
Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints



Abstract
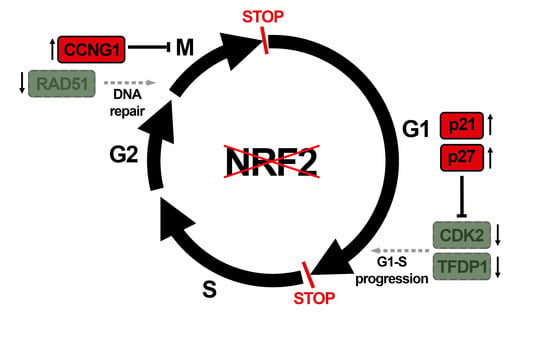
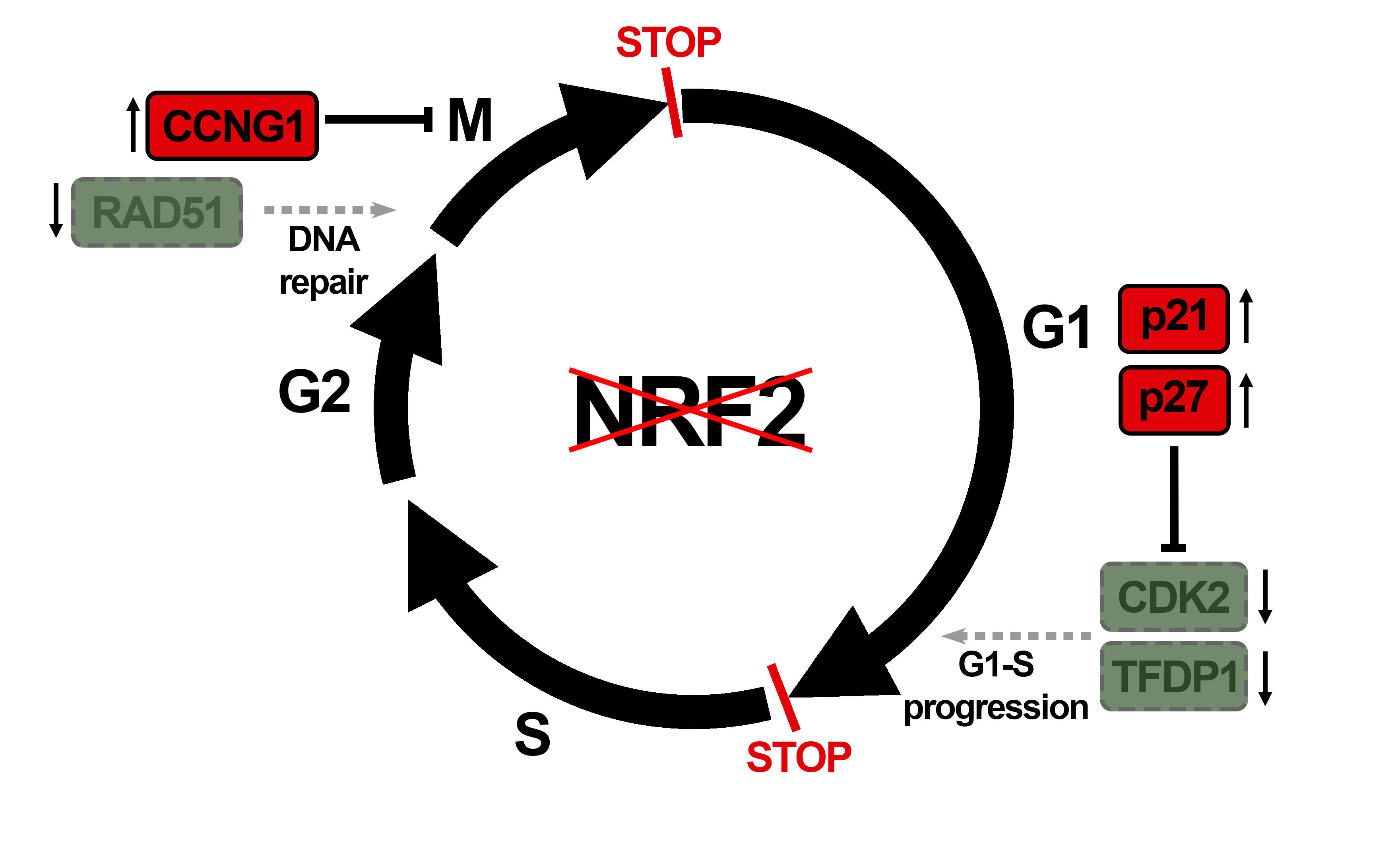
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Reagents and Synchronization
2.2. Lentiviral Vector Production, and Infection
2.3. Immunoblot
2.4. Analysis of mRNA Levels
2.5. MTT and Cell Growth Assays
2.6. Flow Cytometry
2.7. RT2 Profiler PCR Array of Human Cell Cycle
3. Results
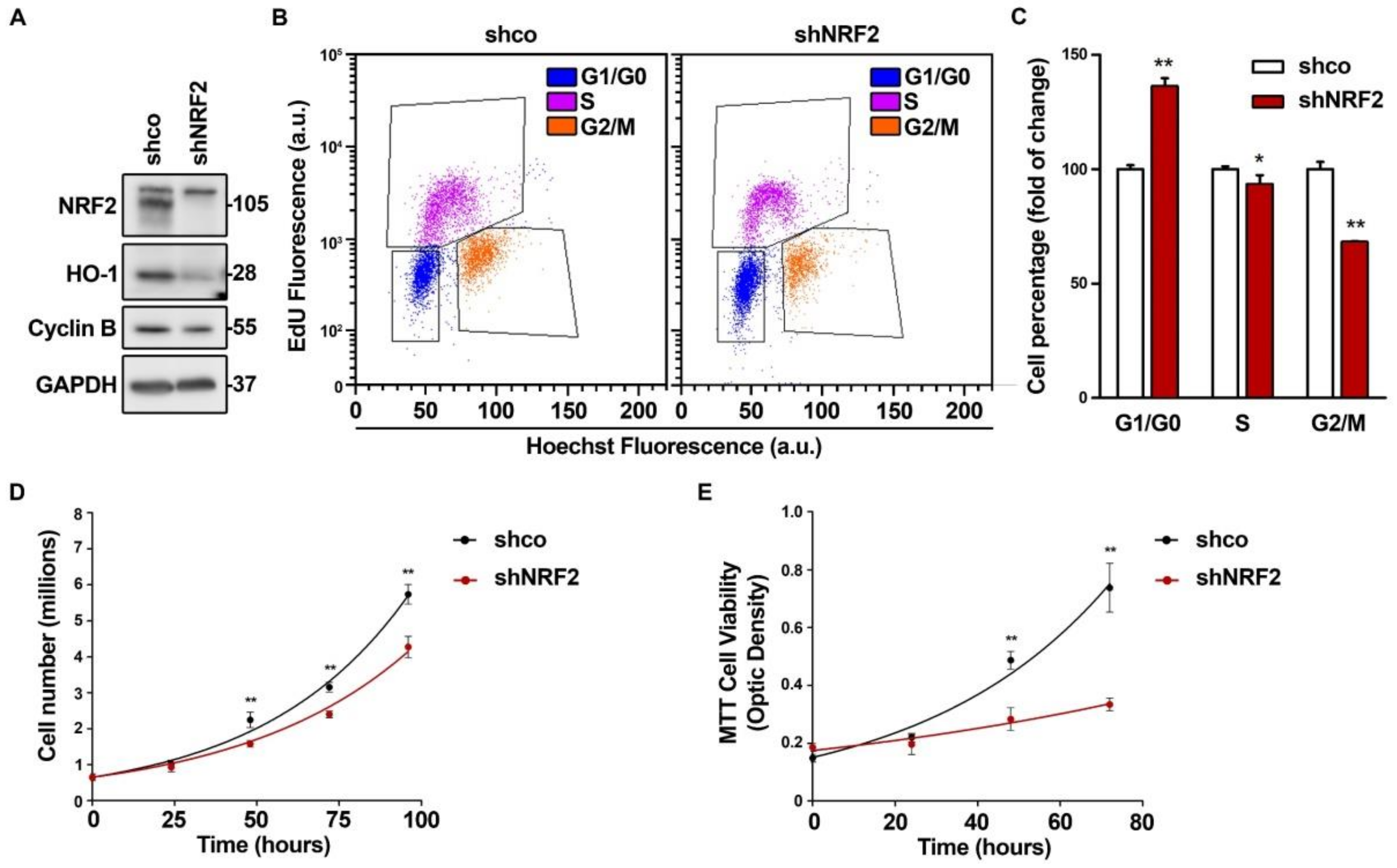
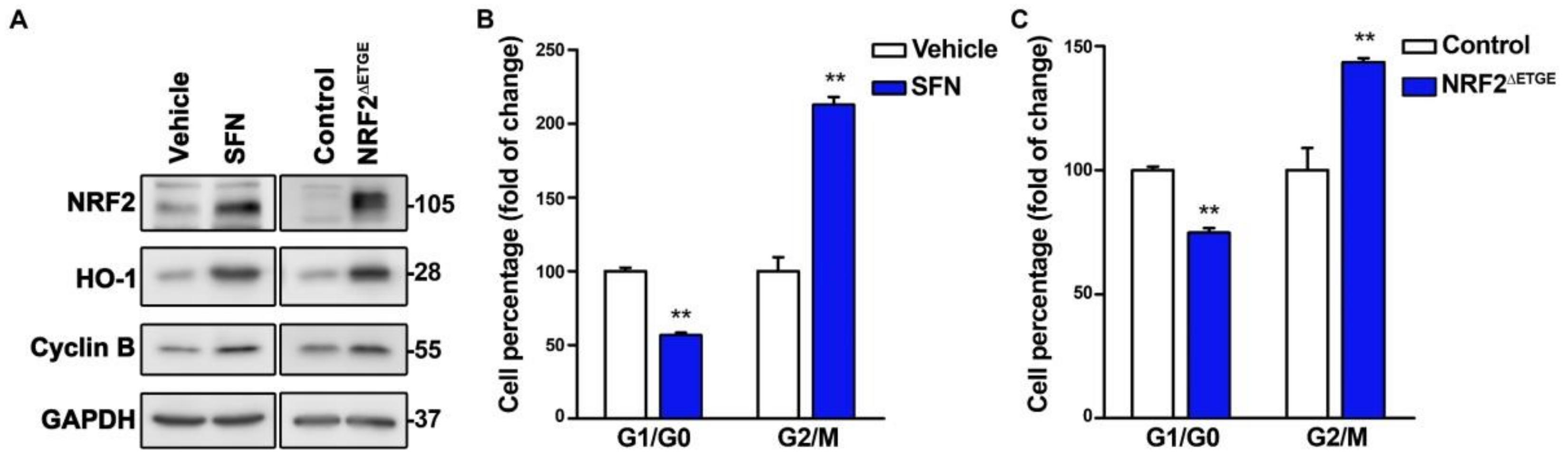
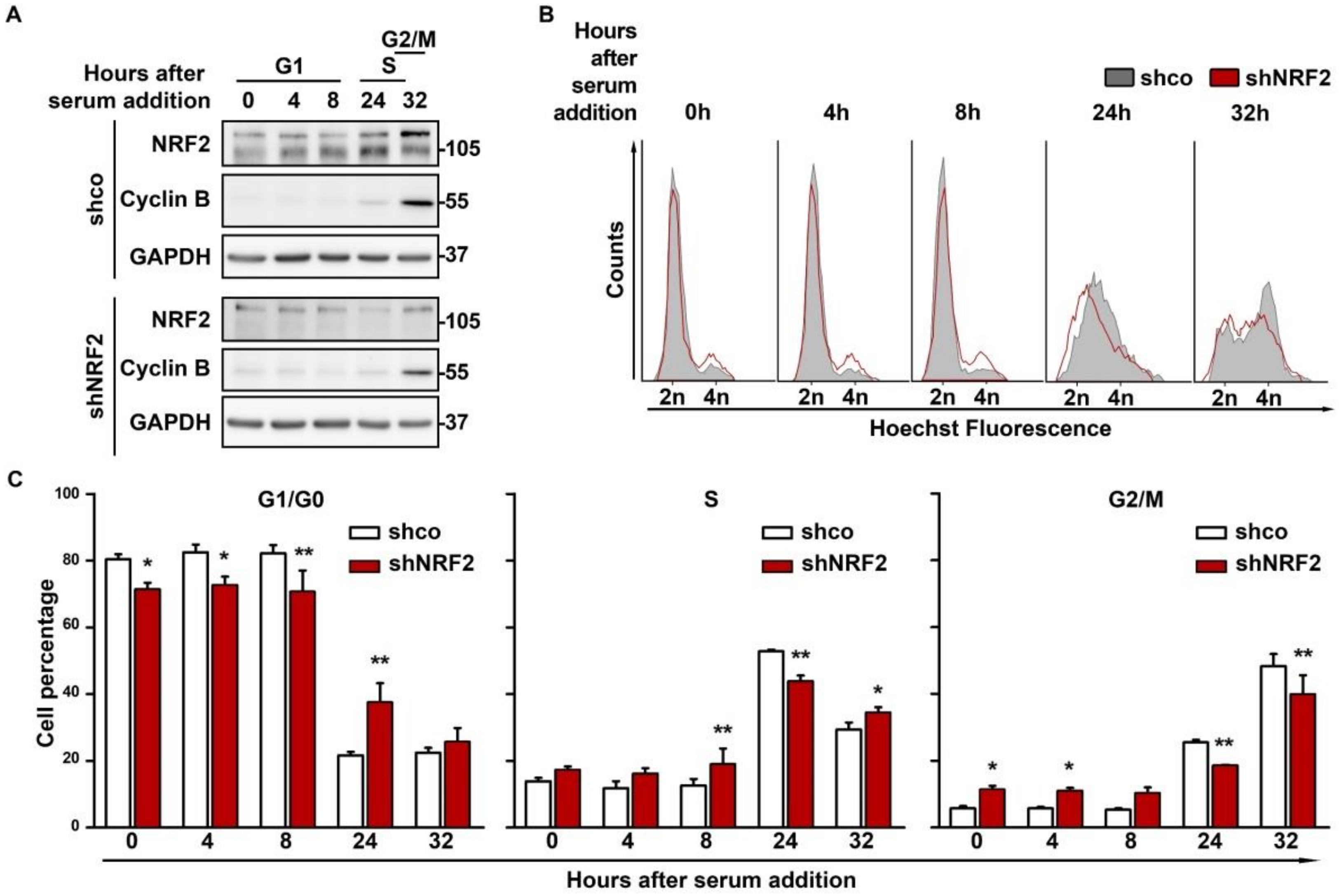
3.1. NRF2 Alters Cell Cycle Progression and Proliferation
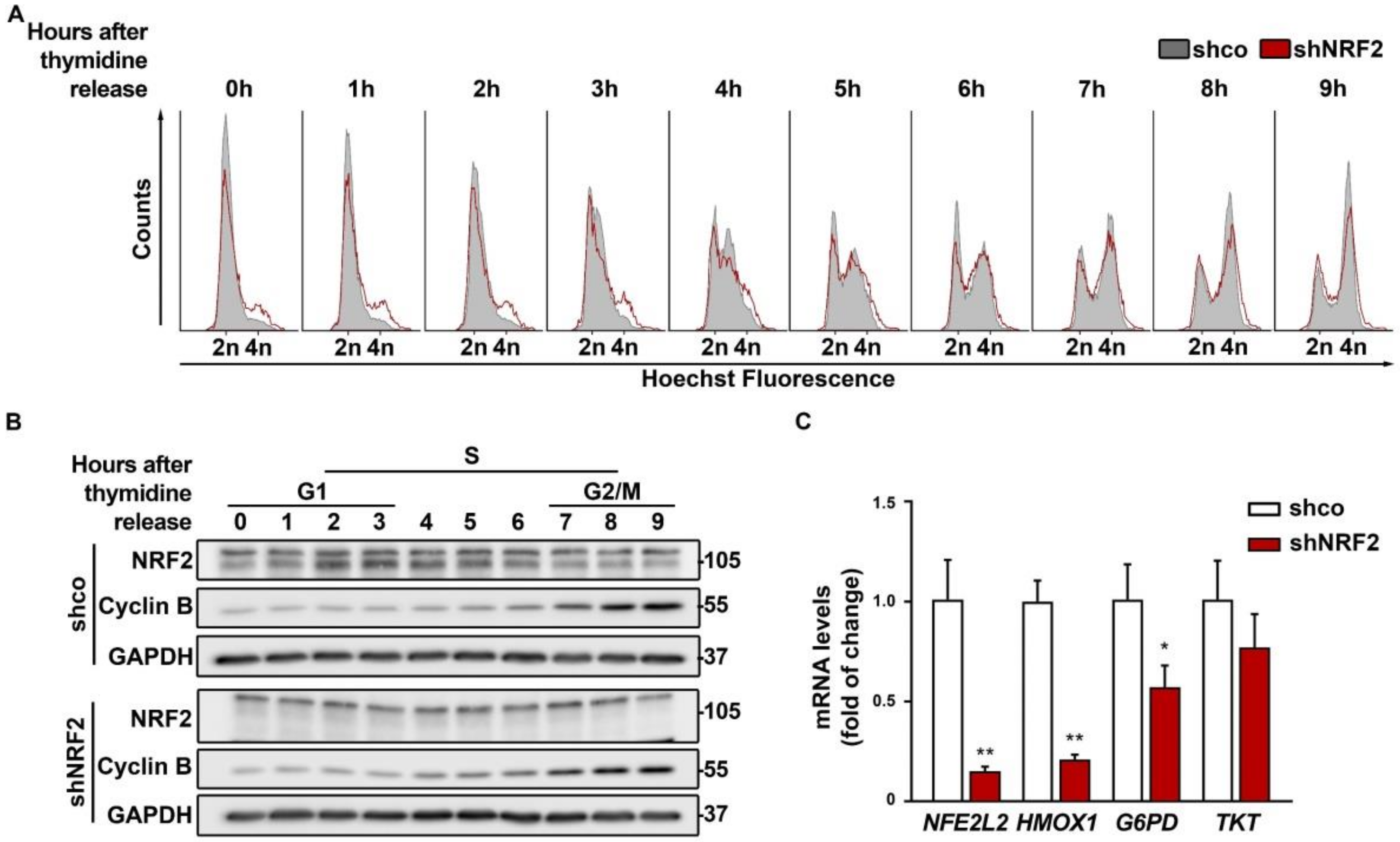
3.2. NRF2 Absence Delays G1 Exit
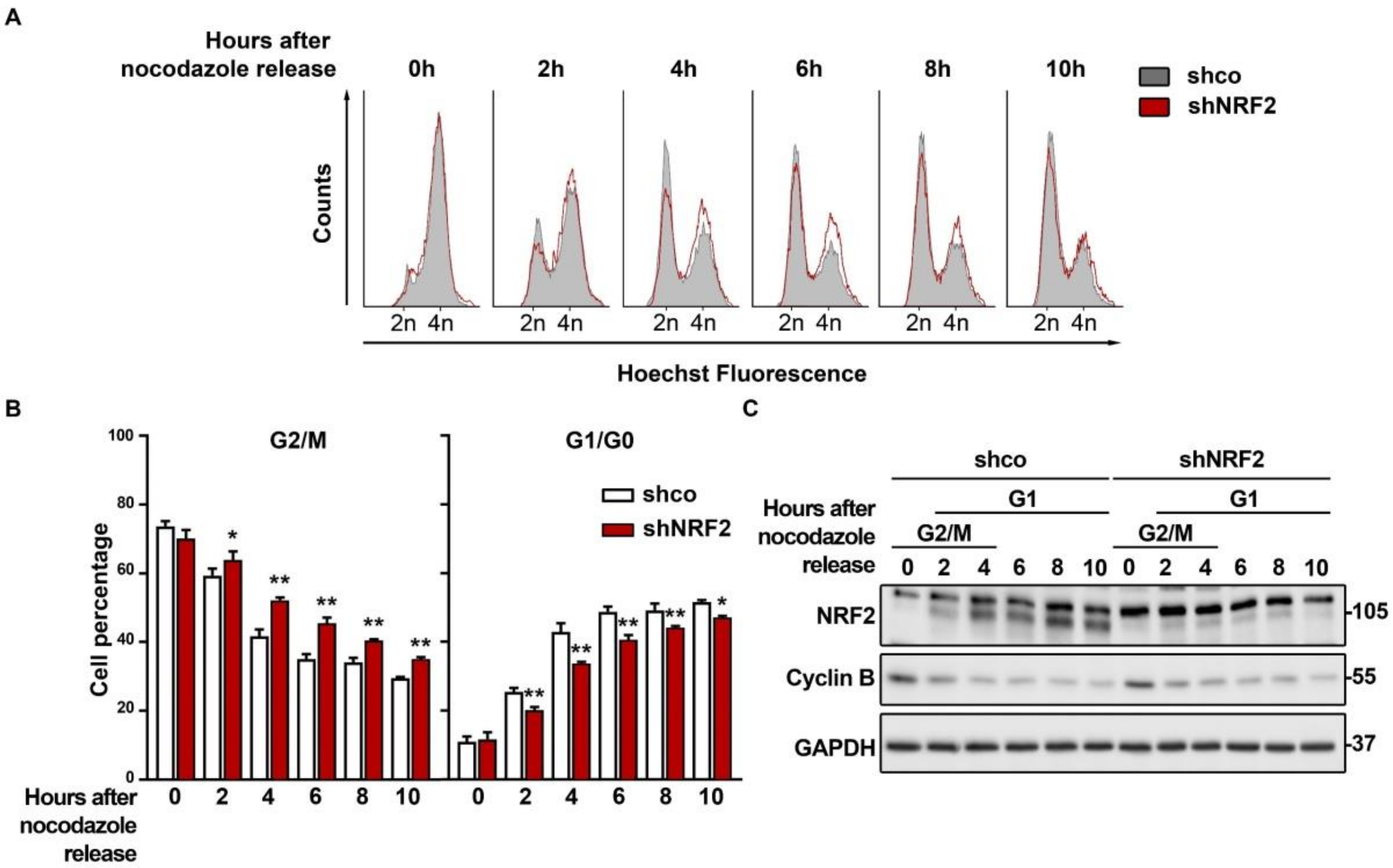
3.3. The Absence of NRF2 Delays Mitosis Withdrawal
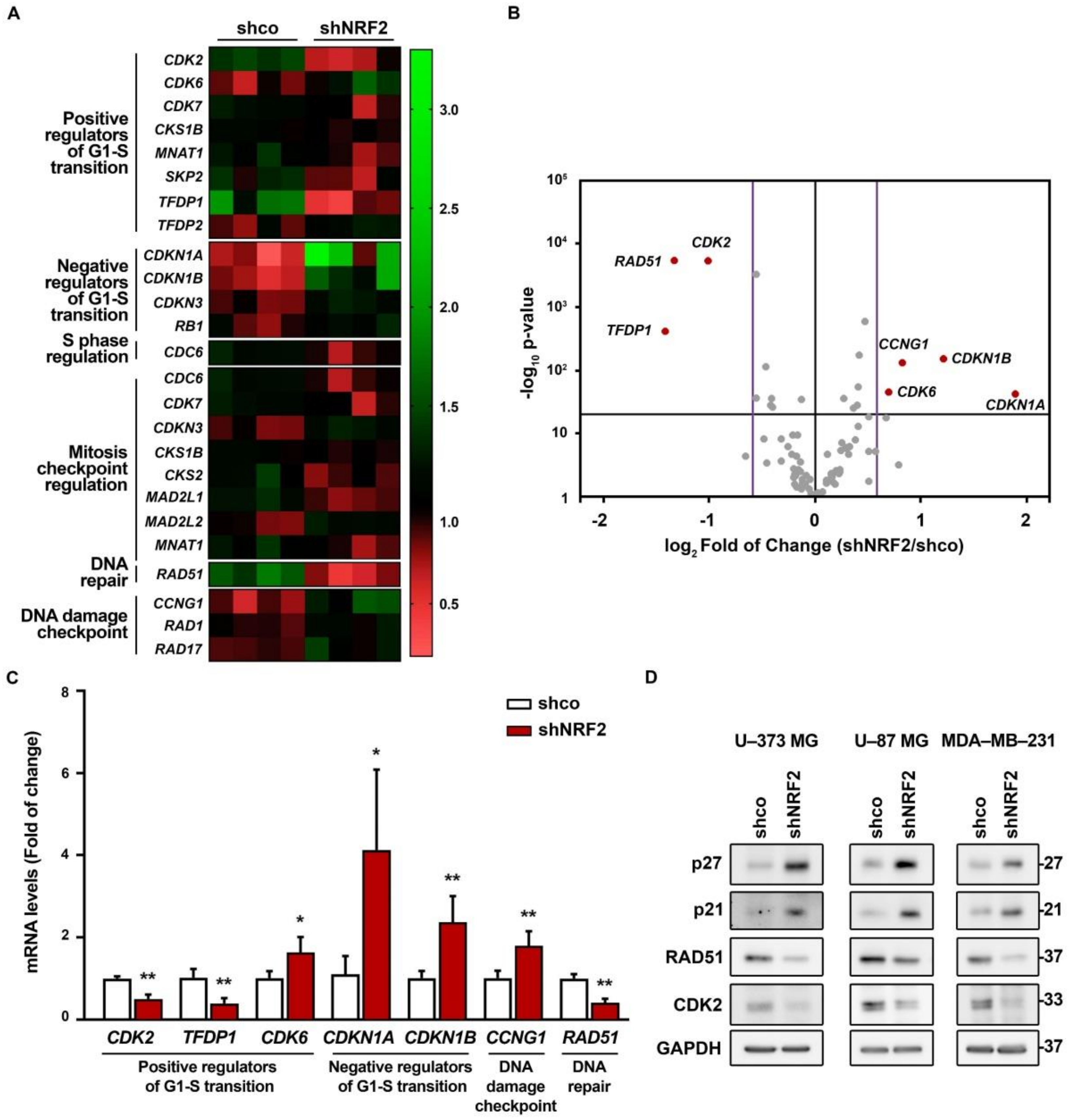
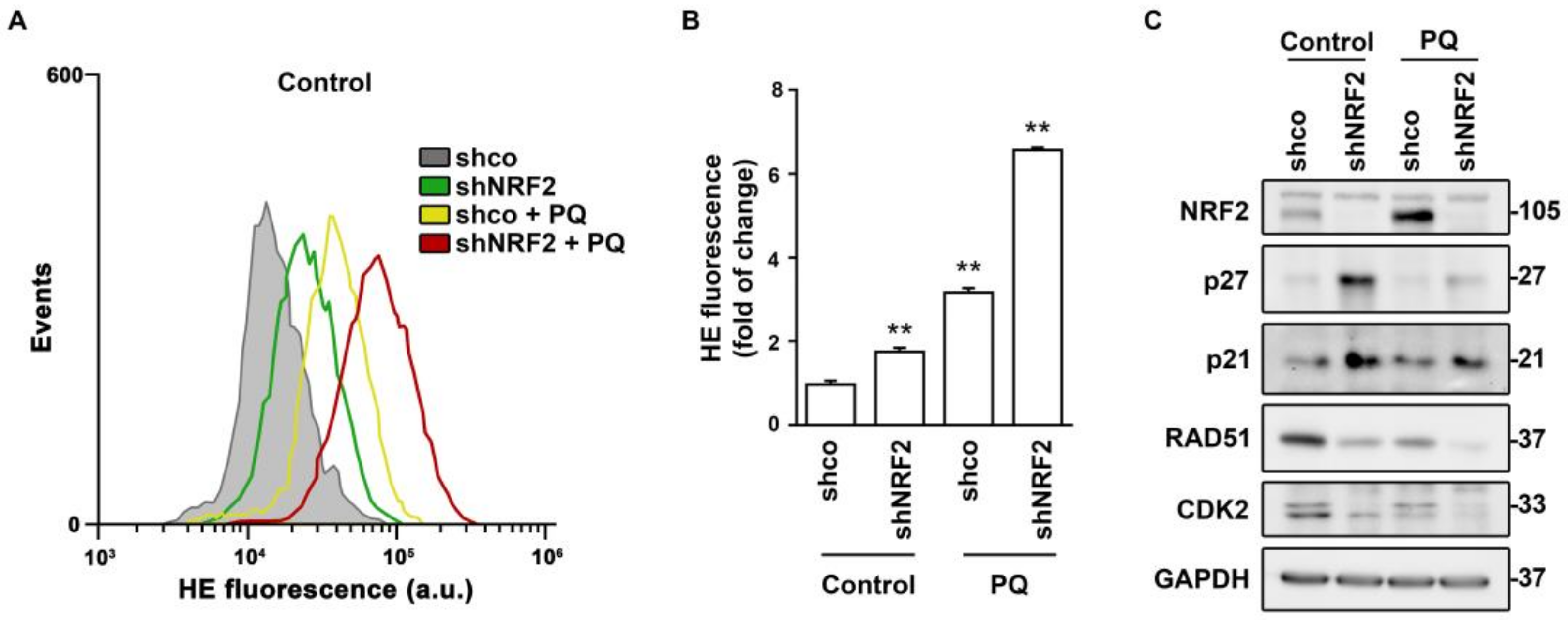
3.4. NRF2 Absence Alters Regulatory Mechanisms of G1–S, Mitosis and DNA Damage Checkpoints
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S. Gain of Nrf2 function in non–small–cell lung cancer cells confers radioresistance. Antioxid. Redox Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Wu, J.; Dodson, M.; Rojo de la Vega, E.M.; Ning, Y.; Zhang, Z.; Yao, M.; Zhang, D.D.; Xu, C.; Yi, X. ABCF2, an Nrf2 target gene, contributes to cisplatin resistance in ovarian cancer cells. Mol. Carcinog. 2017, 56, 1543–1553. [Google Scholar] [CrossRef]
- Lu, B.-C.; Li, J.; Yu, W.-F.; Zhang, G.-Z.; Wang, H.-M.; Ma, H.-M. Elevated expression of Nrf2 mediates multidrug resistance in CD133(+) head and neck squamous cell carcinoma stem cells. Oncol. Lett. 2016, 12, 4333–4338. [Google Scholar] [CrossRef] [Green Version]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. Mol. Mech. Mutagen. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up–regulates antiapoptotic protein Bcl–2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2–induced antiapoptotic Bcl–xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoll, M.; Lastra, D.; Robledinos-Antón, N.; Wandosell, F.; Antón, I.M.; Cuadrado, A. WIP Modulates Oxidative Stress through NRF2/KEAP1 in Glioblastoma Cells. Antioxidants 2020, 9, 773. [Google Scholar] [CrossRef] [PubMed]
- Escoll, M.; Lastra, D.; Pajares, M.; Robledinos-Anton, N.; Rojo, A.I.; Fernandez-Gines, R.; Mendiola, M.; Martinez-Marin, V.; Esteban, I.; Lopez-Larrubia, P.; et al. Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biol. 2020, 30, 101425. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, M.-C.; Lin, P.-L.; Wang, Y.-C.; He, T.-Y.; Lee, M.-C.; Yeh, S.D.; Chen, C.-Y.; Lee, H. Mutant p53 confers chemoresistance in non–small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Marton, M.; Tihanyi, N.; Gyulavari, P.; Banhegyi, G.; Kapuy, O. NRF2–regulated cell cycle arrest at early stage of oxidative stress response mechanism. PLoS ONE 2018, 13, e0207949. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.; Novak, B. Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2001, 2, 908–916. [Google Scholar] [CrossRef]
- Park, M.S.; Koff, A. Overview of the cell cycle. Curr. Protoc. Cell Biol. 2001, 8, 8.1. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β–TrCP promotes glycogen synthase kinase 3–dependent degradation of the Nrf2 transcription factor in a Keap1–independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Rada, P.; Mendiola, M.; Ortega-Molina, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Hardisson, D.; Serrano, M.; Cuadrado, A. The PTEN/NRF2 Axis Promotes Human Carcinogenesis. Antioxid. Redox Signal. 2014, 21, 2498–2514. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.K.; Cook, J.L. Nrf2, a Cap’n’Collar Transcription Factor, Regulates Induction of the Heme Oxygenase–1 Gene *. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Xu, Y.; Luo, Y.; Wang, N.-X.; Xiao, J.-H. Role of Nrf2 in cell senescence regulation. Mol. Cell. Biochem. 2021, 476, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Zhao, M.; Wan, C.; Zhang, Q.; Tang, T.; Liu, J.; Shao, Q.; Yang, B.; He, J.; Jiang, C. Role of tea polyphenols in delaying hyperglycemia–induced senescence in human glomerular mesangial cells via miR–126/Akt–p53–p21 pathways. Int. Urol. Nephrol. 2019, 51, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Patra, K.; Jana, J.; Mandal, D.P.; Bhattacharjee, S. Nrf–2 transcriptionally activates P21(Cip/WAF1) and promotes A549 cell survival against oxidative stress induced by H2O2. Chem. Biol. Interact. 2018, 285, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.M.; Zager, R.A. Mechanisms and consequences of oxidant–induced renal preconditioning: An Nrf2–dependent, P21–independent, anti–senescence pathway. Nephrol. Dial. Transplant. 2018, 33, 1927–1941. [Google Scholar] [CrossRef]
- Yang, F.; Yi, M.; Liu, Y.; Wang, Q.; Hu, Y.; Deng, H. Glutaredoxin–1 Silencing Induces Cell Senescence via p53/p21/p16 Signaling Axis. J. Proteome Res. 2018, 17, 1091–1100. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two–Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Li, J.; Zhang, C.; Xing, Y.; Janicki, J.S.; Yamamoto, M.; Wang, X.L.; Tang, D.-Q.; Cui, T. Up–regulation of p27(kip1) contributes to Nrf2–mediated protection against angiotensin II–induced cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 315–324. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.M.; Kleeberger, S.R.; Bream, J.H.; Fallon, P.G.; Kensler, T.W.; Yamamoto, M.; Reddy, S.P. Genetic disruption of the Nrf2 compromises cell–cycle progression by impairing GSH–induced redox signaling. Oncogene 2008, 27, 5821–5832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, E2949–E2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-C.; Stern, A.; Chiu, D.T.-Y. G6PD: A hub for metabolic reprogramming and redox signaling in cancer. Biomed. J. 2021, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Xu, I.M.-J.; Lai, R.K.-H.; Lin, S.-H.; Tse, A.P.-W.; Chiu, D.K.-C.; Koh, H.-Y.; Law, C.-T.; Wong, C.-M.; Cai, Z.; Wong, C.C.-L.; et al. Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA 2016, 113, E725–E734. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kloska, D.; Kopacz, A.; Cysewski, D.; Aepfelbacher, M.; Dulak, J.; Jozkowicz, A.; Grochot-Przeczek, A. Nrf2 Sequesters Keap1 Preventing Podosome Disassembly: A Quintessential Duet Moonlights in Endothelium. Antioxid. Redox Signal. 2019, 30, 1709–1730. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2–mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Zou, Y.; Nambiar, S.M.; Lee, J.; Yang, Y.; Dai, G. Keap1 modulates the redox cycle and hepatocyte cell cycle in regenerating liver. Cell Cycle 2014, 13, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- Bodrug, T.; Welsh, K.A.; Hinkle, M.; Emanuele, M.J.; Brown, N.G. Intricate Regulatory Mechanisms of the Anaphase–Promoting Complex/Cyclosome and Its Role in Chromatin Regulation. Front. Cell Dev. Biol. 2021, 9, 687515. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence 5′–3′ | Melting Temp. (°C) | Reverse Sequence 5′–3′ | Melting Temp. (°C) |
|---|---|---|---|---|
| GAPDH | CTCTCTGCTCCTCCTGTTCGAC | 66.7 | TGAGCGATGTGGCTCGGCT | 71.9 |
| TBP | TGCACAGGAGCCAAGAGTGAA | 68.3 | CACATCACAGCTCCCCACCA | 69.8 |
| NFE2L2 | AAACCAGTGGATCTGCCAAC | 63.9 | GTGACTGAAACGTAGCCGAAGA | 65.4 |
| HMOX1 | TGCTCAACATCCAGCTCTTTGA | 67.1 | GCAGAATCTTGCACTTTGTTGC | 66.5 |
| G6PD | TGACCTGGCCAAGAAGAAC | 64.9 | CAAAGAAGTCCTCCAGCTTG | 61.6 |
| TKT | ACATCTACCAGAAGCGGTGC | 64.2 | TTCTACCCCCGTGATCCCTC | 67.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lastra, D.; Escoll, M.; Cuadrado, A. Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints. Antioxidants 2022, 11, 946. https://doi.org/10.3390/antiox11050946
Lastra D, Escoll M, Cuadrado A. Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints. Antioxidants. 2022; 11(5):946. https://doi.org/10.3390/antiox11050946
Chicago/Turabian StyleLastra, Diego, Maribel Escoll, and Antonio Cuadrado. 2022. "Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints" Antioxidants 11, no. 5: 946. https://doi.org/10.3390/antiox11050946
APA StyleLastra, D., Escoll, M., & Cuadrado, A. (2022). Transcription Factor NRF2 Participates in Cell Cycle Progression at the Level of G1/S and Mitotic Checkpoints. Antioxidants, 11(5), 946. https://doi.org/10.3390/antiox11050946