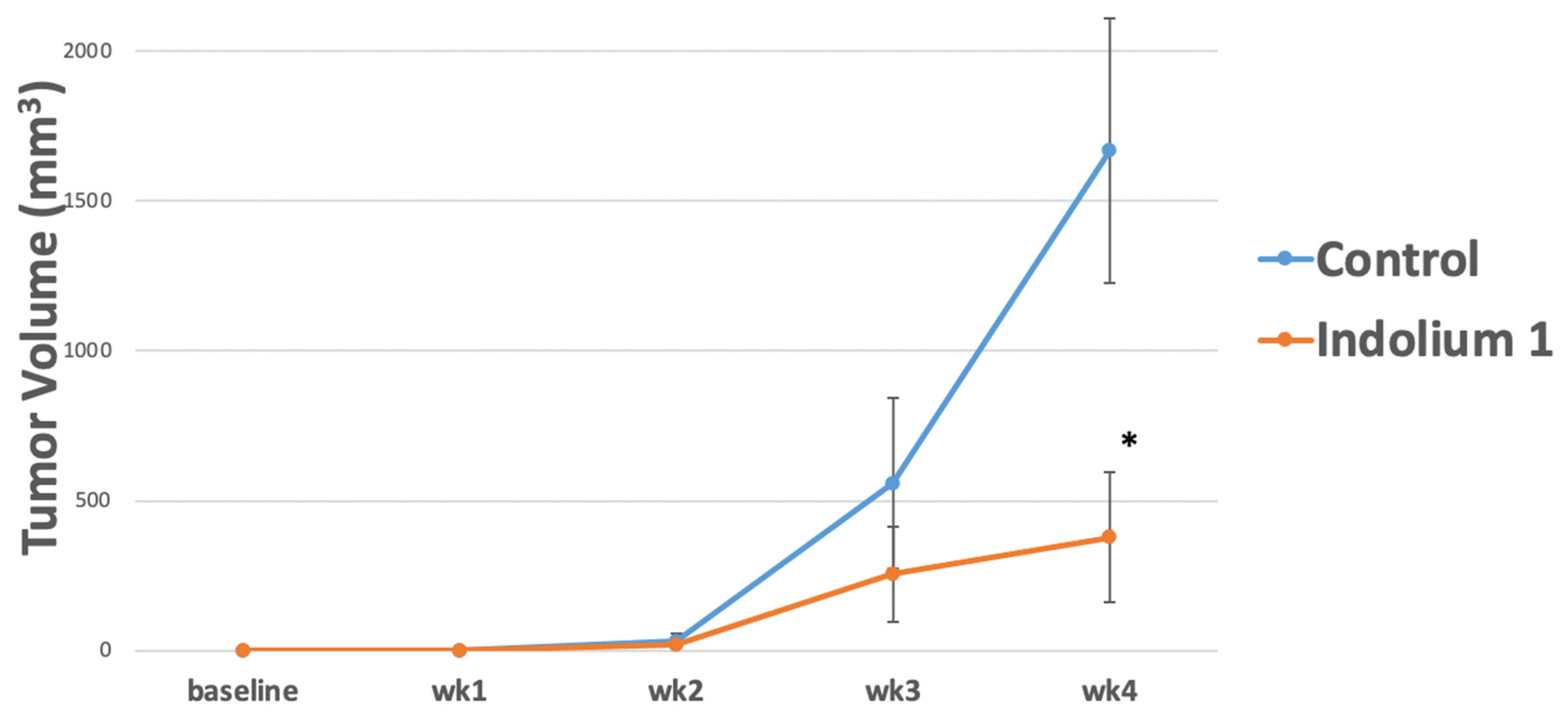
2.5. In Vivo LM36R Xenograft Model
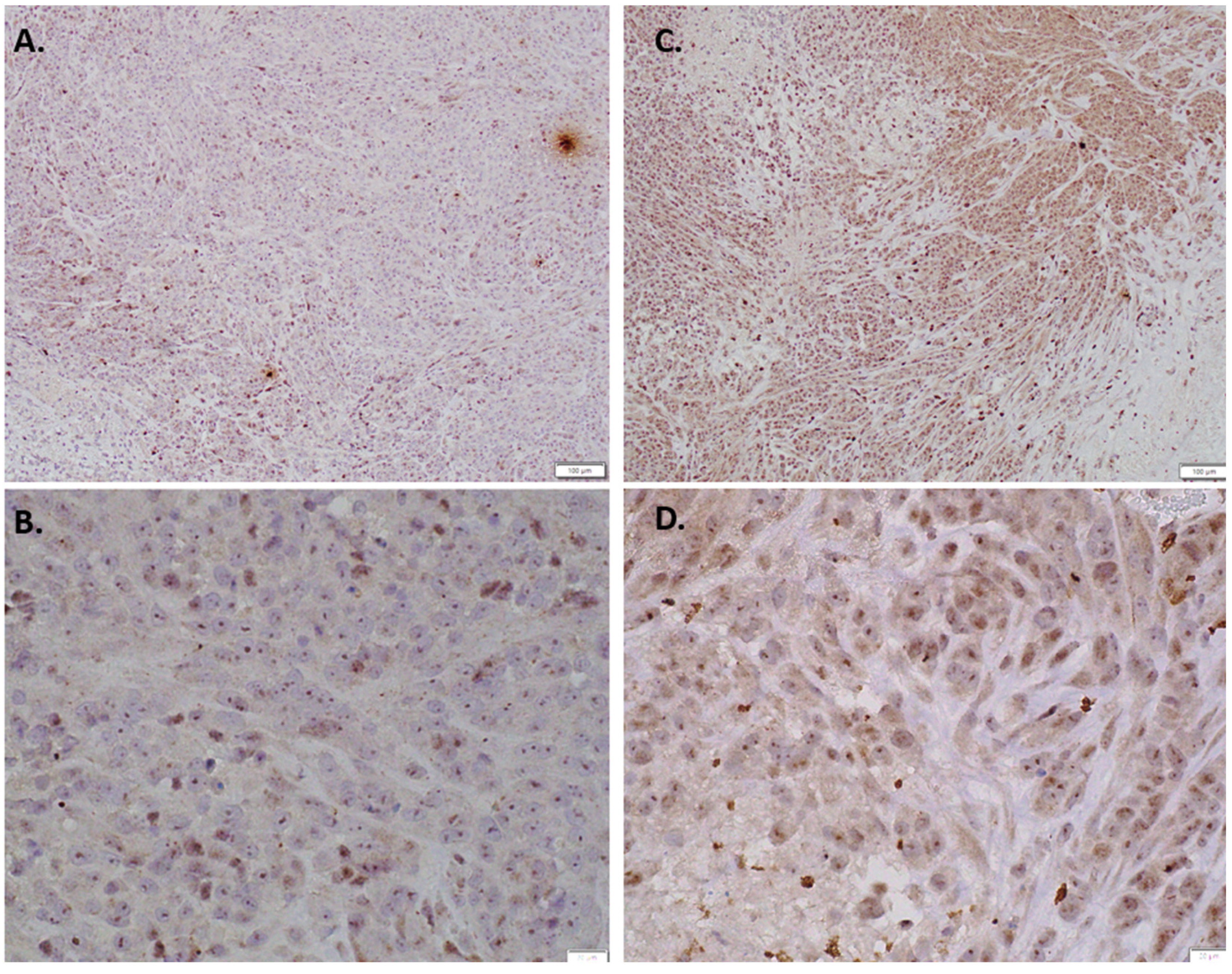
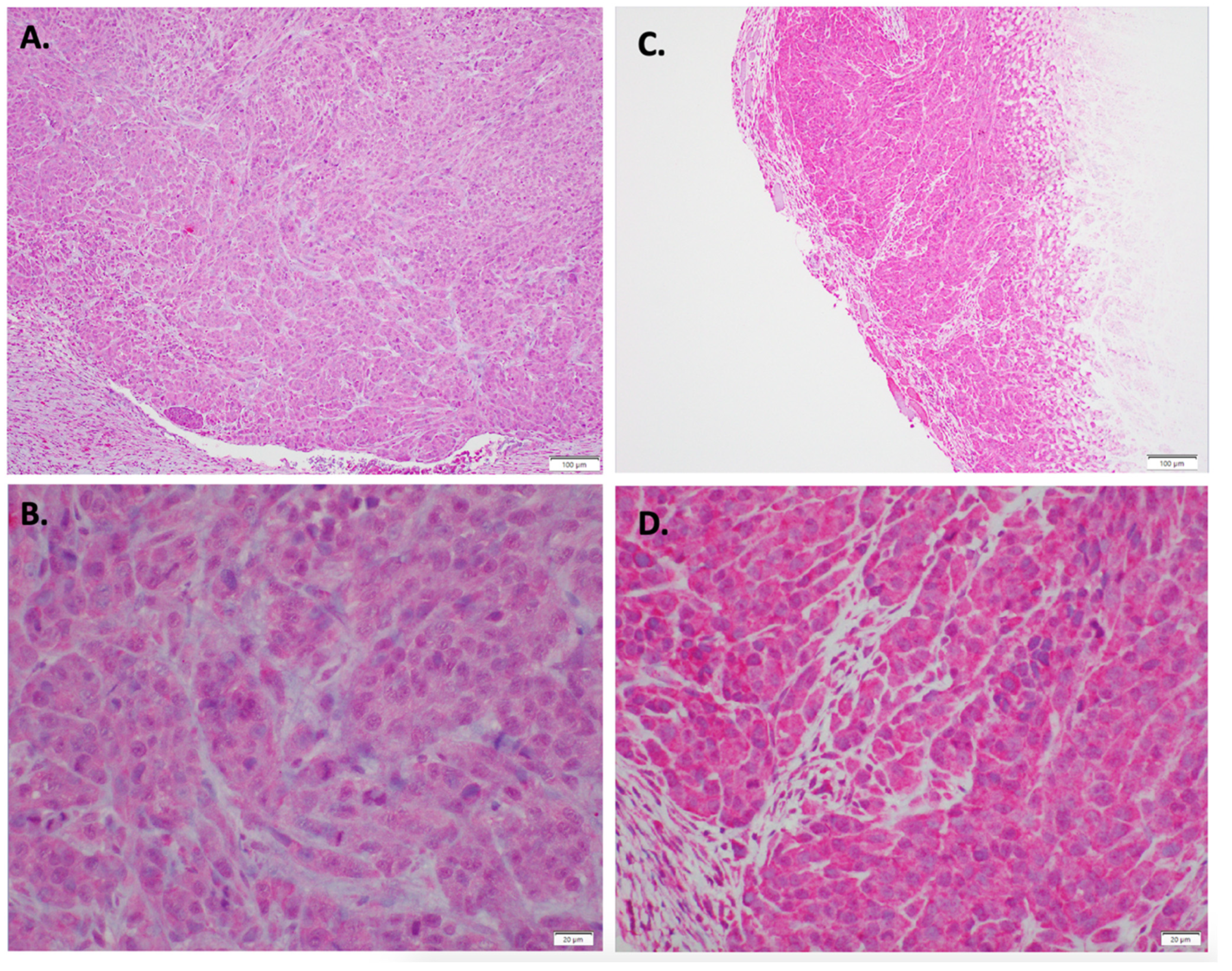
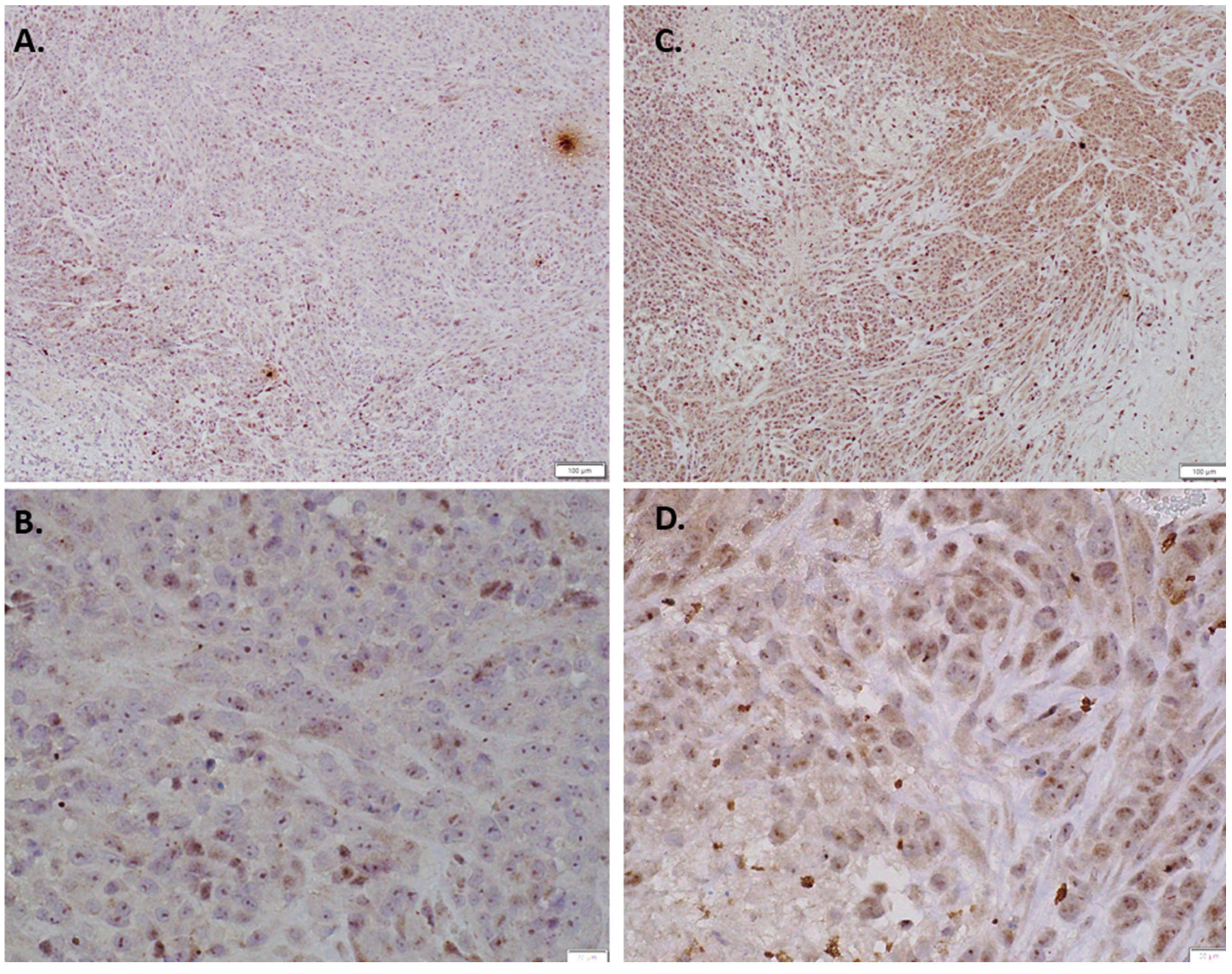
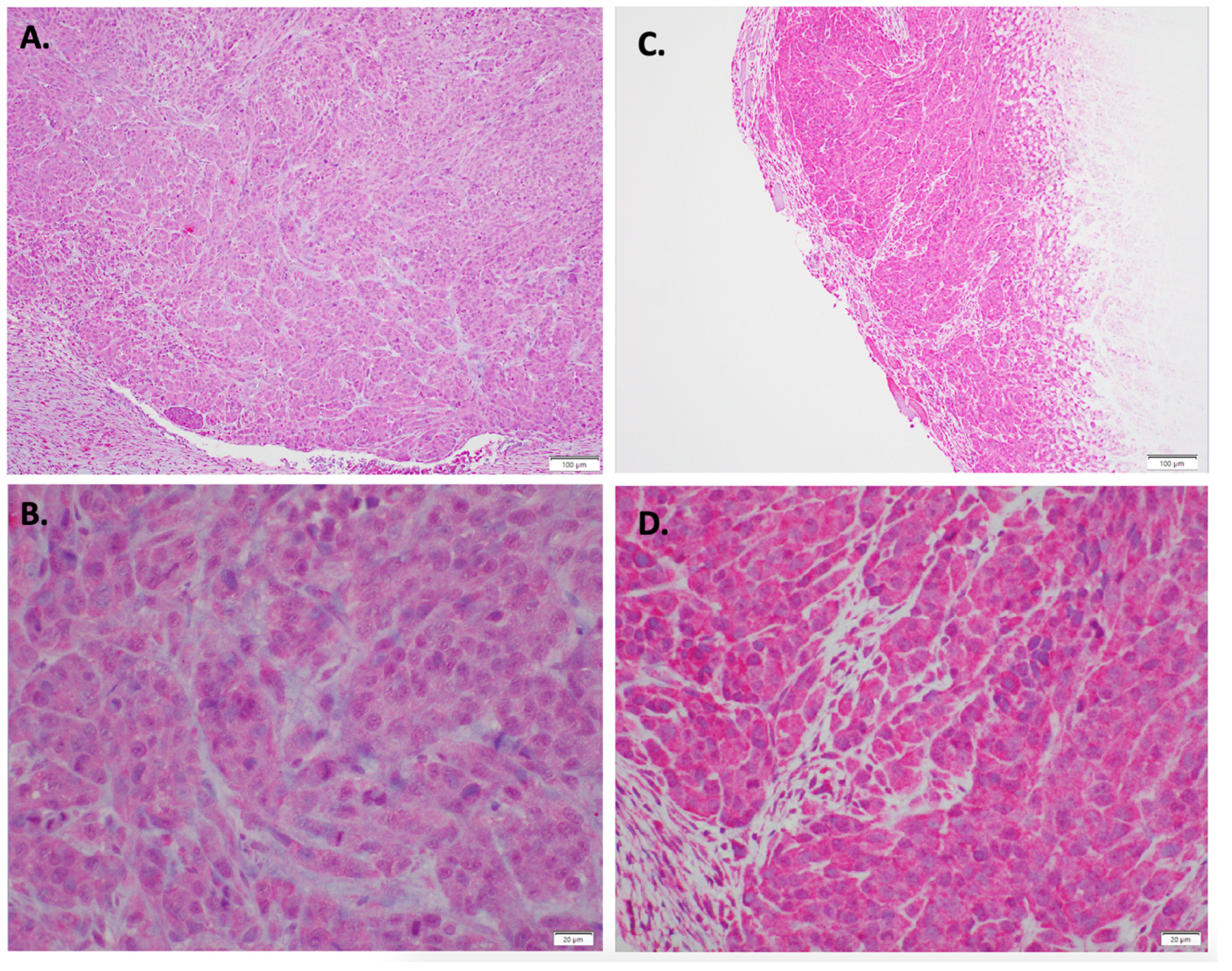
2.6. Immunohistochemistry of In Vivo LM36R Tumor Model
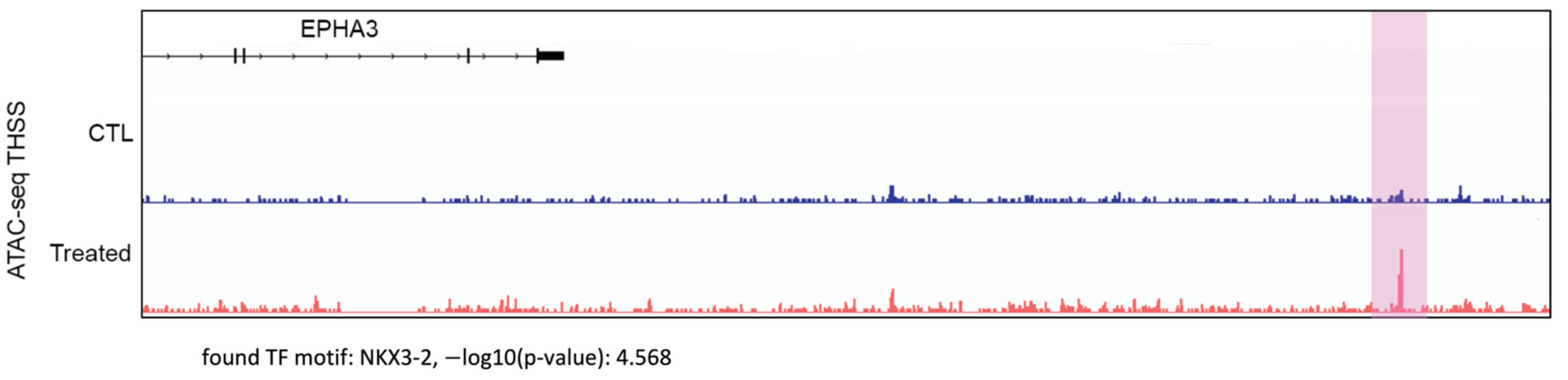
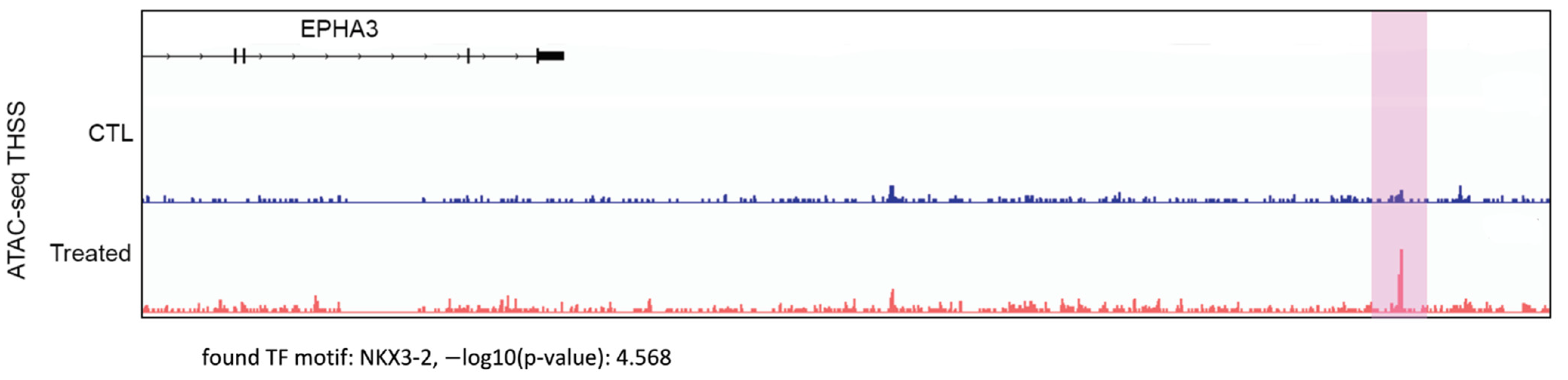
2.7. Assay for Transposase-Accessible Chromatin Using Sequencing (ATAC-seq)
2.8. Analysis of ATAC-seq Data
2.9. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Arbiser, J.L.; Bips, M.; Seidler, A.; Bonner, M.Y.; Kovach, C. Combination therapy of imiquimod and gentian violet for cutaneous melanoma metastases. J. Am. Acad. Dermatol. 2012, 67, e81–e83. [Google Scholar] [CrossRef] [Green Version]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Kappes, J.C.; Katiyar, S.K. Inhibition of NADPH oxidase 1 activity and blocking the binding of cytosolic and membrane-bound proteins by honokiol inhibit migratory potential of melanoma cells. Oncotarget 2016, 7, 7899–7912. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Tian, R.; Lu, N. NADPH oxidase is a primary target for antioxidant effects by inorganic nitrite in lipopolysaccharide-induced oxidative stress in mice and in macrophage cells. Nitric Oxide 2019, 89, 46–53. [Google Scholar] [CrossRef]
- Pietrobono, S.; Morandi, A.; Gagliardi, S.; Gerlini, G.; Borgognoni, L.; Chiarugi, P.; Arbiser, J.L.; Stecca, B. Down-Regulation of SOX2 Underlies the Inhibitory Effects of the Triphenylmethane Gentian Violet on Melanoma Cell Self-Renewal and Survival. J. Investig. Dermatol. 2016, 136, 2059–2069. [Google Scholar] [CrossRef] [Green Version]
- Bonner, M.Y.; Karlsson, I.; Rodolfo, M.; Arnold, R.S.; Vergani, E.; Arbiser, J.L. Honokiol bis-dichloroacetate (Honokiol DCA) demonstrates activity in vemurafenib-resistant melanoma in vivo. Oncotarget 2016, 7, 12857–12868. [Google Scholar] [CrossRef]
- Vergani, E.; Vallacchi, V.; Frigerio, S.; Deho, P.; Mondellini, P.; Perego, P.; Cassinelli, G.; Lanzi, C.; Testi, M.A.; Rivoltini, L.; et al. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia 2011, 13, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Cohen, M.S. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, M.S.; Habiel, D.M.; Espindola, M.S.; Huang, G.; Jones, I.; Narayanan, R.; Coelho, A.L.; Oldham, J.M.; Noth, I.; Ma, S.F.; et al. Antibody-mediated depletion of CCR10+EphA3+ cells ameliorates fibrosis in IPF. JCI Insight 2021, 6, e141061. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, B.; Ma, Q.; Ji, C.D.; Li, J.Z. EphA3 inhibits migration and invasion of esophageal cancer cells by activating the mesenchymalepithelial transition process. Int. J. Oncol. 2019, 54, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisabeth, E.M.; Fernandez, C.; Pasquale, E.B. Cancer somatic mutations disrupt functions of the EphA3 receptor tyrosine kinase through multiple mechanisms. Biochemistry 2012, 51, 1464–1475. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; North, P.E.; Elsey, J.; Bubley, J.; Rao, S.; Jung, Y.; Wu, S.; Zou, M.H.; Pollack, B.P.; Kumar, J.; et al. Propranolol exhibits activity against hemangiomas independent of beta blockade. NPJ Precis. Oncol. 2019, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Jung, Y.; North, P.; Elsey, J.; Choate, K.; Toussaint, M.A.; Huang, C.; Radi, R.; Perricone, A.J.; Corces, V.G.; et al. Introduction of Mutant GNAQ into Endothelial Cells Induces a Vascular Malformation Phenotype with Therapeutic Response to Imatinib. Cancers 2022, 14, 413. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Bleeker, F.E.; Lamba, S.; Rodolfo, M.; Daniotti, M.; Scarpa, A.; van Tilborg, A.A.; Leenstra, S.; Zanon, C.; Bardelli, A. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007, 67, 3545–3550. [Google Scholar] [CrossRef] [Green Version]
- Itoh, T.; Hatano, R.; Horimoto, Y.; Yamada, T.; Song, D.; Otsuka, H.; Shirakawa, Y.; Mastuoka, S.; Iwao, N.; Aune, T.M.; et al. IL-26 mediates epidermal growth factor receptor-tyrosine kinase inhibitor resistance through endoplasmic reticulum stress signaling pathway in triple-negative breast cancer cells. Cell Death Dis. 2021, 12, 520. [Google Scholar] [CrossRef]
- Forse, G.J.; Uson, M.L.; Nasertorabi, F.; Kolatkar, A.; Lamberto, I.; Pasquale, E.B.; Kuhn, P. Distinctive Structure of the EphA3/Ephrin-A5 Complex Reveals a Dual Mode of Eph Receptor Interaction for Ephrin-A5. PLoS ONE 2015, 10, e0127081. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhauser, C.; et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xuan, Z.; Wang, B.; Zhang, D.; Zhang, C.; Wang, J.; Sun, Y. EphA3 Downregulation by Hypermethylation Associated with Lymph Node Metastasis and TNM Stage in Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1514–1522. [Google Scholar] [CrossRef]
- Chiari, R.; Hames, G.; Stroobant, V.; Texier, C.; Maillere, B.; Boon, T.; Coulie, P.G. Identification of a tumor-specific shared antigen derived from an Eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res. 2000, 60, 4855–4863. [Google Scholar]
- Swords, R.T.; Greenberg, P.L.; Wei, A.H.; Durrant, S.; Advani, A.S.; Hertzberg, M.S.; Jonas, B.A.; Lewis, I.D.; Rivera, G.; Gratzinger, D.; et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: Results from a phase 1 study. Leuk. Res. 2016, 50, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Charmsaz, S.; Al-Ejeh, F.; Yeadon, T.M.; Miller, K.J.; Smith, F.M.; Stringer, B.W.; Moore, A.S.; Lee, F.T.; Cooper, L.T.; Stylianou, C.; et al. EphA3 as a target for antibody immunotherapy in acute lymphoblastic leukemia. Leukemia 2017, 31, 1779–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.P.; Stewart, T.; Li, B.; Mulloy, R.; Dimova, D.; Classon, M. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol. Cell. Biol. 2006, 26, 1170–1182. [Google Scholar] [CrossRef] [Green Version]
- Karim, R.Z.; Li, W.; Sanki, A.; Colman, M.H.; Yang, Y.H.; Thompson, J.F.; Scolyer, R.A. Reduced p16 and increased cyclin D1 and pRb expression are correlated with progression in cutaneous melanocytic tumors. Int. J. Surg Pathol. 2009, 17, 361–367. [Google Scholar] [CrossRef]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Slominski, R.M.; Sarna, T.; Płonka, P.M.; Raman, C.; Brożyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar] [CrossRef]
- MacDonald, T.J.; Liu, J.; Yu, B.; Malhotra, A.; Munson, J.; Park, J.C.; Wang, K.; Fei, B.; Bellamkonda, R.; Arbiser, J. Liposome-Imipramine Blue Inhibits Sonic Hedgehog Medulloblastoma In Vivo. Cancers 2021, 13, 1220. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Pietrobono, S.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Stecca, B.; Calorini, L. SOX2 as a novel contributor of oxidative metabolism in melanoma cells. Cell Commun. Signal. 2018, 16, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuławka, Ł.; Donizy, P.; Hałoń, A. Yamanaka’s factors and core transcription factors—The molecular link between embryogenesis and carcinogenesis. Postepy Hig. Med. Dosw. (Online) 2014, 68, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radi, R.; Huang, C.; Elsey, J.; Jung, Y.H.; Corces, V.G.; Arbiser, J.L. Indolium 1 Exerts Activity against Vemurafenib-Resistant Melanoma In Vivo. Antioxidants 2022, 11, 798. https://doi.org/10.3390/antiox11050798
Radi R, Huang C, Elsey J, Jung YH, Corces VG, Arbiser JL. Indolium 1 Exerts Activity against Vemurafenib-Resistant Melanoma In Vivo. Antioxidants. 2022; 11(5):798. https://doi.org/10.3390/antiox11050798
Chicago/Turabian StyleRadi, Rakan, Christina Huang, Justin Elsey, Yoon H. Jung, Victor G. Corces, and Jack L. Arbiser. 2022. "Indolium 1 Exerts Activity against Vemurafenib-Resistant Melanoma In Vivo" Antioxidants 11, no. 5: 798. https://doi.org/10.3390/antiox11050798
APA StyleRadi, R., Huang, C., Elsey, J., Jung, Y. H., Corces, V. G., & Arbiser, J. L. (2022). Indolium 1 Exerts Activity against Vemurafenib-Resistant Melanoma In Vivo. Antioxidants, 11(5), 798. https://doi.org/10.3390/antiox11050798