
Reactive Human Plasma Glutathione Peroxidase Mutant with Diselenide Bond Succeeds in Tetramer Formation

Abstract
:1. Introduction
2. Experimental Procedures
2.1. Plasmids and E. coli Strains
2.2. Cloning Procedures
2.3. Protein Expression and Purification
2.4. SDS−PAGE Analysis
2.5. GPx Activity Assays
2.6. Effect of Temperature and pH on GPx Activity
2.7. Steady−State Kinetics of hGPx3
2.8. Molecular Modelling
3. Results
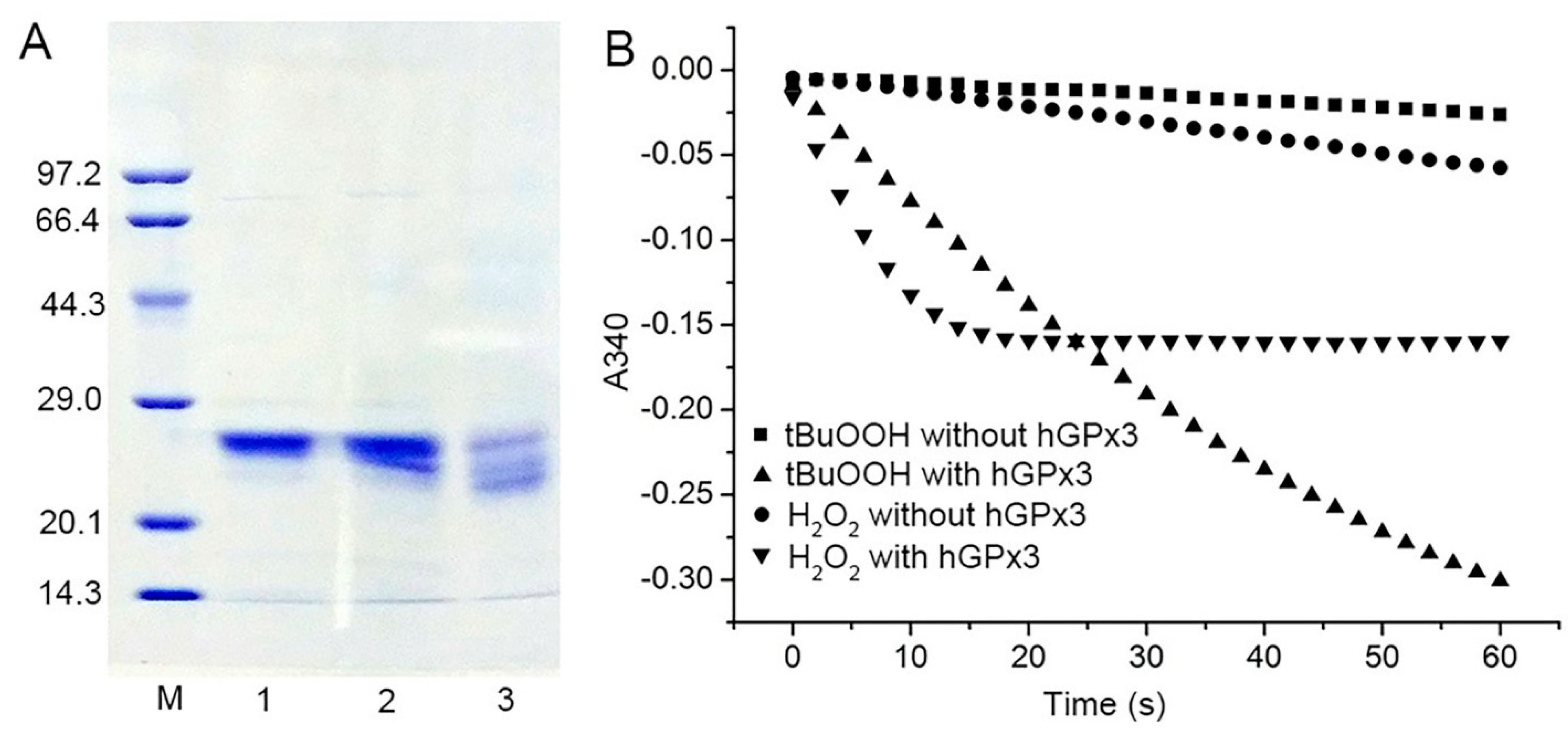
3.1. Expression and Purification of Sec−hGPx3 with Intra−Molecular Disulfide Bond Formation
3.2. Analysis of Disulfide Bond Formation in Sec−hGPx3
3.3. Effect of Cys Mutation on the Catalytic Activity of hGPx3
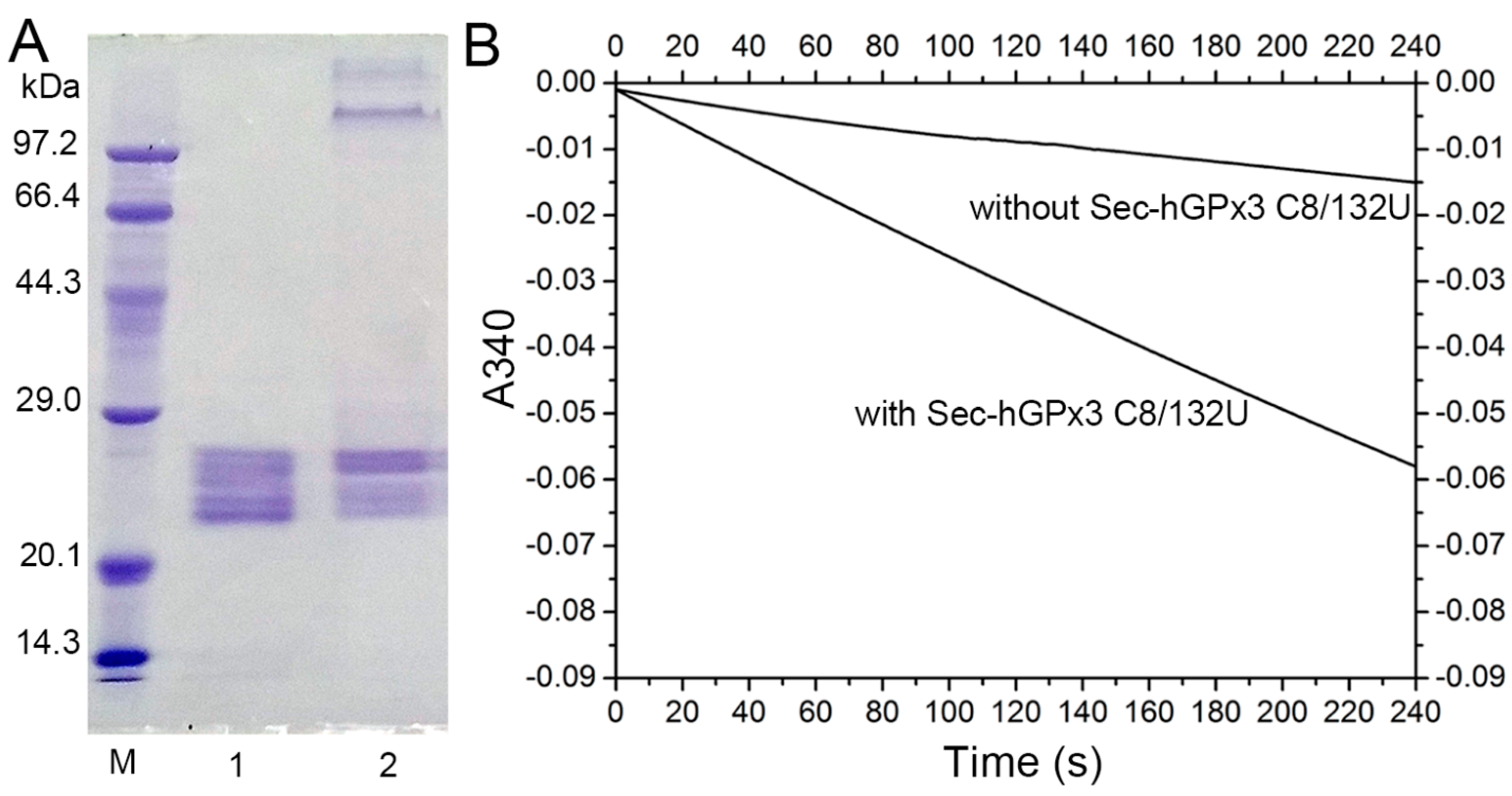
3.4. Mutation of Cys8 and Cys132 to Sec8 and Sec132 Promotes Tetramer Formation
3.5. Characterization of Sec−hGPx3−C8/132U
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Broderick, D.J.; Deagen, J.T.; Whanger, P.D. Properties of Glutathione Peroxidase Isolated from Human Plasma. J. Inorg. Biochem. 1987, 30, 299–308. [Google Scholar] [CrossRef]
- Maddipati, K.; Marnett, L.J. Characterization of the Major Hydroperoxide-Reducing Activity of Human Plasma. Purification and Properties of a Selenium-Dependent Glutathione Peroxidase. J. Biol. Chem. 1987, 262, 17398–17403. [Google Scholar] [CrossRef]
- Takahashi, K.; Avissar, N.; Whitin, J.; Cohen, H. Purification and Characterization of Human Plasma Glutathione Peroxidase: A Selenoglycoprotein Distinct from the Known Cellular Enzyme. Arch. Biochem. Biophys. 1987, 256, 677–686. [Google Scholar] [CrossRef]
- Avissar, N.; Ornt, D.B.; Yagil, Y.; Horowitz, S.; Watkins, R.H.; Kerl, E.A.; Takahashi, K.; Palmer, I.S.; Cohen, H.J.; Ornt, B. Human Kidney Proximal Tubules Are the Main Source of Plasma Glutathione Peroxidase. Am. J. Physiol. 1994, 266, 367–375. [Google Scholar] [CrossRef]
- Whitin, J.C.; Bhamre, S.; Tham, D.M.; Cohen, H.J. Extracellular Glutathione Peroxidase Is Secreted Basolaterally by Human Renal Proximal Tubule Cells. Am. J. Physiol.-Ren. Physiol. 2002, 283, F20–F28. [Google Scholar] [CrossRef]
- Olson, G.E.; Whitin, J.C.; Hill, K.E.; Winfrey, V.P.; Motley, A.K.; Austin, L.M.; Deal, J.; Cohen, H.J.; Burk, R.F. Extracellular Glutathione Peroxidase (Gpx3) Binds Specifically to Basement Membranes of Mouse Renal Cortex Tubule Cells. Am. J. Physiol.-Ren. Physiol. 2010, 298, F1244–F1253. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Yin, D. Glutathione Peroxidase-3 Produced by the Kidney Binds to a Population of Basement Membranes in the Gastrointestinal Tract and in Other Tissues. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G32–G38. [Google Scholar] [CrossRef] [Green Version]
- XuetB, J.; Huangll, W.; Akessonll, B.; Holmgrentll, A. The Thioredoxin and Glutaredoxin Systems Are Efficient Electron Donors to Human Plasma Glutathione Peroxidase. J. Biol. Chem. 1994, 269, 29382–29384. [Google Scholar] [CrossRef]
- Ren, B.; Huang, W.; Åkesson, B.; Ladenstein, R. The Crystal Structure of Seleno-Glutathione Peroxidase from Human Plasma at 2.9 Å Resolution. J. Mol. Biol. 1997, 268, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Avissars, N.; Whitin, J.C.; Allen, P.Z.; Wagner, D.D.; Liegey, P.; Cohen, H.J. Plasma Selenium-Dependent Glutathione Peroxidase. Cell of Origin and Secretion. J. Biol. Chem. 1989, 264, 15850–15855. [Google Scholar] [CrossRef]
- Ottaviano, F.G.; Tang, S.-S.; Handy, D.E.; Loscalzo, J. Regulation of the Extracellular Antioxidant Selenoprotein Plasma Glutathione Peroxidase (GPx-3) in Mammalian Cells. Mol. Cell. Biochem. 2009, 327, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic Mechanisms and Specificities of Glutathione Peroxidases: Variations of a Basic Scheme. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Cozza, G.; Ursini, F. A Comparison of Thiol Peroxidase Mechanisms. Antioxid. Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef]
- Fischer, N.; Paleskava, A.; Gromadski, K.B.; Konevega, A.L.; Wahl, M.C.; Stark, H.; Rodnina, M.V. Towards Understanding Selenocysteine Incorporation into Bacterial Proteins. Biol. Chem. 2007, 388, 1061–1067. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Gladyshev, V.N. How Selenium Has Altered Our Understanding of the Genetic Code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [Green Version]
- Low, S.C.; Berry, M.J. Knowing When Not to Stop: Selenocysteine Incorporation in Eukaryotes. Trends Biochem. Sci. 1996, 21, 203–208. [Google Scholar] [CrossRef]
- Song, J.; Yu, Y.; Xing, R.; Guo, X.; Liu, D.; Wei, J.; Song, H. Unglycosylated Recombinant Human Glutathione Peroxidase 3 Mutant from Escherichia Coli Is Active as a Monomer. Sci. Rep. 2015, 4, 6698. [Google Scholar] [CrossRef] [Green Version]
- Aldag, C.; Bröcker, M.J.; Hohn, M.J.; Prat, L.; Hammond, G.; Plummer, A.; Söll, D. Rewiring Translation for Elongation Factor Tu-Dependent Selenocysteine Incorporation. Angew. Chem.-Int. Ed. 2013, 52, 1441–1445. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Song, J.; Guan, T.; Lv, X.; Wei, J. Efficient Expression of Glutathione Peroxidase with Chimeric TRNA in Amber-Less Escherichia Coli. ACS Synth. Biol. 2018, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Bröcker, M.J.; Prat, L.; Ip, K.; Chirathivat, N.; Feiock, A.; Veszprémi, M.; Söll, D. A Synthetic TRNA for EF-Tu Mediated Selenocysteine Incorporation in Vivo and in Vitro. FEBS Lett. 2015, 589, 2194–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically Recoded Organisms Expand Biological Functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodius, V.A.; Segall-Shapiro, T.H.; Sharon, B.D.; Ghodasara, A.; Orlova, E.; Tabakh, H.; Burkhardt, D.H.; Clancy, K.; Peterson, T.C.; Gross, C.A.; et al. Design of Orthogonal Genetic Switches Based on a Crosstalk Map of Σs, Anti-σs, and Promoters. Mol. Syst. Biol. 2013, 9, 702. [Google Scholar] [CrossRef]
- Guo, X.; Song, J.; Yu, Y.; Wei, J. Can Recombinant Human Glutathione Peroxidase 1 with High Activity Be Efficiently Produced in Escherichia Coli? Antioxid. Redox Signal. 2014, 20, 1524–1530. [Google Scholar] [CrossRef]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A Comparative Study on the Hydroperoxide and Thiol Specificity of the Glutathione Peroxidase Family and Selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Song, J.; Song, Y.; Guo, X.; Han, Y.; Wei, J. Characterization of Catalytic Activity and Structure of Selenocysteine-Containing HGSTZ1c-1c Based on Site-Directed Mutagenesis and Computational Analysis. IUBMB Life 2013, 65, 163–170. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Accelrys Inc. Insight II; Version 98.0; Accelrys Inc.: San Diego, CA, USA, 1998. [Google Scholar]
- Accelrys Inc. Discover 3 User Guide; Accelrys Inc.: San Diego, CA, USA, 1999. [Google Scholar]
- Shchedrina, V.A.; Novoselov, S.V.; Malinouski, M.Y.; Gladyshev, V.N. Identification and Characterization of a Selenoprotein Family Containing a Diselenide Bond in a Redox Motif. Proc. Natl. Acad. Sci. USA 2007, 104, 13919–13924. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Song, J.; Guo, X.; Wang, S.; Yang, X.; Chen, L.; Wei, J. Characterization and Structural Analysis of Human Selenium-Dependent Glutathione Peroxidase 4 Mutant Expressed in Escherichia Coli. Free Radic. Biol. Med. 2014, 71, 332–338. [Google Scholar] [CrossRef]
- Masuda, R.; Kimura, R.; Karasaki, T.; Sase, S.; Goto, K. Modeling the Catalytic Cycle of Glutathione Peroxidase by Nuclear Magnetic Resonance Spectroscopic Analysis of Selenocysteine Selenenic Acids. J. Am. Chem. Soc. 2021, 143, 6345–6350. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef] [PubMed]
- Orian, L.; Mauri, P.; Roveri, A.; Toppo, S.; Benazzi, L.; Bosello-Travain, V.; de Palma, A.; Maiorino, M.; Miotto, G.; Zaccarin, M.; et al. Selenocysteine Oxidation in Glutathione Peroxidase Catalysis: An MS-Supported Quantum Mechanics Study. Free Radic. Biol. Med. 2015, 87, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Orian, L. Proton Transfer and SN2 Reactions as Steps of Fast Selenol and Thiol Oxidation in Proteins: A Model Molecular Study Based on GPx. Chempluschem 2021, 86, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Bortoli, M.; Torsello, M.; Bickelhaupt, F.M.; Orian, L. Role of the Chalcogen (S, Se, Te) in the Oxidation Mechanism of the Glutathione Peroxidase Active Site. ChemPhysChem 2017, 18, 2990–2998. [Google Scholar] [CrossRef]
- Orian, L.; Flohé, L. Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology. Antioxidants 2021, 10, 1560. [Google Scholar] [CrossRef] [PubMed]
- Trofymchuk, O.S.; Zheng, Z.; Kurogi, T.; Mindiola, D.J.; Walsh, P.J. Selenolate Anion as an Organocatalyst: Reactions and Mechanistic Studies. Adv. Synth. Catal. 2018, 360, 1685–1692. [Google Scholar] [CrossRef]
- Flohé, L.; Toppo, S.; Orian, L. The Glutathione Peroxidase Family: Discoveries and Mechanism. Free Radic. Biol. Med. 2022. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Chu, F.F.; Geiger, P.; Girotti, A.W.; Doroshow, J.H. Reactivity of Plasma Glutathione Peroxidase with Hydroperoxide Substrates and Glutathione. Arch. Biochem. Biophys. 1993, 307, 29–34. [Google Scholar] [CrossRef]
- Toppo, S.; Vanin, S.; Bosello, V.; Tosatto, S.C.E. Evolutionary and Structural Insights Into the Multifaceted Glutathione Peroxidase (Gpx) Superfamily. Antioxid. Redox Signal. 2008, 10, 1501–1514. [Google Scholar] [CrossRef]
- Maiorino, M.; Ursini, F.; Bosello, V.; Toppo, S.; Tosatto, S.C.E.; Mauri, P.; Becker, K.; Roveri, A.; Bulato, C.; Benazzi, L.; et al. The Thioredoxin Specificity of Drosophila GPx: A Paradigm for a Peroxiredoxin-like Mechanism of Many Glutathione Peroxidases. J. Mol. Biol. 2007, 365, 1033–1046. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Plasmid | Description | Source |
|---|---|---|
| Strains | ||
| DH5α | Conventional host for plasmid propagation | Invitrogen |
| C321.ΔA.exp | Recoded E.coli MG1655 strain with all of its UAG codon depleted and UAG termination function removed | Marc Lajoie et al. [23] Addgene |
| Plasmids | ||
| pN565 | Expression of T7 RNA polymerase mutant | Christopher Voigt et al. [24] Addgene |
| pACYC−selA/pstk | Expression of SelA and PSTK | Dieter Söll et al. [20] |
| pGFIB−tRNAUTuT6 | Expression of tRNAUTuT6 | Jingyan Wei et al. [21] |
| pUC57−hGPx3 | pUC57 with hGPx349TGA fragment in BamHI/HindIII | This study |
| pRSF−hGPx349TAG and its mutants | Expression of Sec−hGPx3 and its cysteine mutants | This study |
| pRSF−hGPx349TCG and its mutants | Expression of hGPx3 (U49S) and its cysteine mutants | This study |
| pRSF−shGPx49TAG and its mutants | Expression of short isoform of Sec−shGPx3 and its cysteine mutants | This study |
| pRSF−shGPx49TCG and its mutants | Expression of short isoform of shGPx3 (U49S) and its cysteine mutants | This study |
| Protein | Description |
|---|---|
| Sec−hGPx3 | Human GPx3 protein containing Sec49 |
| Sec−hGPx3−C8S | Human GPx3 protein containing Sec49, Ser8 |
| Sec−hGPx3−C8/77S | Human GPx3 protein containing Sec49, Ser8, Ser77 |
| Sec−hGPx3−C8/132S | Human GPx3 protein containing Sec49, Ser8, Ser132 |
| Sec−hGPx3−C77S | Human GPx3 protein containing Sec49, Ser77 |
| Sec−hGPx3−C132S | Human GPx3 protein containing Sec49, Ser132 |
| Sec−hGPx3−C77/132S | Human GPx3 protein containing Sec49, Ser77, Ser132 |
| Sec−hGPx3C8/77/132S | Human GPx3 protein containing Sec49 Ser8, Ser77, Ser132 |
| Sec−shGPx3 | Short isoform of human GPx3 protein containing Sec49 |
| Sec−shGPx3−C77S | Short isoform of human GPx3 protein containing Sec49, Ser77 |
| Sec−shGPx3−C132S | Short isoform of human GPx3 protein containing Sec49, Ser132 |
| Sec−shGPx3−C77/132S | Short isoform of human GPx3 protein containing Sec49, Ser77, Ser132 |
| hGPx3 (U49S)/Ser−hGPx3 | Human GPx3 protein containing Ser49 |
| hGPx3 (U49S)−C8S/Ser−hGPx3−C8S | Human GPx3 protein containing Ser49, Ser8 |
| hGPx3 (U49S)−C8/77S | Human GPx3 protein containing Ser49, Ser8, Ser77 |
| hGPx3 (U49S)−C8/132S | Human GPx3 protein containing Ser49, Ser8, Ser132 |
| hGPx3 (U49S)−C77S | Human GPx3 protein containing Ser49, Ser77 |
| hGPx3 (U49S)−C132S | Human GPx3 protein containing Ser49, Ser132 |
| hGPx3 (U49S)−C77/132S | Human GPx3 protein containing Ser49, Ser77, Ser132 |
| hGPx3 (U49S)−C8/77/132S | Human GPx3 protein containing Ser49, Ser8, Ser77, Ser132 |
| shGPx3 (U49S) | Short isoform of human GPx3 protein containing Ser49 |
| shGPx3 (U49S)−C77S | Short isoform of human GPx3 protein containing Ser49, Ser77 |
| shGPx3 (U49S)−C132S | Short isoform of human GPx3 protein containing Ser49, Ser132 |
| Sec−hGPx3−C8/132U | Human GPx3 protein containing Sec49, Sec8, Sec132 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Z.; Yan, Q.; Song, J.; Wei, J. Reactive Human Plasma Glutathione Peroxidase Mutant with Diselenide Bond Succeeds in Tetramer Formation. Antioxidants 2022, 11, 1083. https://doi.org/10.3390/antiox11061083
Fan Z, Yan Q, Song J, Wei J. Reactive Human Plasma Glutathione Peroxidase Mutant with Diselenide Bond Succeeds in Tetramer Formation. Antioxidants. 2022; 11(6):1083. https://doi.org/10.3390/antiox11061083
Chicago/Turabian StyleFan, Zhenlin, Qi Yan, Jian Song, and Jingyan Wei. 2022. "Reactive Human Plasma Glutathione Peroxidase Mutant with Diselenide Bond Succeeds in Tetramer Formation" Antioxidants 11, no. 6: 1083. https://doi.org/10.3390/antiox11061083
APA StyleFan, Z., Yan, Q., Song, J., & Wei, J. (2022). Reactive Human Plasma Glutathione Peroxidase Mutant with Diselenide Bond Succeeds in Tetramer Formation. Antioxidants, 11(6), 1083. https://doi.org/10.3390/antiox11061083