1. Introduction
The liver is the main organ responsible for maintaining an adequate nitrogen balance in the organism. The combination of ammonia-scavenging pathways guarantees a well-balanced pH regulation and homeostasis of the mitochondrial urea cycle in the periportal region and glutamine synthetase (GLUL) in the perivenous region [
1]. Nevertheless, hyperammonemia is a metabolic condition widely linked to acute and chronic liver diseases [
2,
3], in which a dysregulation in either the urea cycle and/or GLUL leads to perturbations in the homeostasis of the cation. Related to this, our group previously developed a method for staining hepatic ammonia that allows for characterizing hyperammonemia in chronic liver diseases, such as non-alcoholic fatty liver disease (NAFLD) or cirrhosis, and in acute liver injury caused by acetaminophen (APAP) overdose [
4].
Acute liver failure is a severe disorder mainly present in Western countries and caused by the hepatotoxic effect of drugs [
5], where drug-induced liver injury (DILI) is the term normally used to define the condition. With the majority of cases being related to APAP, in the USA, the overdosing of this drug is the leading cause of liver failure [
6], affecting 14 per 100,000 people [
7] and leading annually to 500 deaths, 50,000 visits to the emergency room and 10,000 hospitalizations [
8,
9]. Furthermore, in Europe, APAP overdose comprises 40−70% of all DILI cases [
10]. Currently, the most common treatment for APAP-related liver failure consists of N-acetyl cysteine (NAC) administration, with a probability of 66% for rescuing the liver only if early treatment is provided 8 h after intoxication [
11,
12]. Overall, current therapeutic approaches offer low chances of rescuing the liver from DILI, including the mainstays of early diagnosis and the removal of the suspected drug [
13,
14]. Thus, new approaches are required to improve the prognosis of these patients.
Since their recent approval by the Food and Drugs Agency (FDA), small RNA molecules have entered clinical practice. In this context, a growing number of reports have suggested the significant utility of micro-RNAs (miRNAs) as either biomarkers or drugs, enhancing their use in medical intervention for many diseases [
15]. In this context, a large number of molecules, including miR-34a/b, miR-200c and miR-378, has been related to liver disorders, including fibrosis and viral hepatitis [
16]. Several therapies with compounds targeting miR-34 and miR-122 are currently being evaluated in clinical trials for liver cancer or hepatitis C virus treatment [
15]. Interestingly, our group previously highlighted the potential contribution of miR-873-5p to liver diseases, characterizing its upregulation in NAFLD [
17] and fibrosis [
18]. In these works, miR-873-5p was reported to be an epigenetic regulator of glycine N-methyltransferase (GNMT) for inhibiting its expression.
GNMT is a cytosolic, nuclear and mitochondrial enzyme within hepatocytes that participates in the methionine cycle catalyzing the conversion of S-adenosyl-methionine (AdoMet) to S-adenosyl-homocysteine, being the main enzyme in the liver responsible for the catabolism of excess hepatic AdoMet and synthesis of sarcosine [
19]. The activity of the methionine cycle is essential for correct liver functioning, whereas perturbations in this pathway have been widely linked to liver pathologies [
19,
20]. The intricate metabolic network in the hepatocyte connects the methionine cycle to other pathways such as the folates cycle [
21], the urea cycle [
22] or polyamine synthesis [
23], among others. The modulation of a certain pathway redirects the metabolic flux towards others [
24]. Taking this into consideration, the GNMT expression and methionine cycle recovery mediated by miR-873-5p targeting may modulate the metabolic efflux in the hepatocyte and have an impact on APAP overdose pathophysiology.
The present work evaluates perturbations in ammonia metabolism in the APAP derived hepatotoxicity during DILI. Basing on the existing connection between ammonia homeostasis and the methionine cycle, we demonstrate that GNMT recovery under miR-873-5p knockdown induces a shift from the urea cycle towards polyamine synthesis, preventing DILI development in hepatocytes.
2. Materials and Methods
2.1. Animal Maintenance and Preclinical Studies
All procedures were approved by the CIC bioGUNE Animal Care and Use Committee and the local authority (Diputación de Bizkaia), under the codes P-CBG-CBBA-0218 and P-CBG-CBBA-1421, according to the criteria established by the European Union. Three-month-old C57BL/6J male mice were maintained with ad libitum access to water and a standard chow diet. They were administered 360 mg/kg APAP dissolved in PBS through intraperitoneal injection. Mice were treated 24 h after injection (see below) and sacrificed 24 h later at a final 48 h endpoint. Samples of liver for cryopreservation and paraffin- or O.C.T-embedded and serum were collected.
2.2. Treatment of Primary Mouse Hepatocytes
Primary mouse hepatocytes were obtained from 3-month old C57BL/6J or Glycine-N-methyltransferase lacking (Gnmt−/−) mice by perfusion with Type I collagenase. Hepatocytes were washed three times with Minimal Essential Medium (MEM, ThermoFisher Scientific, Waltham, MA, USA) containing 10% FBS (ThermoFisher Scientific), 1% PSA-G (penicillin-streptamycin-antimycin and glutamine, ThermoFisher Scientific). Cells were seeded over previously collagen I (Corning Inc., Corning, NY, USA)-coated culture plates in MEM 10% FBS, 1% PSA-G. Upon attachment, isolated mouse primary hepatocytes were transfected by overnight incubation with 25 nM anti-miR-873-5p/miR-Ctrl/mimic-miR-873-5p (Horizon Discovery, Waterbeach, Cabridgeshire, UK) using DharmaFECT (GE Healthcare Dharmacon Inc., Lafayette, CO, USA) 1 or jetPRIME (Polyplus-transfection, Strasbourg, Illkrich-Graffenstaden, France). After removing transfection medium 6 h after transfection, hepatocytes were maintained overnight in MEM 0% FBS 1% PSA-G and treated next day with 10mM APAP. Finally, hepatocytes were collected at different times: 0, 1, 3 and 6 h. Different treatments were administered to primary hepatocytes 30 min prior APAP administration: 0.5 μM Dimethylfluoroornithine (DFMO, Sigma-Aldrich, St. Louis, MO, USA), 1 μM SAM486A (Sigma-Aldrich) and 1−2.5 mM ammonium chloride (Sigma-Aldrich) were used.
2.3. Human Subjects
Measurements of serum miR-873-5p and AST, ALT, ALP and TBL were performed in different cohorts recruited at the Hospital Universitario Virgen de la Victoria, Málaga, Spain (
Table 1). Ten human serum samples (six males and four females) with idiosyncratic Drug Induced Liver Injury (DILI) and ten controls (five males and five females) were evaluated for micro-RNA expression levels. Serum samples were obtained at the time of DILI recognition. All the procedures were approved by the Research Ethics Committee of Malaga Hospital (Code AND-HEP-2015-01). The investigators endorse that all patients gave informed consent for the clinical studies, according to the principles embodied in the Declaration of Helsinki.
2.4. miR-873-5p Targeting In Vivo
Mice treated with 360 mg/kg APAP (Sigma-Aldrich) were divided into two groups (n = 4) and administered 60 μg/mouse of an anti-miR-873-5p or miR-Ctrl using Invivofectamine® 3.0 (Invitrogen) Reagent through tail vein injection, which allows a specific silencing in the liver. Mice were sacrificed after 48 h of APAP administration. Samples of serum and liver for cryopreservation and paraffin- or O.C.T-embedded were collected.
2.5. Histological Procedures
All the samples were cut with HistoCore MULTICUT microtome, then deparaffined with Histo-Clear I solution (Fisher Scientific, Hampton, NH, USA) and hydrated through a decreasing concentration of alcohol solutions. Several stainings have been performed: hematoxylin-eosin (H&E) (Sigma-Aldrich), ammonia and immunohistochemistry for glutamine synthetase (GLUL), F4/80, cyclin D1 and proliferating cell nuclear antigen (PCNA). H&E staining: 5 μm sections were subjected to conventional hematoxylin and eosin staining, dehydrating samples and clearing them with histoclear before DPX permanent mounting. Ammonia: 5 μm samples of the paraffin tissue array were incubated for 5 min with 100 mL of Nessler’s reagent (Fisher Scientific) and washed for 10 s with sterile distilled water. Samples were counterstained with Mayer’s hematoxylin, washed with water and dehydrated briefly before clearing with histoclear. Samples were mounted with DPX permanent mounting medium. Nessler’s reagent becomes darker yellow in the presence of ammonia, forming precipitates at higher concentrations. IHC: 5 μm sections were unmasked with 15′ Proteinase K at RT according to the primary antibody used (
Supplementary Table S1) and subjected to endogenous peroxide blocking (3% H
2O
2 in PBS, 10′, RT). For mouse-hosted antibodies, samples were blocked with 2.5% goat anti-mouse Fab fragment (1:10, 1 h, RT) and then blocked with 5% goat serum (30′, RT). Sections were incubated in a humid chamber with the primary antibody in antibody diluent (2% BSA with 0.01% PBS-azide, 1 h, RT) followed by ImmPRESS HRP-conjugated secondary antibodies for Rabbit (Cyclin D1 and GLUL), Rat (F4/80) and Mouse (PCNA) (Vector Laboratories Inc., Burlingame, CA, USA) for 30′ and RT. Colorimetric detection was confirmed with Vector VIP chromogen (Vector Laboratories Inc.) and sections were counterstained with hematoxylin prior to mounting with DPX mounting medium. Images were captured using Leica DM750 optical microscopy with a digital color camera (Leica ICC50W), obtaining 5–10 random images per sample. Stained area percentages of each sample were calculated using FIJI (ImageJ)
https://imagej.net/Fiji, accessed on 10 March 2022.
2.6. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
Total RNA from liver and primary hepatocytes was isolated with Trizol (Invitrogen, Waltham, MA, USA). One to two µg of total RNA were treated with DNAse (Invitrogen) and reverse transcribed into cDNA using M-MLV Reverse Transcriptase (Invitrogen). Quantitative real-time PCR (qPCR) was performed using SYBR
® Select Master Mix (Applied Biosystems, Waltham, MA, USA) and the Viia7/QS6 Real-Time PCR System (Applied Biosystems). The Ct values were compared with a certain group (Ctrl or APAP + siCtrl), and data were then normalized to the housekeeping expression of
Arp. Primers sequences are described in
Supplementary Table S2.
2.7. Metabolite Analysis
Metabolites from tissue/serum were extracted in methanol/water (50/50% v/v) with 10 mM acetic acid and 10 μM stable labelled 13CD3-methionine (methionine-SL) (Cambridge Isotope Laboratories, Tewksbury, MA, USA) as internal standard. Chilled extracts were evaporated with a SpeedVac for approximately 2 h. Pellets were dissolved in water/acetonitrile (MeCN)/formic acid (FA) (39.9/60/0.1% v/v/v). Samples were measured with a UPLC system (Acquity, Waters Inc., Manchester, UK) coupled to a Time of Flight mass spectrometer (ToF MS, SYNAPT G2, Waters Inc.). In a 2.1 × 100 mm, 1.7 μm BEH amide column (Waters Inc.), samples were separated in different solvents (previously stabilized at 40 °C): solvent A (aqueous phase) consisted of 99.5% water, 0.5% FA and 20 mM ammonium formate, while solvent B (organic phase) consisted of 29.5% water, 70% MeCN, 0.5% FA and 1 mM ammonium formate. Extracted ion traces were obtained for AdoMet (m/z = 3 99.145), 13CD3-methionine (m/z = 154.0796), spermine (m/z = 203.2236), spermidine (m/z = 146.1657), GSH (m/z = 308.0916) and GSSG (m/z = 613.1598) in a 20 mDa window for the most abundant isotopes and subsequently smoothed and integrated with QuanLynx software (Waters, Manchester, UK). Metabolite levels were normalized using mg of liver tissue taken.
2.8. Protein Isolation and Western Blotting
Total protein extracts from primary hepatocytes and hepatic tissue were resolved in sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes (GE Healthcare). After 1 h blocking with 5% milk diluted in tris-buffer saline with 0.1% tween-20 (TBS-T) at room temperature, membranes were incubated overnight at 4 °C with a primary antibody diluted in 5% milk in TBS-T. As secondary antibodies, we used anti-rabbit-IgG-HRP-linked (Cell Signaling Technology, Danvers, MA, USA) and anti-mouse IgG-HRP-linked (Cell Signaling) diluted in 5% milk in TBS-T for 1 h at room temperature. The antibodies and conditions used for Western Blotting are described in
Supplementary Table S3.
2.9. Hepatic microRNA Quantitative Real-Time PCR
Total RNA was from liver and primary hepatocytes isolated with Trizol (Invitrogen) using TaqMan
® MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific, 4366597) procedure using 100 ng of total RNA and using specific oligos for hsa-miR-873-5p (Thermo Fisher Scientific, 002356) and U6 (Thermo Fisher Scientific, 001973). Quantitative real-time PCR was performed using TaqMan
® Universal PCR Master Mix, no AmpErase
® (Thermo Fisher Scientific, 432408), following manufacturer’s procedure. Expression levels were normalized using U6 snRNA. Sequences are described in
Supplementary Table S4.
2.10. Serum microRNA Quantification
Micro-RNAs were isolated from serum with the miRNeasy Serum/Plasma Kit (QIAGEN, Hilden, Mettmann, Germany) following manufacturer’s procedure, adding miR-39 as a Spike-In Control. miR-873-5p-specific RT/q-PCR were performed as described and miR-873-5p levels were normalized to the Spike-In.
2.11. TUNEL Assay for Cell Death Detection
Apoptotic cells were detected by TUNEL staining using in situ cell death detection kit (Roche, city, State, country). Cells were fixed in 4% paraformaldehyde for 10 min, washed twice in PBS and treated with 3% H
2O
2 (Panreac Applichem, Darmstadt, Germany) diluted in MeOH (Merk Millipore, Basilea, Schwitzerland) for 5 min and citrate pH 7.4 for 3 min, then, cells were treated with an enzyme:FITC:buffer mixture (1:9:40) and mounted with Fluoroshield
TM with DAPI (Sigma-Aldrich). Five random images per sample were taken and the percentage of TUNEL positive cells was calculated using FIJI (FIJI is just ImageJ)
https://imagej.net/Fiji, accessed on 10 March 2022.
2.12. GNMT Immunofluorescence
Cells previously fixed in 4% paraformaldehyde (ThermoFisher Scientific, J19943) were blocked and permeabilized with 0.1% BSA (Sigma-Aldrich, A3912), 10% GS (Sigma) with 0.01% Triton X-100 (Sigma-Aldrich, T9284). Then, samples were incubated overnight at 4 °C with anti-GNMT homemade antibody diluted 1:1000 in 0.1% BSA and 10% GS. The next day, samples previously washed with PBS were developed with secondary Rabbit-Cy3 (1:200; 1 h; RT in 0.1% BSA and 10% GS). Pictures were taken with an Axio Imager D1 Upright Fluorescence Microscope (Carl Zeiss AG, Jena, Germany).
2.13. Transaminases Determination in Serum
The levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in serum were determined by a Selectra Junior Spinlab 100 analyzer (Vital Scientific, Dieren, The Netherlands) according to the manufacturers’ suggested protocol.
2.14. Enzyme-Linked Immunosorbent Assay (ELISA)
The amount of serum tumor necrosis factor (TNF) in serum was measured using 20 µL of samples by paired antibodies. Samples were analyzed by ELISA using the DuoSet II kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s recommendations.
2.15. SOD Assay in Liver Homogenates
Superoxide dismutase activity was followed through a colorimetric assay by using a SOD Determination Kit (Sigma-Aldrich). The procedure was realized according to the manufacturer’s instructions.
2.16. Determination of Mitochondrial Reactive Oxygen Species (ROS)
Mitochondrial ROS production in primary hepatocytes was assessed using MitoSOX Red mitochondrial superoxide indicator (ThermoFisher Scientific). The cells were loaded with 2 μM MitoSOX Red for 10 min at 37 °C in a CO2 incubator. The cells were then washed three times with PBS. Fluorescence was read at 510 nm (excitation) and 595 nm (emission) using a plate reader SpectraMax M2 (bioNova Científica SL, Madrid, Comunidad de Madrid, Spain).
2.17. Respiration Studies in Primary Hepatocytes
The respiration activity in primary hepatocytes was measured at 37 °C by high-resolution respirometry with the Seahorse Bioscience XF24-3 Extracellular Flux Analyzer. Succinate (10 mM) and rotenone (2 μM) were used as substrates to quantify State 2. State 3 was initiated with ADP, State 4 induced with the addition of oligomycin (State 4o), and FCCP-induced maximal uncoupler-stimulated respiration (State 3u) were sequentially measured. The normalized data are expressed as pmol of O2 per minute or milli-pH units (mpH) per minute, per µg total protein. The dual-ATP production rate was assessed using Seahorse XFe24 Analyzer (Agilent Technologies, Santa Clara, CA, USA); simultaneous reads of ATP production from glycolysis and mitochondria were performed using a label-free technology XF Real-Time ATP Rate Assay kit, as described in the User Guide (Agilent Technologies).
2.18. Mitochondrial Labelling
The relative number of functional mitochondria was determined by using a MitoTracker
TM Green FM (ThermoFisher) probe. Cells were stained according to manufacturer’s instructions and then fixed with 4% paraformaldehyde for 10 min. Five pictures per sample were acquired randomly using an Axioimager D1 upright fluorescence microscope (Leica Biosystems, Nußloch, Baden-Wurttemberg, Germany), and the relative amount of fluorescence was determined by FIJI (ImageJ)
https://imagej.net/Fiji, accessed on 10 March 2022.
2.19. Intracellular ATP Determination
Intracellular ATP levels were determined in primary hepatocytes by using an ATPliteTM luminescence ATP detection kit (Perkin Elmer, Waltham, MA, USA) following manufacturer’s recommendation. In brief, 50 μL of the mammalian cell lysis solution were added per well with 100 μL of MEM without serum and incubated on an orbital shaker (700 rpm, 5′, RT). Then, 35 μL of the lysate were incubated in the white plate’s wells (Gibco, ThermoFisher Scientific) containing 100 μL of MEM 0% previously added, and 50 μL of the substrate solution were then added and incubated (700 rpm, 5′, RT). The plate was adapted to the dark for 10′ and the luminescence was measured in a luminometer. The obtained values were normalized to total protein concentration.
2.20. Carbamoyl-Phosphate 1 Synthetase (CPS1) Activity Determination
The amount of hepatic CPS1 activity was determined by using a previously described method [
25]. Briefly, 20 mg liver samples were lysed in 200 μL of mitochondrial lysis buffer (10 mM HEPES pH 7.4, 0.5% triton X-100 and 2 mM DTT) and samples normalized to 0.1 μg/μL. Next, 10 μL of sample were mixed with 90 μL and then incubated with 25 μL 50 mM ornithine, 25 μL 2.7M triethanolamine and 25 μL 150 mM carbamoylphosphate. A 0 to 100 nmoles standard curve was also incubated with the ornithine: triethanolamine carbamoylphosphate mixture. After 30 min at 37 °C incubation protected from light, samples were added 80 μL phosphoric acid: sulfuric acid 3:1 and 20 μL butanedione monoxime. The plate was shacked for 30 s and then incubated at 95 °C during 30 min prior absorbance determination at 490 nm wavelength.
2.21. Ornithine Transcarbamylase (OTC) Activity Determination
To start, 20 mg liver samples were lysed in 200 μL of mitochondrial lysis buffer (10 mM HEPES pH 7.4, 0.5% triton X-100 and 2 mM DTT) and samples normalized to 2 μg/μL. A 20 μL sample was incubated at 37 °C during 10 min with 40 μL of an enzymatic reaction mixture containing 10 μM NH4HCO3, 1 μM ATP, 2 μM magnesium acetate, 1 μM N-acetyl-L-glutamic, 0.2 μM DTT and 10 μM triethanolamine. A standard curve of 60 μL from 0 to 50 nmol was also incubated. Then, 6 μL of 2M hydroxylamine were added and the plate was incubated at 95 °C for 10 min. Next, 240 μL were added of a stop/developer solution consisting on equal parts of 850 mg antipyrine in 100 mL of 40% sulfuric acid and 625 mg of 2,3-butanedione monoxime in 5% acetic acid. The plate was incubated at 95 °C for 15 min and absorbance was determined at 450 nm wavelength.
2.22. Metabolic Flux Determination in Primary Hepatocytes
For the relative quantification with high resolution chromatography-coupled Time-of-Flight mass spectrometry analysis in primary hepatocytes, 30 μg/mL of L-methionine (13C5, 99%, Cambridge Isotope Laboratories, Inc.) were added to methionine-free cell media (ThermoFisher Scientific); when the experiment was finished, plates were immediately frozen and the subsequent analysis was performed. For the metabolite extraction, 500 μL ice cold methanol (80%) containing 200 mM acetic acid was added to the wells of the culture plates, then, buffer was transferred to Eppendorf tubes and centrifuged at 3750 rpm at 4 °C during 30 min. After, 200 μL of the chilled supernatants were evaporated with a SpeedVac in approximately 1.5 h. The resulting pellets were resuspended in 150 μL water/acetonitrile (MeCN) (40/60/v/v). Measurements were made with a UPLC system (Acquity, Waters Inc.) coupled to a Time-of-Flight mass spectrometer (ToF MS, SYNAPT G2, Waters Inc.). A 2.1 × 100 mm, 1.7 μm BEH amide column (Waters Inc.), thermostated at 40 °C, was used to separate the analytes before entering the MS. Solvent A (aqueous phase) consisted of 99.5% water, 0.5% formic acid and 20 mM ammonium formate while solvent B (organic phase) consisted of 29.5% water, 70% acetonitrile, 0.5% formic acid and 1mM ammonium formate. Samples were injected following a gradient, and every eight injections, a QC sample was injected.
2.23. Subcellular Protein Extraction
Cytosolic, membrane and nuclear fractions’ lysates from frozen liver tissue samples were obtained using the Subcellular Proteome Extraction Kit (Calbiochem) following manufacturer’s procedure. The lysates were quantified by BCA protein assay (ThermoFisher Scientific). Mitochondrial isolation from frozen liver tissue samples was performed using the Mitochondria/Cytosol Fractionation Kit (Abcam, Cambridge, UK) as indicated by the manufacturer. Briefly, frozen livers were ground in mortar previously cooled with liquid nitrogen. Then, they were resuspended in the cytosolic buffer and mechanically homogenized, always in cold. Cytosols were centrifuged (13,000 rpm, 10′) three times. Pellets obtained from the sequential centrifugations were collected as crude mitochondria and finally mixed and lysed in the mitochondrial buffer for BCA quantification.
2.24. Statistical Analysis
All the experiments were performed at least in triplicate, with n = 3 (in vitro) and n = 4 (in vivo). The data are expressed as mean ± SEM and represent the fold change vs. control mean value when indicated. Statistical significance was determined using Prism 8 (GraphPad Software, Dotmatics, Bishop’s Stortford, East Hertfordshire, UK). Groups were compared by one-way analysis of variance (ANOVA) followed by post hoc Bonferroni tests (for three or more groups) or Student´s t-tests (for two groups). Correlations were calculated by using Pearson’s correlation coefficient from Prism 8 (GraphPad Software).
4. Discussion
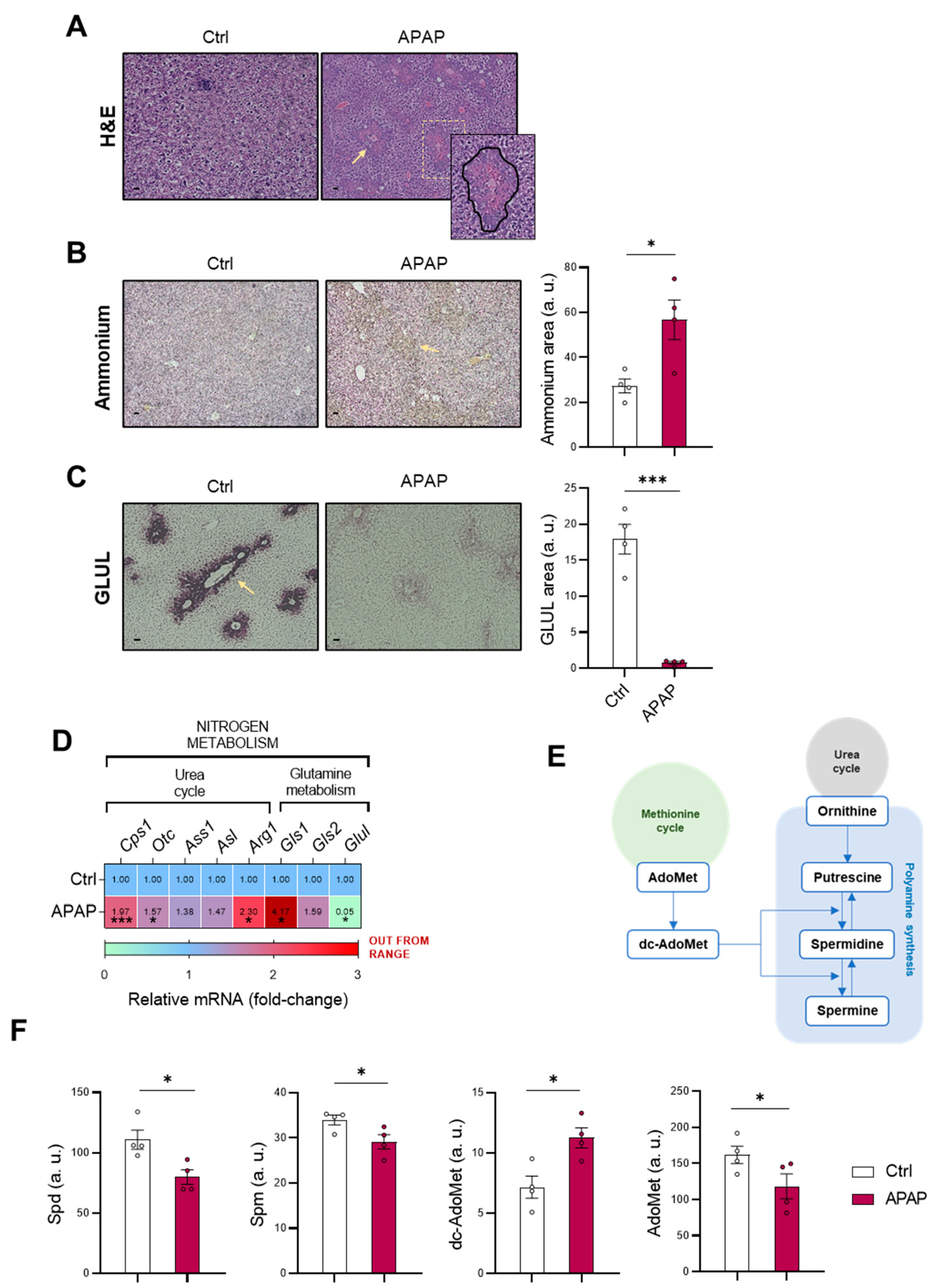
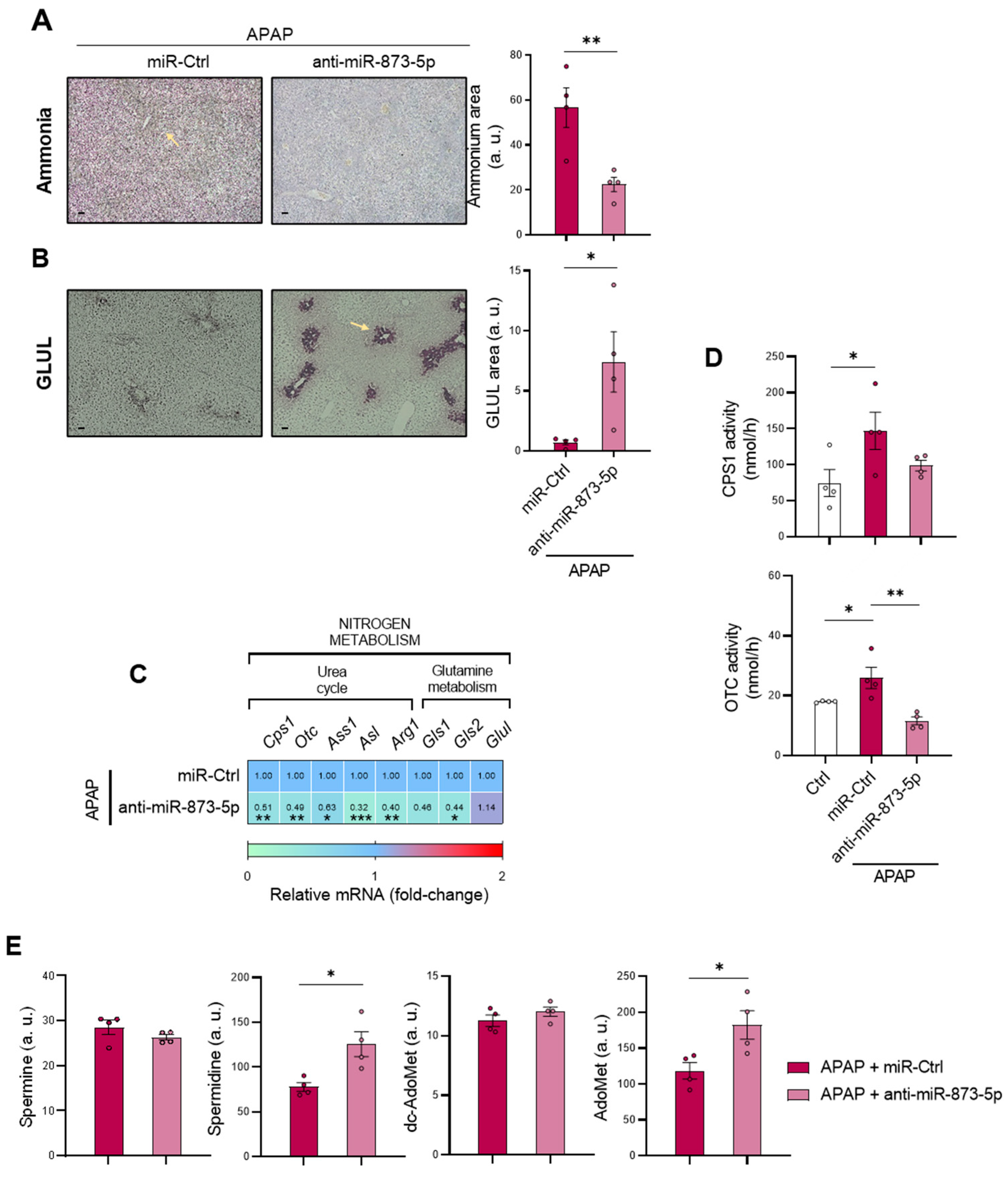
This work highlights the relevance of the interconnection between nitrogen metabolism, with the urea cycle and polyamine synthesis pathways, and the methionine cycle in DILI development. Related to the maintenance of ammonia in the liver, hepatocytes play a key role in scavenging the cation by either: (i) the action of CPS1 from the urea cycle in the periportal region or (ii) the GLUL-mediated glutamine synthesis in the perivenous region [
1]. Although the liver achieves a normal homeostasis of the cation, during liver pathologies, perturbations in both ammonia production and/or scavenging lead to hyperammonemia [
38,
39]. Indeed, in a previously developed staining method for ammonia, our group already characterized hepatic hyperammonemia in chronic and acute liver diseases [
4]. Correlated with this, the present study characterizes the accumulation of the cation in those mice treated with 360 mg/kg APAP for 48 h accompanied by a GLUL depletion, already reported to occur [
40]. Otherwise, the increased mRNA expression of the urea cycle-limiting enzymes
Cps1 and
Otc in those mice administered APAP suggested enhanced urea cycle activity that might attempt to compensate the hepatotoxic ammonia accumulation during DILI.
As cited above, there is an existing connection between the urea cycle and other pathways such as polyamine synthesis and the methionine cycle [
22,
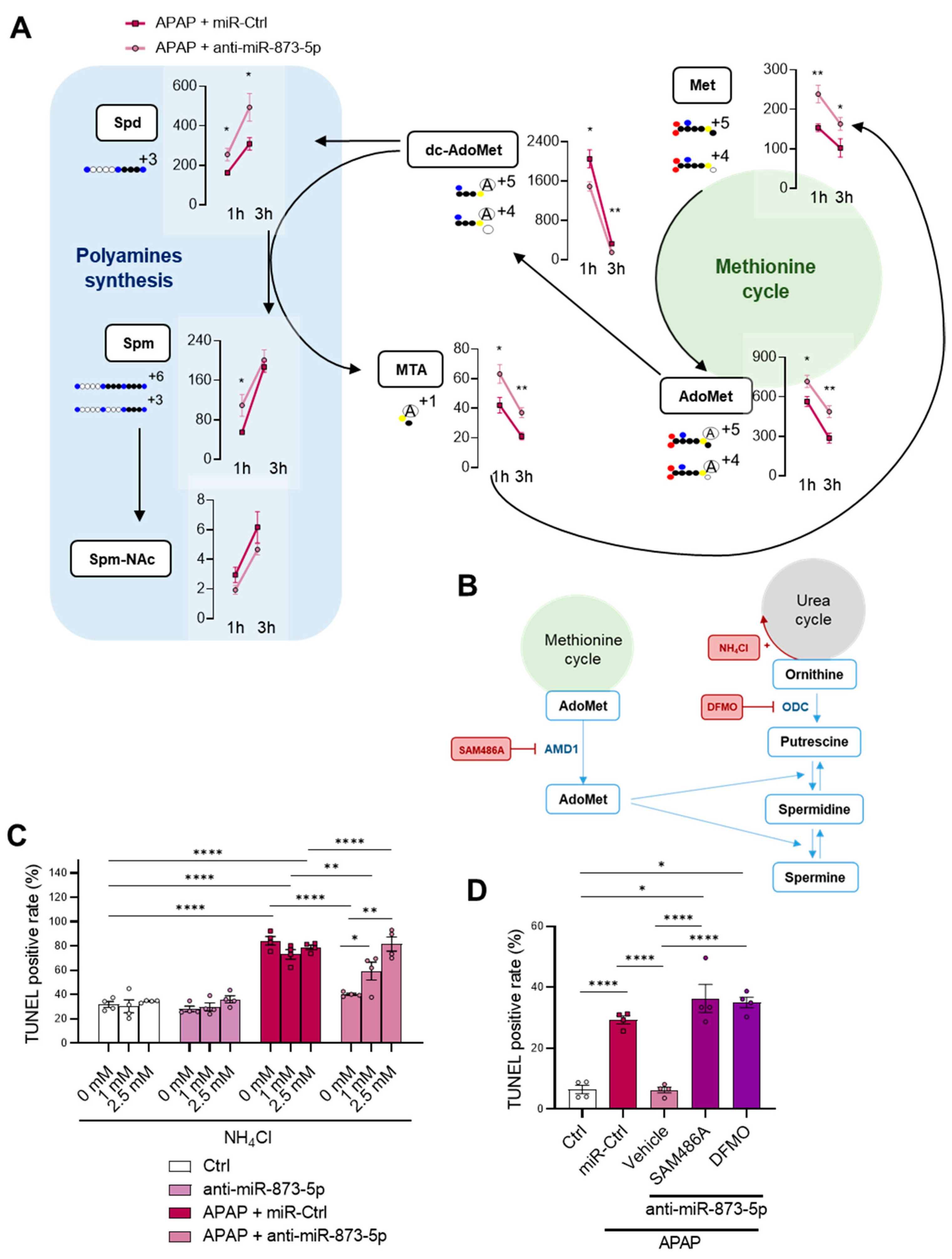
23]. Related to this, the metabolomics determination of polyamines Spd and Spm revealed a decrease in those mice under APAP, together with a decreased hepatic AdoMet content as characterized in previous studies [
41]. AdoMet has already been reported as an indicator of the methionine cycle activity [
41], which appeared to be downregulated during DILI. AdoMet anabolism is mediated in the liver by MATI/III, while several studies have already provided evidence for the formation of peroxynitrite during APAP hepatotoxicity [
42] with S-nitrosylation of MATI/III associated with marked inactivation of the enzyme. Additionally, MATI/III inactivation is reversed by increased levels of GSH [
43]. Although our data revealed overexpression of MATI/III in mice overdosed with APAP, lower AdoMet levels indicated an inhibition of this enzyme, which potentially could be reversed when oxidative stress and GSH levels are restored under anti-miR-873-5p treatment. GNMT, responsible of AdoMet catabolism, was downregulated in our study under APAP overdose according to previous findings already reported in many chronic liver pathologies. Indeed, mice lacking the gene spontaneously develop steatosis and hepatocellular carcinoma [
19]. Thus, the increased hepatic dc-AdoMet content in DILI observed in our study may be a consequence of the combination of a reduced AdoMet metabolism and decreased polyamine synthesis, where Spm has been reported to negatively regulate AdoMet decarboxylase [
44]. These results point towards not only perturbations in ammonium homeostasis during DILI, but also a decreased methionine cycle activity. The recovery of the methionine cycle may be essential to diminish the hepatotoxic effects of APAP during DILI development.
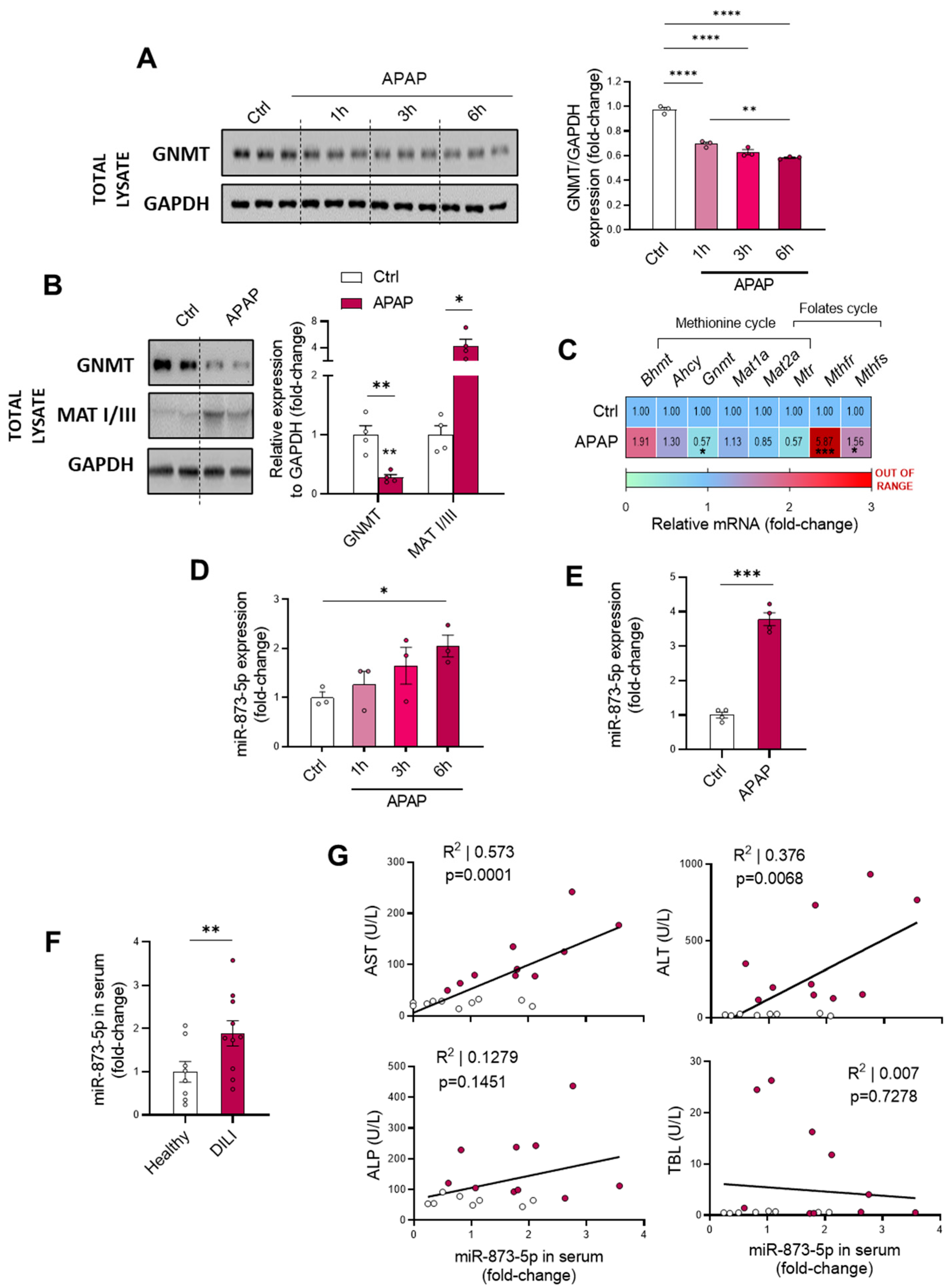
This study focuses on the miR-873-5p-mediated regulation of GNMT expression for restoring the methionine cycle and preventing the pathology. This miRNA has already been reported by our group to act as an epigenetic modulator of GNMT, finding it overexpressed in NAFLD [
17] and fibrosis [
18] and being inversely correlated with the expression of the enzyme. Similar to previous findings, GNMT downregulation was accompanied by an increased miR-873-5p expression in both primary wild-type hepatocytes under different periods of APAP overdose and liver from C57BL/6J mice treated with APAP. Remarkably, the presence of the miRNA was found in the serum from DILI patients, being increased when compared to healthy individuals and finding it correlated with ALT and AST and liver damage parameters.
The GNMT restoration, according to protein levels and mRNA expression, provided by an anti-miR-873-5p-based therapy prevented the development of DILI in primary wild-type hepatocytes at different times of APAP, while in treated mice it reduced the appearance of necrotic areas in the liver and transaminases levels in the serum. On the contrary, the overexpression of miR-873-5p by using a mimic-miRNA aggravated the hepatotoxic effect of APAP overdose. Although the loss of
Gnmt mRNA was not completely prevented by the anti-miRNA, it must be taken into account that the epigenetic regulation of
Gnmt is not only determined by the miR-873-5p but also by DNA hypermethylation. This plays an important role in the repression of GNMT, at least in liver cancer, meaning that the changes by the anti-miR-873-5p were expected to be mild [
45]. Nevertheless, the protein determination in vitro by Western blot and immunofluorescence revealed a significant increase in the anti-miR-873-5p-treated groups at different times and at both cytoplasmic and nuclear levels. The importance of GNMT restoration by anti-miRNA treatment was further demonstrated in primary hepatocytes isolated from mice lacking
Gnmt (
Gnmt-/-), where the protective effect of anti-miR therapy was lost. Likewise, the transient GNMT overexpression in primary wild-type hepatocytes also protected the cells from APAP at 3 and 6 h of incubation. However, this effect over a longer period (9 h) was not achieved, highlighting the efficacy of miRNA-based therapies as an improved therapeutic approach for DILI [
15,
46].
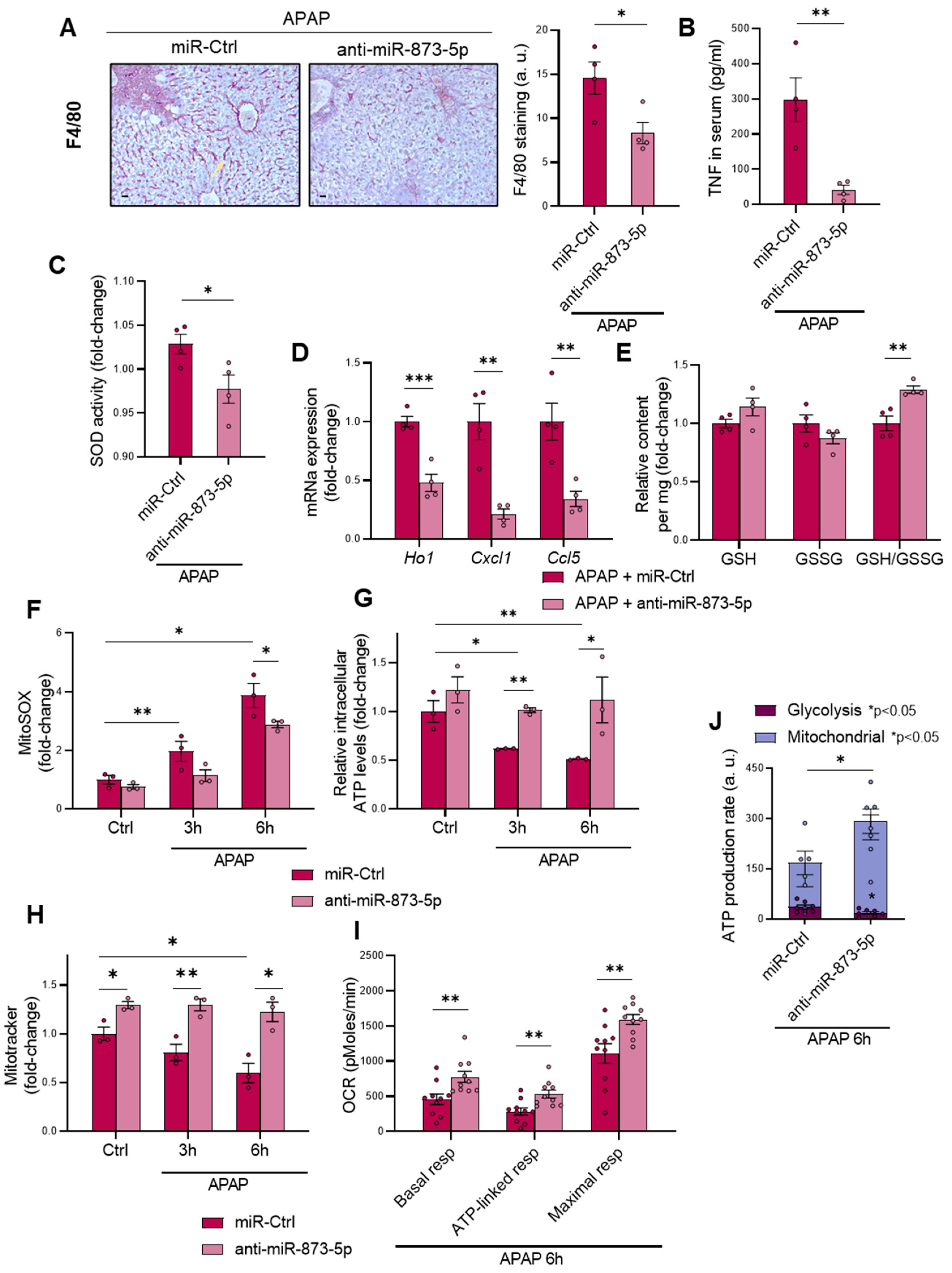
Apart from the appearance of necrotic areas in the liver, another DILI hallmark is the development of mitochondrial dysfunction and a pro-inflammatory stage due to glutathione depletion by produced NAPQI [
30,
31]. The anti-miR873-5p therapy was also effective in reducing macrophage infiltration, measured by F4/80 staining and inflammation development. The presence of TNF in the serum and SOD activity were also reduced by the therapy, together with a reduced mRNA expression of pro-inflammatory cytokines. Although TNF might be essential for liver regeneration [
47], in the context when it is measured, when liver regeneration has already occurred, such reduction in the serum might be an indicator readout of the reduced injury under the therapy [
32]. The characteristic glutathione depletion was also prevented by the anti-miR-873-5p, as reflected by the observed increase in the GSH/GSSG ratio. The mRNA expression of glutathione synthesis enzymes suggested that GSH restoration was not due to an enhanced production, but to reduced oxidative stress development. This was further confirmed by mitoSOX staining, where ROS production increased upon APAP stimulation and was reduced by the anti-miR-873-5p at different times. Mitochondrial functionality was improved by the therapy, as reflected in the increased MitoTracker staining that was accompanied by the prevention of ATP loss by APAP. The OCR measurement also showed an increased respiration of primary hepatocytes treated with the anti-miRNA, where the ATP source was mainly from oxidative phosphorylation. The interaction with GNMT, restored under anti-miR-873-5p and mitochondrial Complex II [
17], might lead to the observed reduction in mitochondrial dysfunction independently of other key antioxidant enzymes, as is shown in the case of SOD activity [
48].
Considering the increased ammonium content and the loss of GLUL expression in the perivenous region upon APAP overdose, the restoration of ammonium levels in both the liver and serum and GLUL protein expression may be a consequence of the observed reduction in mitochondrial dysfunction and subsequent hepatocyte protection. Indeed, hyperammonemia has already been linked to mitochondrial dysfunction and, in the meantime, reported to induce, among others, a senescence phenotype [
49]. The observed GLUL recovery may suggest miR-873-5p as an upstream regulator of its gene. However, the mRNA determination in primary hepatocytes did not show an increased expression under anti-miR-873-5p therapy in wild-type cells, whereas
Gnmt-/- did. Thus, the
Glul expression cannot be proposed to be directly regulated by miR-873-5p, as its expression might be indirectly modulated by the damage suffered by the hepatocyte. Likewise, the reduced ammonium content could also be a consequence of reduced cell death, as urea cycle enzymes’
Cps1 and
Otc mRNA expression and activities were reduced by the anti-miR-873-5p, despite reducing hepatic hyperammonemia. Considering this fact, the less active urea cycle might lead to an increased ornithine availability, which would be then converted into putrescine and act as a substrate, together with dc-AdoMet, for the synthesis of polyamines. This could explain the increased Spd levels in the liver from mice treated with the anti-miR-873-5p, a metabolite with a role in improving mitochondrial functionality in other cell types [
50,
51] and reported to be essential for hepatocyte proliferation [
52]. The rewiring in the hepatocyte metabolism from the urea cycle, increased under APAP overdose, towards polyamine synthesis for hepatocyte protection was further confirmed by the fluxomic approach performed. The administration of labelled methionine to hepatocytes resulted in an increased Spd, Spm and Spm-NAc content with decreased dc-AdoMet levels. The recovery of methionine cycle when targeting miR-873-5p is depicted by both: (i) increased GNMT expression at protein and mRNA levels and (ii) hepatic AdoMet restoration observed by metabolomics. Thus, AdoMet restoration may be due to the recovered methionine cycle, while the increased
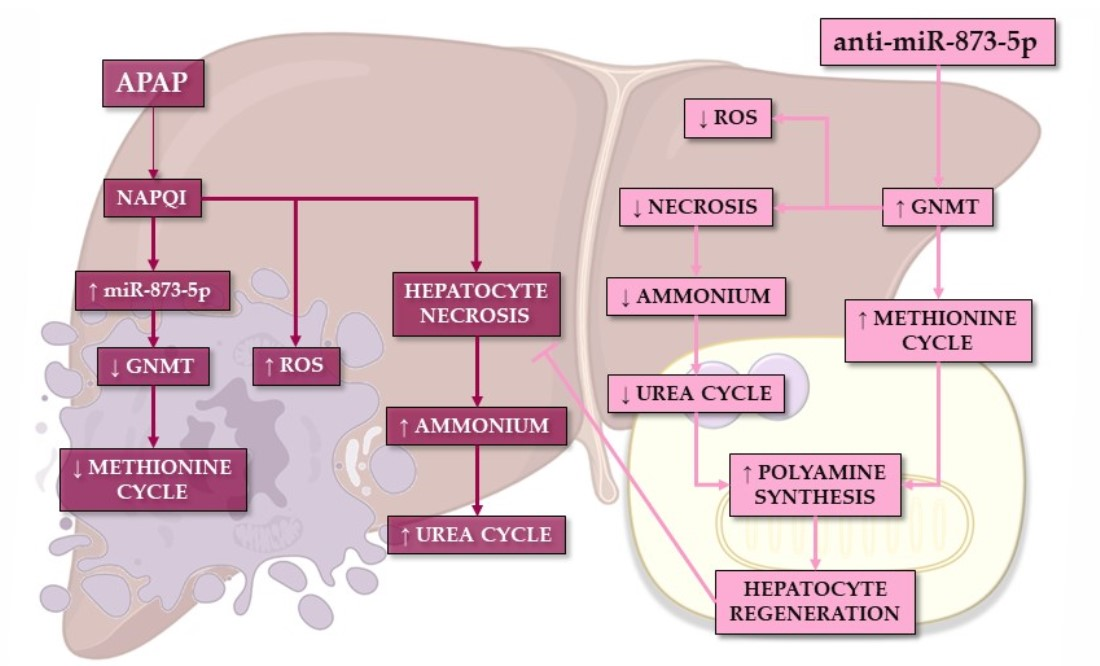
Mtr mRNA expression observed by the anti-miR-873-5p suggested an increased homocysteine recycling and subsequent higher methionine levels. The scavenging of the urea cycle by increasing ammonium content in culture medium prevented the hepatoprotective effect by the anti-miR-873-5p therapy, while the inhibition of polyamine synthesis had the same effect, thus demonstrating the relevance of the proposed metabolic shifting with the therapy.
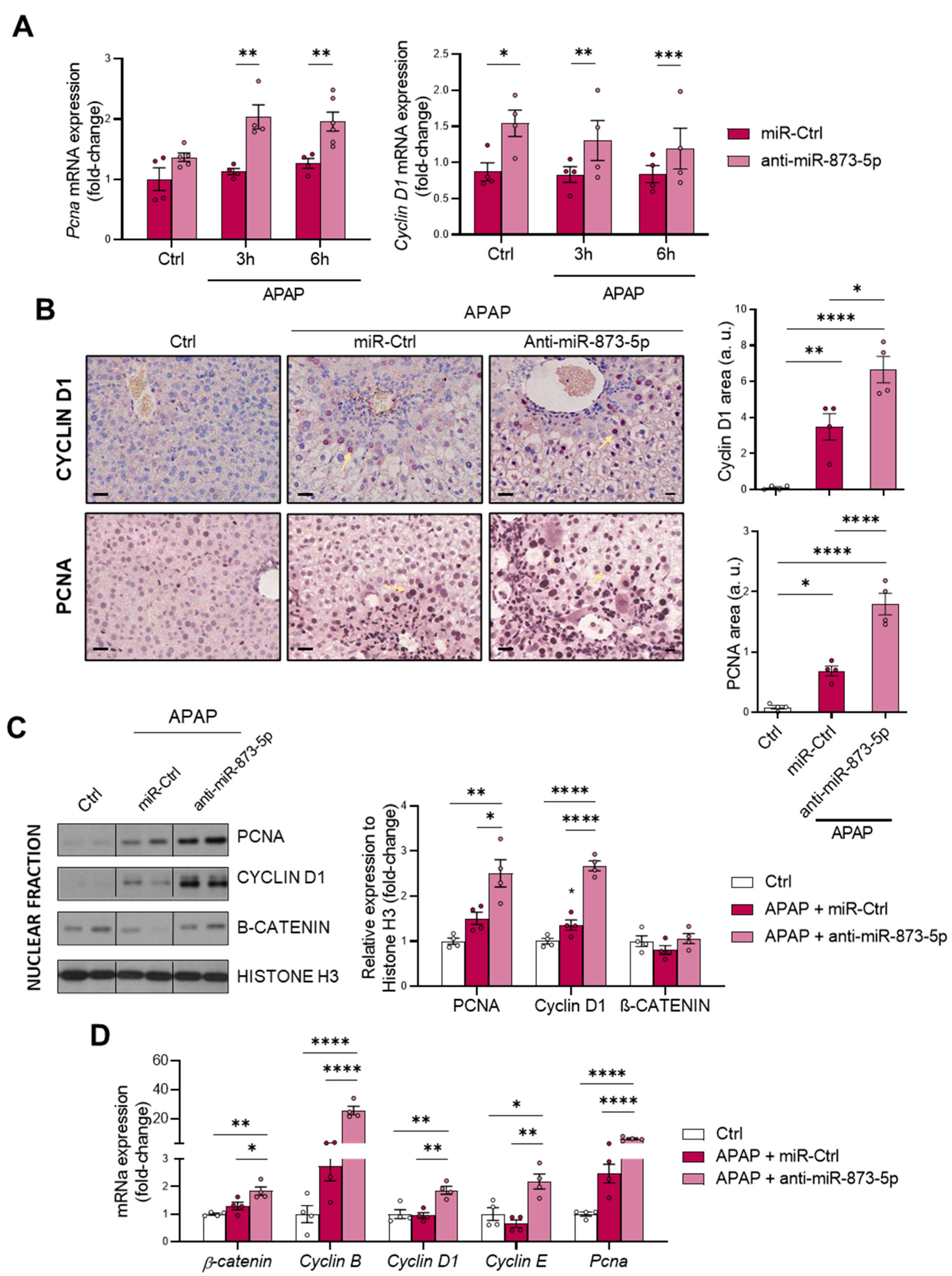
As a consequence of the decreased urea cycle and higher ornithine availability leading to increased polyamine synthesis, the proliferative response of hepatocytes was increased. This was observed in vitro by the higher mRNA expression of the proliferation markers
Pcna and
Cyclin D1. Correlated with this, the tissue expression of both markers and mRNA expression was also higher in liver from mice treated with the anti-miR-873-5p. Although, during APAP overdose, there is a higher expression in an attempt to compensate for the hepatotoxic damage, this may not be sufficient to reduce the appearance of necrotic areas. Moreover, in isolated fractions of liver tissue from treated mice, an increased nuclear expression of such proliferation markers and β-catenin was also observed. The mRNA expression of other proliferation markers, such as
Cyclin B and
E isoforms, was also higher in the anti-miR-treated group. Therefore, the role that polyamines have in mitochondrial functionality [
50,
51] and hepatocyte proliferation [
52], together with the requirements of adequate ATP levels in liver regeneration [
36,
37], may enhance the regenerative response for preventing APAP-derived hepatotoxicity.
In summary, the present work demonstrates the preventive effect of an anti-miR-873-5p-based therapy on the hepatotoxic effect of APAP overdose. The methionine cycle recovery due to the increased GNMT expression when targeting the miRNA leads to an enhanced mitochondrial functionality, thus diminishing the effect of APAP. Meanwhile, the reduced ammonium accumulation may lead to reduced urea cycle activity and a subsequent ornithine availability. In combination with the recovery of hepatic AdoMet content, ornithine might be the substrate of the enhanced polyamine synthesis observed when targeting the miRNA. The role that polyamines have in mitochondrial functionality and liver regeneration might modulate the regenerative response upon APAP overdose while preventing the cytotoxic effect of the compound.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}