
Antioxidant and Anticancer Functions of Protein Acyltransferase DHHC3

Abstract
:1. Background
2. DHHC3 and Oxidative Stress Regulation
2.1. DHHC3 Ablation Enhances OS
2.2. DHHC3 Substrates Include Several Oxidative Stress Regulators
3. DHHC3 and Cancer
4. DHHC3 Impact on Innate Anticancer Immunity
5. DHHC3 Mediated Regulation of Adaptive Anticancer Immunity
6. DHHC3 Regulation of Metastasis
7. DHHC3 Mediated OS and Anticancer Drug Efficacy
7.1. Anticancer Drugs and Oxidative Stress
7.2. DHHC3 Disruption Enhances OS-Inducing Drug Effects
8. Other DHHC3 Substrates May Affect Cancer Independent of OS
9. DHHC3 as a Potential Drug Target
9.1. Reasons Why DHHC3 May Be an Excellent Target for Therapeutic Intervention
9.2. Implications of Targeting DHHC3
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Vasudevan, A.; Linder, M.E.; Deschenes, R.J. Thematic review series: Lipid Posttranslational Modifications. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J. Lipid Res. 2006, 47, 1118–1127. [Google Scholar] [CrossRef] [Green Version]
- Linder, M.E.; Deschenes, R. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef]
- Gottlieb, C.D.; Linder, M.E. Structure and function of DHHC protein S-acyltransferases. Biochem. Soc. Trans. 2017, 45, 923–928. [Google Scholar] [CrossRef]
- Blanc, M.; David, F.; Abrami, L.; Migliozzi, D.; Armand, F.; Bürgi, J.; Van Der Goot, F.G. SwissPalm: Protein Palmitoylation database. F1000Research 2015, 4, 261. [Google Scholar] [CrossRef] [Green Version]
- Stix, R.; Lee, C.-J.; Faraldo-Gómez, J.D.; Banerjee, A. Structure and Mechanism of DHHC Protein Acyltransferases. J. Mol. Biol. 2020, 432, 4983–4998. [Google Scholar] [CrossRef]
- Rana, M.S.; Kumar, P.; Lee, C.-J.; Verardi, R.; Rajashankar, K.R.; Banerjee, A. Fatty acyl recognition and transfer by an integral membrane S-acyltransferase. Science 2018, 359. [Google Scholar] [CrossRef]
- Sharma, C.; Rabinovitz, I.; Hemler, M.E. Palmitoylation by DHHC3 is critical for the function, expression, and stability of integrin α6β4. Exp. Cell Mol. Life Sci. 2012, 69, 2233–2244. [Google Scholar] [CrossRef] [Green Version]
- Ko, P.; Dixon, S.J. Protein palmitoylation and cancer. EMBO Rep. 2018, 19, e46666. [Google Scholar] [CrossRef]
- Won, S.J.; Kit, M.C.S.; Martin, B.R. Protein depalmitoylases. Crit. Rev. Biochem. Mol. Biol. 2017, 53, 83–98. [Google Scholar] [CrossRef]
- Le, X.; Mu, J.; Peng, W.; Tang, J.; Xiang, Q.; Tian, S.; Feng, Y.; He, S.; Qiu, Z.; Ren, G.; et al. DNA methylation downregulated ZDHHC1 suppresses tumor growth by altering cellular metabolism and inducing oxidative/ER stress-mediated apoptosis and pyroptosis. Theranostics 2020, 10, 9495–9511. [Google Scholar] [CrossRef]
- Shen, L.-F.; Chen, Y.-J.; Liu, K.-M.; Haddad, A.N.S.; Song, I.-W.; Roan, H.-Y.; Chen, L.-Y.; Yen, J.J.Y.; Wu, J.-Y.; Chen, Y.-T. Role of S-Palmitoylation by ZDHHC13 in Mitochondrial function and Metabolism in Liver. Sci. Rep. 2017, 7, 2182. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Wang, H.-X.; Li, Q.; Knoblich, K.; Reisenbichler, E.; Richardson, A.L.; Hemler, M.E. Protein Acyltransferase DHHC3 Regulates Breast Tumor Growth, Oxidative Stress, and Senescence. Cancer Res. 2017, 77, 6880–6890. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Yang, W.; Steen, H.; Freeman, M.R.; Hemler, M.E. Antioxidant functions of DHHC3 suppress anti-cancer drug activities. Cell. Mol. Life Sci. 2020, 78, 2341–2353. [Google Scholar] [CrossRef]
- Mahmood, D.D.F.; Abderrazak, A.; EL Hadri, K.; Simmet, T.; Rouis, M. The Thioredoxin System as a Therapeutic Target in Human Health and Disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef]
- Zhang, X.; Gibhardt, C.S.; Will, T.; Stanisz, H.; Körbel, C.; Mitkovski, M.; Stejerean, I.; Cappello, S.; Pacheu-Grau, D.; Dudek, J.; et al. Redox signals at the ER –mitochondria interface control melanoma progression. EMBO J. 2019, 38, e100871. [Google Scholar] [CrossRef]
- Ding, C.; Fan, X.; Wu, G. Peroxiredoxin 1—An antioxidant enzyme in cancer. J. Cell. Mol. Med. 2016, 21, 193–202. [Google Scholar] [CrossRef]
- Meng, Y.; Qian, Y.; Gao, L.; Cai, L.-B.; Cui, Y.-G.; Liu, J.-Y. Downregulated Expression of Peroxiredoxin 4 in Granulosa Cells from Polycystic Ovary Syndrome. PLoS ONE 2013, 8, e76460. [Google Scholar] [CrossRef]
- Kropotov, A.; Usmanova, N.; Serikov, V.; Zhivotovsky, B.; Tomilin, N. Mitochondrial targeting of human peroxiredoxin V protein and regulation of PRDX5 gene expression by nuclear transcription factors controlling biogenesis of mitochondria. FEBS J. 2007, 274, 5804–5814. [Google Scholar] [CrossRef]
- Resh, M.D. Palmitoylation of proteins in cancer. Biochem. Soc. Trans. 2017, 45, 409–416. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, B.; Yin, C.; Liu, W.; Han, C.; Chen, B.; Liu, T.; Li, X.; Chen, X.; Li, C.; et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017, 549, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Lukk, M.; Kapushesky, M.; Nikkilä, J.; Parkinson, H.; Goncalves, A.; Huber, W.; Ukkonen, E.; Brazma, A. A global map of human gene expression. Nat. Biotechnol. 2010, 28, 322–324. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.-P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L.; et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 2019, 3, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Bartholomew, J.N.; Volonte, D.; Galbiati, F. Oxidative Stress Induces Premature Senescence by Stimulating Caveolin-1 Gene Transcription through p38 Mitogen-Activated Protein Kinase/Sp1–Mediated Activation of Two GC-Rich Promoter Elements. Cancer Res. 2006, 66, 10805–10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Gogvadze, V.; Norberg, E.; Venkatesh, R.; Orrenius, S.; Zhivotovsky, B. Reactive oxygen species generated in different compartments induce cell death, survival, or senescence. Free Radic. Biol. Med. 2013, 57, 176–187. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, X.; Saredy, J.; Yuan, Z.; Yang, X.; Wang, H. Innate-adaptive immunity interplay and redox regulation in immune response. Redox Biol. 2020, 37, 101759. [Google Scholar] [CrossRef]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [Green Version]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; De Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hsu, J.-M.; Sun, L.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Wei, Y.; Xia, W.; Hou, J.; Qiu, Y.; et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2018, 29, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2015, 8, 68–78. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.-J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Sun, W.; Song, H.; Wang, C.; Li, Q.; Li, C.; Wei, D.; Zhao, Y.; Li, C.; Zhang, H. NOL6 promotes the proliferation and migration of endometrial cancer cells by regulating TWIST1 expression. Epigenomics 2021, 13, 1571–1585. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Song, M.; Wu, X.; Wang, W. NOL6, a new founding oncogene in human prostate cancer and targeted by miR-590-3p. Cytotechnology 2020, 72, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ma, Z.; Li, Z.; Zhuang, H.; Liu, C.; Gong, Y.; Huang, S.; Zhang, C.; Hou, B. CMTM3 Overexpression Predicts Poor Survival and Promotes Proliferation and Migration in Pancreatic Cancer. J. Cancer 2021, 12, 5797–5806. [Google Scholar] [CrossRef]
- Li, Q.; Pan, Y.; Cao, Z.; Zhao, S. Comprehensive Analysis of Prognostic Value and Immune Infiltration of Chromobox Family Members in Colorectal Cancer. Front. Oncol. 2020, 10, 1901. [Google Scholar] [CrossRef]
- Yu, Y.-H.; Chiou, G.-Y.; Huang, P.-I.; Lo, W.-L.; Wang, C.-Y.; Lu, K.-H.; Yu, C.-C.; Alterovitz, G.; Huang, W.-C.; Lo, J.-F.; et al. Network Biology of Tumor Stem-like Cells Identified a Regulatory Role of CBX5 in Lung Cancer. Sci. Rep. 2012, 2, 584. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Jiang, L.; Zhong, X.; Hochwald, S.N.; Wang, Y.; Huang, L.; Nie, Q.; Huang, H.; Xu, J.-F. Discovery of Aberrant Alteration of Genome in Colorectal Cancer by Exome Sequencing. Am. J. Med. Sci. 2019, 358, 340–349. [Google Scholar] [CrossRef]
- Beaulieu, J.F. Integrin α6β4 in Colorectal Cancer: Expression, Regulation, Functional Alterations and Use as a Biomarker. Cancers 2019, 12, 41. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Li, H.; Wang, M.; Ju, S.; Li, F.; Chen, P.; Lu, H.; Han, X.; Ren, J. PSMC2/ITGA6 axis plays critical role in the development and progression of hepatocellular carcinoma. Cell Death Discov. 2021, 7, 217. [Google Scholar] [CrossRef]
- Bigoni-Ordóñez, G.D.; Czarnowski, D.; Parsons, T.; Madlambayan, G.J.; Villa-Diaz, L. Integrin α6 (CD49f), The Microenvironment and Cancer Stem Cells. Curr. Stem Cell Res. Ther. 2019, 14, 428–436. [Google Scholar] [CrossRef]
- Kilpatrick, C.L.; Murakami, S.; Feng, M.; Wu, X.; Lal, R.; Chen, G.; Du, K.; Luscher, B. Dissociation of Golgi-associated DHHC-type Zinc Finger Protein (GODZ)- and Sertoli Cell Gene with a Zinc Finger Domain-beta (SERZ-beta)-mediated Palmitoylation by Loss of Function Analyses in Knock-out Mice. J. Biol. Chem. 2016, 291, 27371–27386. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Liu, S.; Li, X.; Qin, Z.; Wang, K.; Wang, J.; Song, Y.; Yang, K. Recent progress of palmitoyl transferase DHHC3 as a novel antitumor target. Future Med. Chem. 2022, 14, 443–455. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 3, 27–40. [Google Scholar] [CrossRef]
- Draper, J.M.; Smith, C.D. Palmitoyl acyltransferase assays and inhibitors (Review). Mol. Membr. Biol. 2009, 26, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Jennings, B.C.; Linder, M.E. DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J. Biol. Chem. 2012, 287, 7236–7245. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Delalande, C.; Dickinson, B.C. Inhibitors of DHHC family proteins. Curr. Opin. Chem. Biol. 2021, 65, 118–125. [Google Scholar] [CrossRef]
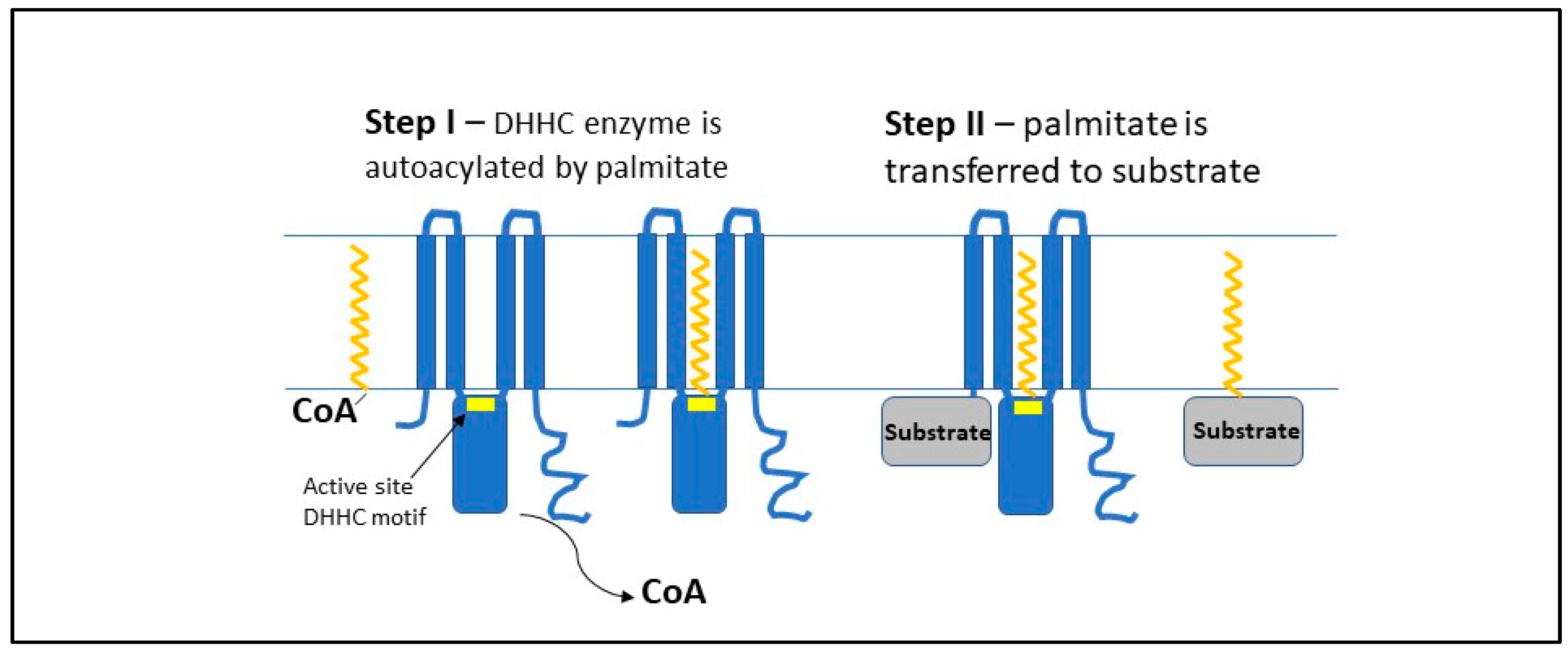
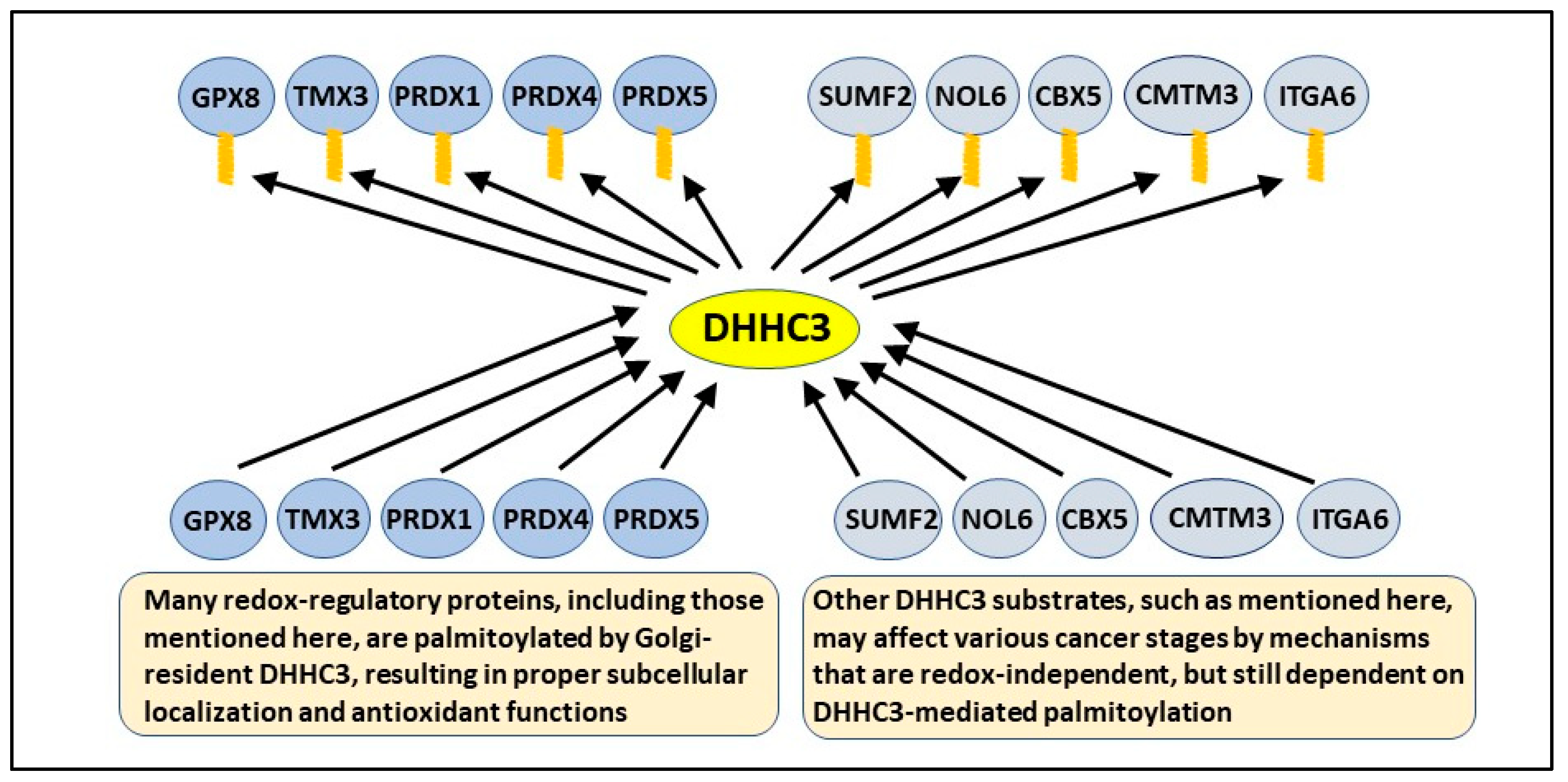

{kind=link}
{kind=link}
| Result | References |
|---|---|
| Enhanced CellRox fluorescent dye detection | [18,19] |
| Diminished activity of select tyrosine phosphatases | [18] |
| Effects of DHHC3 ablation partially reversed by OS inhibitors | [18] |
| Upregulated appearance of TXNIP | [18] |
| Changes in gene expression consistent with elevated OS | [18] |
| Diminished palmitoylation of Redox/Antioxidant regulators | [19] |
| Increased senescence, innate immune cells in tumors | [18] |
| Increased efficacy of OS-inducing anticancer drugs | [19] |
| Result | References |
|---|---|
| Upregulated ZDHHC3 gene levels and diminished patient survival | [18] |
| DHHC3 upregulated in breast, prostate, and colon cancers | [18,30] |
| Xenograft tumor growth reduced upon DHHC3 ablation | [18,19,31] |
| Tumor metastasis is reduced for DHHC3-ablated cells | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, C.; Hemler, M.E. Antioxidant and Anticancer Functions of Protein Acyltransferase DHHC3. Antioxidants 2022, 11, 960. https://doi.org/10.3390/antiox11050960
Sharma C, Hemler ME. Antioxidant and Anticancer Functions of Protein Acyltransferase DHHC3. Antioxidants. 2022; 11(5):960. https://doi.org/10.3390/antiox11050960
Chicago/Turabian StyleSharma, Chandan, and Martin E. Hemler. 2022. "Antioxidant and Anticancer Functions of Protein Acyltransferase DHHC3" Antioxidants 11, no. 5: 960. https://doi.org/10.3390/antiox11050960
APA StyleSharma, C., & Hemler, M. E. (2022). Antioxidant and Anticancer Functions of Protein Acyltransferase DHHC3. Antioxidants, 11(5), 960. https://doi.org/10.3390/antiox11050960