
Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls

 ,
,  ,
,  and
and 
Abstract
1. Introduction to Chronic Kidney Disease
2. Introduction to the Nrf2 Pathway
3. Preclinical Data of Salutary, Ambiguous, or Disadvantageous Effects of Nrf2 Activation in CKD
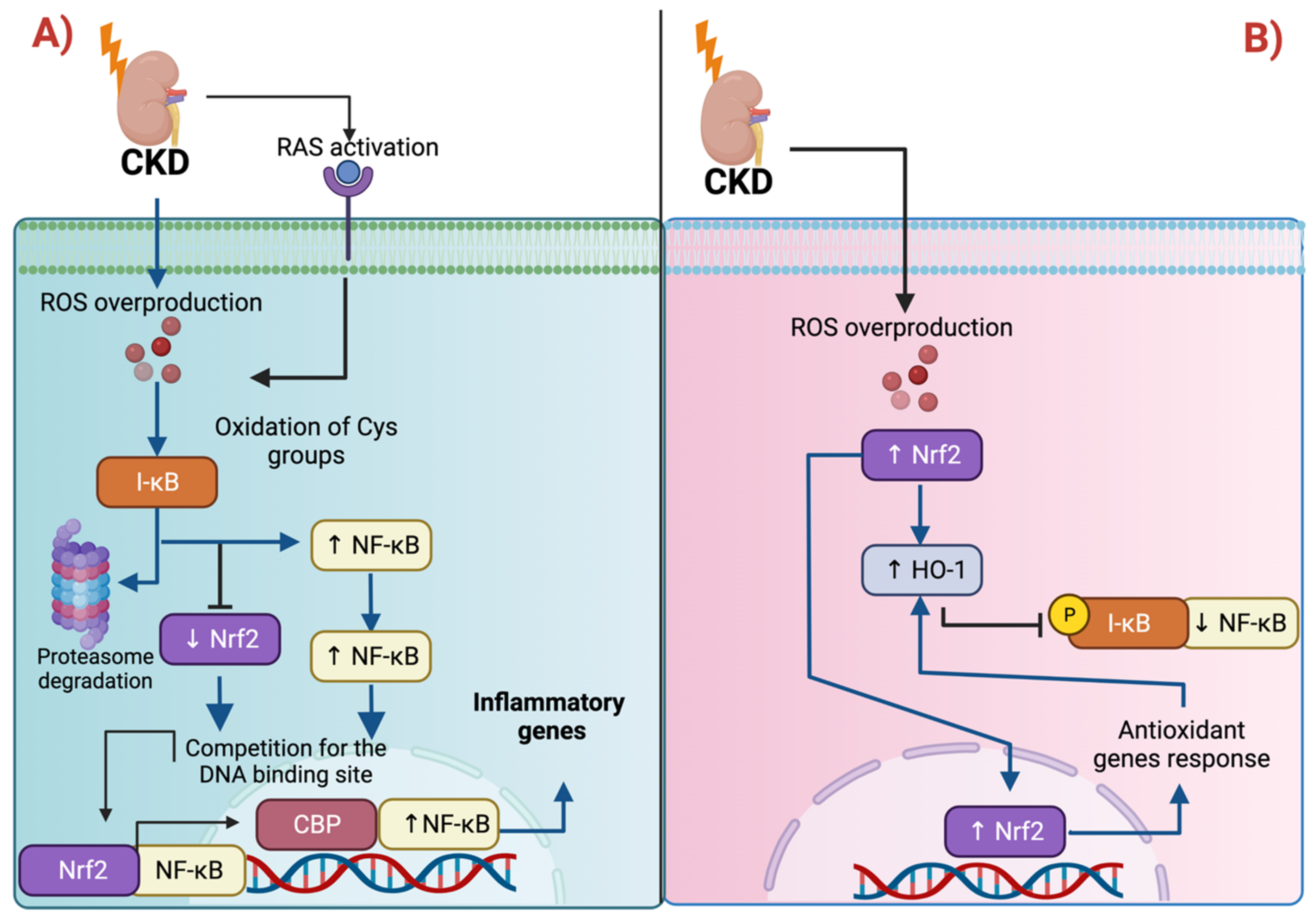
3.1. Nrf2 and Inflammation: The Intimate Relation with NF-κB
3.2. Nrf2 and Fibrosis: The Role of TGF-β1
3.3. Nrf2 Targets Mitochondrial Proteins
3.4. Nrf2 in Non-CKD
4. Clinical Data of Nrf2 Activation in Human CKD
4.1. Endogenous Nrf2 Activation in Human CKD
4.1.1. Mechanisms Relevant for Endogenous Nrf2 Activation in Human CKD
Oxidative Stress
Uremic Toxins
Nuclear Factor κ-Light-Chain Enhancer of Activated B Cells (NF-κB)
4.1.2. Nrf2 Activation in Patients with CKD According to Cause of CKD, CKD Stage, Comorbidity, and Investigated Cell Type
Nrf2 Activation in Renal Cells of Human CKD
Acute-Kidney-Injury (AKI)-to-CKD Progression
Diabetes Mellitus and DKD
Lupus Nephritis
Nrf2 Repression in Renal Cells of Human CKD
Nrf2 Status in Non-Renal Cells of Human CKD
4.2. Pharmacological Nrf2 Activation in Human CKD
4.2.1. Bardoxolone Methyl
4.2.2. Sulforaphane
4.2.3. Resveratrol
4.2.4. Curcumin
4.3. Future Directions in Nrf2-Targeted Therapies in Human CKD
4.3.1. Targeting Nrf2-System Disturbances in CKD More Specifically
4.3.2. Pharmacological Keap1 Inhibition
4.3.3. Reduction in Factors Responsible for Endogenous Nrf2 Activation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Inter. 2013, 3, 1–150. [Google Scholar]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Tetzner, F.; Scholze, A.; Wittstock, A.; Zidek, W.; Tepel, M. Impaired vascular reactivity in patients with chronic kidney disease. Am. J. Nephrol. 2008, 28, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Hörl, W.H. Immune dysfunction in uremia—An update. Toxins 2012, 4, 962–990. [Google Scholar] [CrossRef]
- Espi, M.; Koppe, L.; Fouque, D.; Thaunat, O. Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins 2020, 12, 300. [Google Scholar] [CrossRef]
- Akchurin, O.; Patino, E.; Dalal, V.; Meza, K.; Bhatia, D.; Brovender, S.; Zhu, Y.S.; Cunningham-Rundles, S.; Perelstein, E.; Kumar, J.; et al. Interleukin-6 Contributes to the Development of Anemia in Juvenile CKD. Kidney Int. Rep. 2018, 4, 470–483. [Google Scholar] [CrossRef]
- Gwozdzinski, K.; Pieniazek, A.; Gwozdzinski, L. Reactive Oxygen Species and Their Involvement in Red Blood Cell Damage in Chronic Kidney Disease. Oxid. Med. Cell Longev. 2021, 2021, 6639199. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 Activation Promotes the Recurrence of Dormant Tumour Cells through Regulation of Redox and Nucleotide Metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 Is Controlled by Two Distinct β-TrCP Recognition Motifs in Its Neh6 Domain, One of Which Can Be Modulated by GSK-3 Activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, X.; Miao, J.; Yuan, H.; Liu, E.; Liang, T.; Li, Q. Farrerol Directly Targets GSK-3β to Activate Nrf2-ARE Pathway and Protect EA.Hy926 Cells against Oxidative Stress-Induced Injuries. Oxidative Med. Cell. Longev. 2020, 2020, e5967434. [Google Scholar] [CrossRef] [PubMed]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and Non-Canonical Mechanisms of Nrf2 Activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2015, 36, 271–284. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 Degradation by Autophagy for the Maintenance of Redox Homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 Defines a Physiologically Important Stress Response Mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric Oxide Regulates Mitochondrial Fatty Acid Metabolism Through Reversible Protein S-Nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Ortega-Lozano, A.J.; Pedraza-Chaverri, J. Redox Signaling Pathways in Unilateral Ureteral Obstruction (UUO)-Induced Renal Fibrosis. Free Radic. Biol. Med. 2021, 172, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi Tosi, M.E.; Bocanegra, V.; Manucha, W.; Gil Lorenzo, A.; Vallés, P.G. The Nrf2–Keap1 Cellular Defense Pathway and Heat Shock Protein 70 (Hsp70) Response. Role in Protection against Oxidative Stress in Early Neonatal Unilateral Ureteral Obstruction (UUO). Cell Stress Chaperones 2011, 16, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Mohan, T.; Narasimhan, K.K.S.; Ravi, D.B.; Velusamy, P.; Chandrasekar, N.; Chakrapani, L.N.; Srinivasan, A.; Karthikeyan, P.; Kannan, P.; Tamilarasan, B.; et al. Role of Nrf2 Dysfunction in the Pathogenesis of Diabetic Nephropathy: Therapeutic Prospect of Epigallocatechin-3-Gallate. Free Radic. Biol. Med. 2020, 160, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Bianco, M.; Lopes, J.A.; Beiral, H.J.V.; Filho, J.D.D.; Frankenfeld, S.P.; Fortunato, R.S.; Gattass, C.R.; Vieyra, A.; Takiya, C.M. The Contralateral Kidney Presents with Impaired Mitochondrial Functions and Disrupted Redox Homeostasis after 14 Days of Unilateral Ureteral Obstruction in Mice. PLoS ONE 2019, 14, e0218986. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-Deficient Female Mice Develop Lupus-like Autoimmune Nephritis. Kidney Int. 2001, 60, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, L.M.; Stockler-Pinto, M.B.; Leite, M.; Mafra, D. Nrf2–Keap1 System versus NF-ΚB: The Good and the Evil in Chronic Kidney Disease? Biochimie 2012, 94, 2461–2466. [Google Scholar] [CrossRef]
- Liebman, S.E.; Le, T.H. Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease. Nutrients 2021, 13, 266. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic Potential of Nrf2 Activators in Streptozotocin-Induced Diabetic Nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane Attenuation of Experimental Diabetic Nephropathy Involves GSK-3 Beta/Fyn/Nrf2 Signaling Pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Wang, X.; Liu, X.; Wang, L.; Ren, F.; Wang, X.; Leng, X. Hippuric Acid Promotes Renal Fibrosis by Disrupting Redox Homeostasis via Facilitation of NRF2-KEAP1-CUL3 Interactions in Chronic Kidney Disease. Antioxidants 2020, 9, 783. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Beltrán, C.E.; Calderón-Oliver, M.; Martínez-Abundis, E.; Tapia, E.; Zarco-Márquez, G.; Zazueta, C.; Pedraza-Chaverri, J. Protective Effect of Sulforaphane against Cisplatin-Induced Mitochondrial Alterations and Impairment in the Activity of NAD(P)H: Quinone Oxidoreductase 1 and γ Glutamyl Cysteine Ligase: Studies in Mitochondria Isolated from Rat Kidney and in LLC-PK1 Cells. Toxicol. Lett. 2010, 199, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Stidham, R.D.; Bumeister, R.; Trevino, I.; Winters, A.; Sprouse, M.; Ding, M.; Ferguson, D.A.; Meyer, C.J.; Wigley, W.C.; et al. The Synthetic Triterpenoid, RTA 405, Increases the Glomerular Filtration Rate and Reduces Angiotensin II–Induced Contraction of Glomerular Mesangial Cells. Kidney Int. 2013, 83, 845–854. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk Factors for Heart Failure in Patients with Type 2 Diabetes Mellitus and Stage 4 Chronic Kidney Disease Treated with Bardoxolone Methyl. J. Card. Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef]
- Rush, B.M.; Bondi, C.D.; Stocker, S.D.; Barry, K.M.; Small, S.A.; Ong, J.; Jobbagy, S.; Stolz, D.B.; Bastacky, S.I.; Chartoumpekis, D.V.; et al. Genetic or Pharmacologic Nrf2 Activation Increases Proteinuria in Chronic Kidney Disease in Mice. Kidney Int. 2021, 99, 102–116. [Google Scholar] [CrossRef]
- Zhao, S.; Ghosh, A.; Lo, C.-S.; Chenier, I.; Scholey, J.W.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.-L.; Chan, J.S.D. Nrf2 Deficiency Upregulates Intrarenal Angiotensin-Converting Enzyme-2 and Angiotensin 1-7 Receptor Expression and Attenuates Hypertension and Nephropathy in Diabetic Mice. Endocrinology 2018, 159, 836–852. [Google Scholar] [CrossRef]
- Maher, J.; Yamamoto, M. The Rise of Antioxidant Signaling—The Evolution and Hormetic Actions of Nrf. Toxicol. Appl. Pharmacol. 2010, 244, 4–15. [Google Scholar] [CrossRef]
- Okamura, D.M.; Pennathur, S. The Balance of Powers: Redox Regulation of Fibrogenic Pathways in Kidney Injury. Redox Biol. 2015, 6, 495–504. [Google Scholar] [CrossRef]
- Kim, H.J.; Vaziri, N.D. Contribution of Impaired Nrf2-Keap1 Pathway to Oxidative Stress and Inflammation in Chronic Renal Failure. Am. J. Physiol.-Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef]
- Ziady, A.G.; Sokolow, A.; Shank, S.; Corey, D.; Myers, R.; Plafker, S.; Kelley, T.J. Interaction with CREB Binding Protein Modulates the Activities of Nrf2 and NF-ΚB in Cystic Fibrosis Airway Epithelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1221–L1231. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Miao, N.; Xu, J.; Gan, X.; Xu, D.; Zhou, L.; Xue, H.; Zhang, W.; Lu, L. N-Acetylcysteine Alleviates Angiotensin II-Mediated Renal Fibrosis in Mouse Obstructed Kidneys. Acta Pharm. Sin. 2016, 37, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating Cell-Signalling Pathways with NF-ΚB and IKK Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, H.; Li, J.; Ma, T.; Zhou, S.; Zhang, Z.; Miao, L.; Cai, L. Sulforaphane Prevents Type 2 Diabetes-Induced Nephropathy via AMPK-Mediated Activation of Lipid Metabolic Pathways and Nrf2 Antioxidative Function. Clin. Sci. 2020, 134, 2469–2487. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl Sulfate, a Uremic Toxin, Downregulates Renal Expression of Nrf2 through Activation of NF-ΚB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shen, G.; Chen, C.; Gélinas, C.; Kong, A.-N.T. Suppression of NF-ΚB and NF-ΚB-Regulated Gene Expression by Sulforaphane and PEITC through IκBα, IKK Pathway in Human Prostate Cancer PC-3 Cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-ΚB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Korn, S.H.; Wouters, E.F.M.; Vos, N.; Janssen-Heininger, Y.M.W. Cytokine-Induced Activation of Nuclear Factor-ΚB Is Inhibited by Hydrogen Peroxide through Oxidative Inactivation of IκB Kinase. J. Biol. Chem. 2001, 276, 35693–35700. [Google Scholar] [CrossRef]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-ΚB Nuclear Translocation via HO-1 Activation Underlies α-Tocopheryl Succinate Toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef]
- Seldon, M.P.; Silva, G.; Pejanovic, N.; Larsen, R.; Gregoire, I.P.; Filipe, J.; Anrather, J.; Soares, M.P. Heme Oxygenase-1 Inhibits the Expression of Adhesion Molecules Associated with Endothelial Cell Activation via Inhibition of NF-KappaB RelA Phosphorylation at Serine. J. Immunol. 2007, 179, 7840–7851. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, S.-Y.; Li, J.-S.; Zhang, Q.-F.; Wang, Y.-X.; Zhao, S.-L.; Yu, J.; Wang, C.; Qin, Y.; Wei, Q.-J.; et al. Overexpression of Heme Oxygenase-1 Prevents Renal Interstitial Inflammation and Fibrosis Induced by Unilateral Ureter Obstruction. PLoS ONE 2016, 11, e0147084. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Han, S.H.; Li, J.J.; Lee, S.H.; Jung, D.-S.; Kwak, S.-J.; Kim, S.H.; Kim, D.K.; Yoo, T.-H.; Kim, J.H.; et al. Induction of Heme Oxygenase-1 Protects against Podocyte Apoptosis under Diabetic Conditions. Kidney Int. 2009, 76, 838–848. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The Protective Role of Nrf2 in Streptozotocin-Induced Diabetic Nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Lai, T.Y.; Chien, C.T.; Yu, H.J. Activating Nrf-2 Signaling Depresses Unilateral Ureteral Obstruction-Evoked Mitochondrial Stress-Related Autophagy, Apoptosis and Pyroptosis in Kidney. PLoS ONE 2012, 7, e47299. [Google Scholar] [CrossRef] [PubMed]
- Antar, S.A.; Abdo, W.; Taha, R.S.; Farage, A.E.; El-Moselhy, L.E.; Amer, M.E.; Abdel Monsef, A.S.; Abdel Hamid, A.M.; Kamel, E.M.; Ahmeda, A.F.; et al. Telmisartan Attenuates Diabetic Nephropathy by Mitigating Oxidative Stress and Inflammation, and Upregulating Nrf2/HO-1 Signaling in Diabetic Rats. Life Sci. 2022, 291, 120260. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Muruve, D.A. The Inflammasomes in Kidney Disease. JASN 2011, 22, 1007–1018. [Google Scholar] [CrossRef]
- Mulay, S.R. Multifactorial Functions of the Inflammasome Component NLRP3 in Pathogenesis of Chronic Kidney Diseases. Kidney Int. 2019, 96, 58–66. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, X.; Sullivan, M.A.; Shi, C.; Deng, B. Protective Effect of Yi Shen Pai Du Formula against Diabetic Kidney Injury via Inhibition of Oxidative Stress, Inflammation, and Epithelial-to-Mesenchymal Transition in Db/Db Mice. Oxid. Med. Cell Longev. 2021, 2021, 7958021. [Google Scholar] [CrossRef]
- Ram, C.; Gairola, S.; Syed, A.M.; Kulhari, U.; Kundu, S.; Mugale, M.N.; Murty, U.S.; Sahu, B.D. Biochanin A Alleviates Unilateral Ureteral Obstruction-Induced Renal Interstitial Fibrosis and Inflammation by Inhibiting the TGF-Β1/Smad2/3 and NF-KB/NLRP3 Signaling Axis in Mice. Life Sci. 2022, 298, 120527. [Google Scholar] [CrossRef]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 Is Essential for Cholesterol Crystal-Induced Inflammasome Activation and Exacerbation of Atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wu, W.; Sheng, M.; Yang, S.; Tan, J. Amygdalin Inhibits Renal Fibrosis in Chronic Kidney Disease. Mol. Med. Rep. 2013, 7, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.T.; Melo, S.A.; Özdemir, B.C.; Kato, N.; Revuelta, I.; Miller, C.A.; Gattone, V.H.; LeBleu, V.S.; Kalluri, R. TGF-Β1–Containing Exosomes from Injured Epithelial Cells Activate Fibroblasts to Initiate Tissue Regenerative Responses and Fibrosis. JASN 2013, 24, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, H.; Huang, L.; Zhu, X.; Sha, J.; Li, G.; Ma, G.; Zhang, W.; Gu, M.; Guo, Y. Nrf2 Signaling Attenuates Epithelial-to-Mesenchymal Transition and Renal Interstitial Fibrosis via PI3K/Akt Signaling Pathways. Exp. Mol. Pathol. 2019, 111, 104296. [Google Scholar] [CrossRef]
- Kie, J.-H.; Kapturczak, M.H.; Traylor, A.; Agarwal, A.; Hill-Kapturczak, N. Heme Oxygenase-1 Deficiency Promotes Epithelial-Mesenchymal Transition and Renal Fibrosis. JASN 2008, 19, 1681–1691. [Google Scholar] [CrossRef]
- Brezniceanu, M.-L.; Liu, F.; Wei, C.-C.; Tran, S.; Sachetelli, S.; Zhang, S.-L.; Guo, D.-F.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S.D. Catalase Overexpression Attenuates Angiotensinogen Expression and Apoptosis in Diabetic Mice. Kidney Int. 2007, 71, 912–923. [Google Scholar] [CrossRef]
- Brezniceanu, M.-L.; Liu, F.; Wei, C.-C.; Chénier, I.; Godin, N.; Zhang, S.-L.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S.D. Attenuation of Interstitial Fibrosis and Tubular Apoptosis in Db/Db Transgenic Mice Overexpressing Catalase in Renal Proximal Tubular Cells. Diabetes 2008, 57, 451–459. [Google Scholar] [CrossRef]
- Shi, Y.; Lo, C.-S.; Chenier, I.; Maachi, H.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.-L.; Chan, J.S.D. Overexpression of Catalase Prevents Hypertension and Tubulointerstitial Fibrosis and Normalization of Renal Angiotensin-Converting Enzyme-2 Expression in Akita Mice. Am. J. Physiol. Ren. Physiol. 2013, 304, F1335–F1346. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear Activators and Coactivators in Mammalian Mitochondrial Biogenesis. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Wu, K.L.H.; Wu, C.-W.; Chao, Y.-M.; Hung, C.-Y.; Chan, J.Y.H. Impaired Nrf2 Regulation of Mitochondrial Biogenesis in Rostral Ventrolateral Medulla on Hypertension Induced by Systemic Inflammation. Free Radic. Biol. Med. 2016, 97, 58–74. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme Oxygenase-1 Regulates Cardiac Mitochondrial Biogenesis via Nrf2-Mediated Transcriptional Control of Nuclear Respiratory Factor. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Meo, S. The Role of Reactive Oxygen Species in the Life Cycle of the Mitochondrion. Int. J. Mol. Sci. 2020, 21, 2173. [Google Scholar] [CrossRef] [PubMed]
- MacGarvey, N.C.; Suliman, H.B.; Bartz, R.R.; Fu, P.; Withers, C.M.; Welty-Wolf, K.E.; Piantadosi, C.A. Activation of Mitochondrial Biogenesis by Heme Oxygenase-1–Mediated NF-E2–Related Factor-2 Induction Rescues Mice from Lethal Staphylococcus Aureus Sepsis. Am. J. Respir. Crit. Care Med. 2012, 185, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 System Prevents Onset of Diabetes Mellitus. Mol. Cell Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Uribe, A.P.; Bellido, B.; Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Hernández-Santos, J.A.; Fernández-Valverde, F.; Hernández-Cruz, E.Y.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Temporal Characterization of Mitochondrial Impairment in the Unilateral Ureteral Obstruction Model in Rats. Free Radic. Biol. Med. 2021, 172, 358–371. [Google Scholar] [CrossRef]
- Prieto-Carrasco, R.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; Rojas-Morales, P.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Tapia, E.; Pedraza-Chaverri, J. Progressive Reduction in Mitochondrial Mass Is Triggered by Alterations in Mitochondrial Biogenesis and Dynamics in Chronic Kidney Disease Induced by 5/6 Nephrectomy. Biology 2021, 10, 349. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kojima-Yuasa, A.; Tadano, H.; Mizuno, A.; Kon, A.; Norikura, T. Ursolic Acid Improves the Indoxyl Sulfate-Induced Impairment of Mitochondrial Biogenesis in C2C12 Cells. Nutr. Res. Pr. 2022, 16, 147–160. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. P53 Orchestrates the PGC-1α-Mediated Antioxidant Response Upon Mild Redox and Metabolic Imbalance. Antioxid. Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef]
- Mohammad, R.S.; Lokhandwala, M.F.; Banday, A.A. Age-Related Mitochondrial Impairment and Renal Injury Is Ameliorated by Sulforaphane via Activation of Transcription Factor NRF. Antioxidants 2022, 11, 156. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Cheng, X.; Zheng, X.; Song, Y.; Qu, L.; Tang, J.; Meng, L.; Wang, Y. Apocynin Attenuates Renal Fibrosis via Inhibition of NOXs-ROS-ERK-Myofibroblast Accumulation in UUO Rats. Free Radic. Res. 2016, 50, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.; Monsalve, M.; Ramos, A.; Sanchez-Niño, M.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Chertow, G.M.; Appel, G.B.; Block, G.A.; Chin, M.P.; Coyne, D.W.; Goldsberry, A.; Kalantar-Zadeh, K.; Meyer, C.J.; Molitch, M.E.; Pergola, P.E.; et al. Effects of Bardoxolone Methyl on Body Weight, Waist Circumference and Glycemic Control in Obese Patients with Type 2 Diabetes Mellitus and Stage 4 Chronic Kidney Disease. J. Diabetes Its Complicat. 2018, 32, 1113–1117. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxidative Med. Cell. Longev. 2020, 2020, e4678252. [Google Scholar] [CrossRef]
- Foresti, R.; Bucolo, C.; Platania, C.M.B.; Drago, F.; Dubois-Randé, J.-L.; Motterlini, R. Nrf2 Activators Modulate Oxidative Stress Responses and Bioenergetic Profiles of Human Retinal Epithelial Cells Cultured in Normal or High Glucose Conditions. Pharmacol. Res. 2015, 99, 296–307. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Lizano, M. Cellular Redox, Cancer and Human Papillomavirus. Virus Res. 2018, 246, 35–45. [Google Scholar] [CrossRef]
- Daroui, P.; Desai, S.D.; Li, T.-K.; Liu, A.A.; Liu, L.F. Hydrogen Peroxide Induces Topoisomerase I-Mediated DNA Damage and Cell Death. J. Biol. Chem. 2004, 279, 14587–14594. [Google Scholar] [CrossRef]
- Bernier, J.; Hall, E.J.; Giaccia, A. Radiation Oncology: A Century of Achievements. Nat. Rev. Cancer 2004, 4, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Martínez-Ramírez, I.; Pedraza-Chaverri, J.; Lizano, M. Reprogramming of Energy Metabolism in Response to Radiotherapy in Head and Neck Squamous Cell Carcinoma. Cancers 2019, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Muthusamy, V.R.; Whitehead, K.J.; Wang, L.; Gomes, A.V.; Litwin, S.E.; Kensler, T.W.; Abel, E.D.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 Deficiency Prevents Reductive Stress-Induced Hypertrophic Cardiomyopathy. Cardiovasc. Res. 2013, 100, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the Regulation of CD36 and Stress Protein Expression in Murine Macrophages: Activation by Oxidatively Modified LDL and 4-Hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine Inhibits Autophagic Flux by Decreasing Autophagosome-Lysosome Fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015, 88 Pt B, 108–146. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Ryan, D.G.; Knatko, E.V.; Casey, A.M.; Hukelmann, J.L.; Dayalan Naidu, S.; Brenes, A.J.; Ekkunagul, T.; Baker, C.; Higgins, M.; Tronci, L.; et al. Nrf2 activation reprograms macrophage intermediary metabolism and suppresses the type I interferon response. iScience 2022, 25, 103827. [Google Scholar] [CrossRef]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef]
- Pedraza-Chaverri, J.; Sánchez-Lozada, L.G.; Osorio-Alonso, H.; Tapia, E.; Scholze, A. New Pathogenic Concepts and Therapeutic Approaches to Oxidative Stress in Chronic Kidney Disease. Oxid. Med. Cell Longev. 2016, 2016, 6043601. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Tanaka, T.; Yoshida, Y.; Inagi, R. Physiological and pathophysiological role of reactive oxygen species and reactive nitrogen species in the kidney. Clin. Exp. Pharm. Physiol. 2018, 45, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Pieniazek, A.; Bernasinska-Slomczewska, J.; Gwozdzinski, L. Uremic Toxins and Their Relation with Oxidative Stress Induced in Patients with CKD. Int. J. Mol. Sci. 2021, 22, 6196. [Google Scholar] [CrossRef] [PubMed]
- Ebert, T.; Neytchev, O.; Witasp, A.; Kublickiene, K.; Stenvinkel, P.; Shiels, P.G. Inflammation and Oxidative Stress in Chronic Kidney Disease and Dialysis Patients. Antioxid Redox Signal. 2021, 35, 1426–1448. [Google Scholar] [CrossRef]
- Tepel, M.; Echelmeyer, M.; Orie, N.N.; Zidek, W. Increased intracellular reactive oxygen species in patients with end-stage renal failure: Effect of hemodialysis. Kidney Int. 2000, 58, 867–872. [Google Scholar] [CrossRef]
- Sanner, B.M.; Meder, U.; Zidek, W.; Tepel, M. Effects of glucocorticoids on generation of reactive oxygen species in platelets. Steroids 2002, 67, 715–719. [Google Scholar] [CrossRef]
- Schulz, A.M.; Terne, C.; Jankowski, V.; Cohen, G.; Schaefer, M.; Boehringer, F.; Tepel, M.; Kunkel, D.; Zidek, W.; Jankowski, J.; et al. Modulation of NADPH oxidase activity by known uraemic retention solutes. Eur. J. Clin. Investig. 2014, 44, 802–811. [Google Scholar] [CrossRef]
- Mihajlovic, M.; Krebber, M.M.; Yang, Y.; Ahmed, S.; Lozovanu, V.; Andreeva, D.; Verhaar, M.C.; Masereeuw, R. Protein-Bound Uremic Toxins Induce Reactive Oxygen Species-Dependent and Inflammasome-Mediated IL-1β Production in Kidney Proximal Tubule Cells. Biomedicines 2021, 9, 1326. [Google Scholar] [CrossRef]
- Gnemmi, V.; Li, Q.; Ma, Q.; De Chiara, L.; Carangelo, G.; Li, C.; Molina-Van den Bosch, M.; Romagnani, P.; Anders, H.J.; Steiger, S. Asymptomatic Hyperuricemia Promotes Recovery from Ischemic Organ Injury by Modulating the Phenotype of Macrophages. Cells 2022, 11, 626. [Google Scholar] [CrossRef]
- Krueger, K.; Koch, K.; Jühling, A.; Tepel, M.; Scholze, A. Low expression of thiosulfate sulfurtransferase (rhodanese) predicts mortality in hemodialysis patients. Clin. Biochem. 2010, 43, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Scholze, A.; Krueger, K.; Diedrich, M.; Räth, C.; Torges, A.; Jankowski, V.; Maier, A.; Thilo, F.; Zidek, W.; Tepel, M. Superoxide dismutase type 1 in monocytes of chronic kidney disease patients. Amino Acids 2011, 41, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Stępniewska, J.; Dołęgowska, B.; Cecerska-Heryć, E.; Gołembiewska, E.; Malinowska-Jędraszczyk, A.; Marchelek-Myśliwiec, M.; Ciechanowski, K. The activity of antioxidant enzymes in blood platelets in different types of renal replacement therapy: A cross-sectional study. Int. Urol. Nephrol. 2016, 48, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Krueger, K.; Shen, J.; Maier, A.; Tepel, M.; Scholze, A. Lower Superoxide Dismutase 2 (SOD2) Protein Content in Mononuclear Cells Is Associated with Better Survival in Patients with Hemodialysis Therapy. Oxid. Med. Cell Longev. 2016, 2016, 7423249. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.M.; Guillem, A.; Keyser, J.; Singh, B. A Pharmacologic “Stress Test” for Assessing Select Antioxidant Defenses in Patients with CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 633–642. [Google Scholar] [CrossRef]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef]
- Hou, F.; Wang, G.; Zhou, Z.; Zhang, X. Enhanced oxidant stress in synovial vessels of patients on hemodialysis. Chin. Med. J. 2000, 113, 934–937. [Google Scholar]
- Vanholder, R.; Nigam, S.K.; Burtey, S.; Glorieux, G. What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis. Toxins 2022, 14, 221. [Google Scholar] [CrossRef]
- Kim, H.Y.; Yoo, T.H.; Cho, J.Y.; Kim, H.C.; Lee, W.W. Indoxyl sulfate-induced TNF-α is regulated by crosstalk between the aryl hydrocarbon receptor, NF-κB, and SOCS2 in human macrophages. FASEB J. 2019, 33, 10844–10858. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Alvarenga, L.; Cardozo, L.F.M.F.; Leal, V.O.; Kemp, J.A.; Saldanha, J.F.; Ribeiro-Alves, M.; Meireles, T.; Nakao, L.S.; Mafra, D. Can resveratrol supplementation reduce uremic toxins plasma levels from the gut microbiota in non-dialyzed chronic kidney disease patients? J. Ren. Nutr. 2022, S1051-2276(22)00010-3. [Google Scholar] [CrossRef]
- Juul-Nielsen, C.; Shen, J.; Stenvinkel, P.; Scholze, A. Systematic review of the nuclear factor erythroid 2-related factor 2 (NRF2) system in human chronic kidney disease: Alterations, interventions, and relation to morbidity. Nephrol. Dial. Transplant. 2022, 37, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Emerging Glycation-Based Therapeutics-Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors. Int. J. Mol. Sci. 2022, 23, 2453. [Google Scholar] [CrossRef] [PubMed]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Sapienza, M.; Puleo, E.; Calafato, S.; Cornelius, C.; Finocchiaro, M.; Mangiameli, A.; Di Mauro, M.; Stella, A.M.; et al. Oxidative stress and cellular stress respons.se in diabetic nephropathy. Cell Stress Chaperones 2007, 12, 299–306. [Google Scholar] [CrossRef]
- Sharma, M.; Mehndiratta, M.; Gupta, S.; Kalra, O.P.; Shukla, R.; Gambhir, J.K. Genetic association of NAD(P)H quinone oxidoreductase (NQO1*2) polymorphism with NQO1 levels and risk of diabetic nephropathy. Biol. Chem. 2016, 397, 725–730. [Google Scholar] [CrossRef]
- Van Raaij, S.; van Swelm, R.; Bouman, K.; Cliteur, M.; van den Heuvel, M.C.; Pertijs, J.; Patel, D.; Bass, P.; van Goor, H.; Unwin, R.; et al. Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci. Rep. 2018, 8, 9353. [Google Scholar] [CrossRef]
- Zhao, S.; Lo, C.S.; Miyata, K.N.; Ghosh, A.; Zhao, X.P.; Chenier, I.; Cailhier, J.F.; Ethier, J.; Lattouf, J.B.; Filep, J.G.; et al. Overexpression of Nrf2 in Renal Proximal Tubular Cells Stimulates Sodium-Glucose Cotransporter 2 Expression and Exacerbates Dysglycemia and Kidney Injury in Diabetic Mice. Diabetes 2021, 70, 1388–1403. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Srivastava, A.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Mulay, S.R.; Scholze, A. Involvement of Inflammasome Components in Kidney Disease. Antioxidants 2022, 11, 246. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Leal, V.O.; Saldanha, J.F.; Stockler-Pinto, M.B.; Cardozo, L.F.; Santos, F.R.; Albuquerque, A.S.; Leite, M., Jr.; Mafra, D. NRF2 and NF-κB mRNA expression in chronic kidney disease: A focus on nondialysis patients. Int. Urol. Nephrol. 2015, 47, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, S.; Strom, J.; Purdom-Dickinson, S.; Chen, Q.M. NAD(P)H:quinone oxidoreductase 1 is induced by progesterone in cardiomyocytes. Cardiovasc. Toxicol. 2012, 12, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.V.; Cattani-Cavalieri, I.; de Brito-Gitirana, L.; Fautrel, A.; Lagente, V.; Schmidt, M.; Porto, L.C.; Romana-Souza, B.; Valença, S.S.; Lanzetti, M. Propolis reversed cigarette smoke-induced emphysema through macrophage alternative activation independent of Nrf. Bioorg. Med. Chem. 2017, 25, 5557–5568. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.C.; Lee, H.T.; Chien, P.J.; Huang, Y.H.; Chang, M.Y.; Lee, Y.C.; Chang, W.W. NAD(P)H:quinone oxidoreductase 1 determines radiosensitivity of triple negative breast cancer cells and is controlled by long non-coding RNA NEAT. Int. J. Med. Sci. 2020, 17, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Jeong, M.G.; Oh, S.; Jang, E.J.; Kim, H.K.; Hwang, E.S. A FoxO1-dependent, but NRF2-independent induction of heme oxygenase-1 during muscle atrophy. FEBS Lett. 2014, 588, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.S.; Park, J.J.; Choi, J.Y.; Lee, D.H.; Yun, S.J.; Lee, J.B.; Lee, S.C. Nrf2-dependent and Nrf2-independent induction of phase 2 detoxifying and antioxidant enzymes during keratinocyte differentiation. Arch. Derm. Res. 2012, 304, 387–395. [Google Scholar] [CrossRef]
- Wright, M.M.; Kim, J.; Hock, T.D.; Leitinger, N.; Freeman, B.A.; Agarwal, A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem. J. 2009, 422, 353–361. [Google Scholar] [CrossRef]
- Kong, W.; Fu, J.; Liu, N.; Jiao, C.; Guo, G.; Luan, J.; Wang, H.; Yao, L.; Wang, L.; Yamamoto, M.; et al. Nrf2 deficiency promotes the progression from acute tubular damage to chronic renal fibrosis following unilateral ureteral obstruction. Nephrol. Dial. Transplant. 2018, 33, 771–783. [Google Scholar] [CrossRef]
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3β-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275. [Google Scholar] [CrossRef]
- Gu, X.; Liu, Y.; Wang, N.; Zhen, J.; Zhang, B.; Hou, S.; Cui, Z.; Wan, Q.; Feng, H. Transcription of MRPL12 regulated by Nrf2 contributes to the mitochondrial dysfunction in diabetic kidney disease. Free Radic. Biol. Med. 2021, 164, 329–340. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, L.; Yang, Y.; Chen, Y.; Song, Y.; Lu, X.; Liang, Y. The inhibition of Nrf2 accelerates renal lipid deposition through suppressing the ACSL1 expression in obesity-related nephropathy. Ren. Fail. 2019, 41, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Sun, Y.; Liu, Z.; Lu, Y.; Zhu, X.; Lan, B.; Mi, Z.; Dang, L.; Li, N.; Zhan, W.; et al. Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci. Transl. Med. 2020, 12, eaba3613. [Google Scholar] [CrossRef] [PubMed]
- Rhone, E.T.; Bardhi, E.; Bontha, S.V.; Walker, P.D.; Almenara, J.A.; Dumur, C.I.; Cathro, H.; Maluf, D.; Mas, V. An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients. Int. J. Mol. Sci. 2021, 22, 5414. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wang, B.; Sui, W.; Zou, G.; Chen, H.; Xie, S.; Zou, H. Expression of GSK-3β in renal allograft tissue and its significance in pathogenesis of chronic allograft dysfunction. Diagn. Pathol. 2012, 7, 5. [Google Scholar] [CrossRef]
- Girndt, M.; Trojanowicz, B.; Ulrich, C. Monocytes in Uremia. Toxins 2020, 12, 340. [Google Scholar] [CrossRef]
- Cantero-Navarro, E.; Rayego-Mateos, S.; Orejudo, M.; Tejedor-Santamaria, L.; Tejera-Muñoz, A.; Sanz, A.B.; Marquez-Exposito, L.; Marchant, V.; Santos-Sanchez, L.; Egido, J.; et al. Role of Macrophages and Related Cytokines in Kidney Disease. Front. Med. 2021, 8, 688060. [Google Scholar] [CrossRef]
- Scholze, A.; Thies, C.; Cheikhalfraj, M.; Wittstock, A.; Pommer, W.; Zidek, W.; Tepel, M. Mortality risk in hemodialysis patients with increased arterial stiffness is reduced by attainment of classical clinical performance measures. Am. J. Nephrol. 2009, 29, 598–606. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E.; Price, S.R. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 138–152. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.S.; Dominic, E.A.; Pai, A.; Osman, F.; Morgan, M.; Pickett, G.; Shah, V.O.; Ferrando, A.; Moseley, P. Skeletal muscle, cytokines, and oxidative stress in end-stage renal disease. Kidney Int. 2005, 68, 2338–2344. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pedruzzi, L.M.; Cardozo, L.F.; Daleprane, J.B.; Stockler-Pinto, M.B.; Monteiro, E.B.; Leite, M., Jr.; Vaziri, N.D.; Mafra, D. Systemic inflammation and oxidative stress in hemodialysis patients are associated with down-regulation of Nrf. J. Nephrol. 2015, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Rasmussen, M.; Dong, Q.R.; Tepel, M.; Scholze, A. Expression of the NRF2 Target Gene NQO1 Is Enhanced in Mononuclear Cells in Human Chronic Kidney Disease. Oxid. Med. Cell Longev. 2017, 2017, 9091879. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.L.; Baker, L.A.; Wilkinson, T.J.; Gould, D.W.; Graham-Brown, M.P.M.; Major, R.W.; Ashford, R.U.; Philp, A.; Smith, A.C. Reductions in skeletal muscle mitochondrial mass are not restored following exercise training in patients with chronic kidney disease. FASEB J. 2020, 34, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-κB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- Chin, M.P.; Bakris, G.L.; Block, G.A.; Chertow, G.M.; Goldsberry, A.; Inker, L.A.; Heerspink, H.J.L.; O’Grady, M.; Pergola, P.E.; Wanner, C.; et al. Bardoxolone Methyl Improves Kidney Function in Patients with Chronic Kidney Disease Stage 4 and Type 2 Diabetes: Post-Hoc Analyses from Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes Study. Am. J. Nephrol. 2018, 47, 40–47. [Google Scholar] [CrossRef]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef]
- Lewis, J.H.; Jadoul, M.; Block, G.A.; Chin, M.P.; Ferguson, D.A.; Goldsberry, A.; Meyer, C.J.; O’Grady, M.; Pergola, P.E.; Reisman, S.A.; et al. Effects of Bardoxolone Methyl on Hepatic Enzymes in Patients with Type 2 Diabetes Mellitus and Stage 4 CKD. Clin. Transl. Sci. 2021, 14, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.N.; Dillon, J.F.; Tapper, E.B. Gamma-Glutamyl Transferase (γ-GT)—An old dog with new tricks? Liver Int. 2022, 42, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Agarwal, R.; Audhya, P.; Bakris, G.L.; Chin, M.; Krauth, M.; Lambers Heerspink, H.J.; Meyer, C.J.; McMurray, J.J.; et al. Rationale and trial design of Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes: The Occurrence of Renal Events (BEACON). Am. J. Nephrol. 2013, 37, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Tayek, J.A.; Kalantar-Zadeh, K. The extinguished BEACON of bardoxolone: Not a Monday morning quarterback story. Am. J. Nephrol. 2013, 37, 208–211. [Google Scholar] [CrossRef]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef]
- Saldanha, J.F.; Leal, V.O.; Rizzetto, F.; Grimmer, G.H.; Ribeiro-Alves, M.; Daleprane, J.B.; Carraro-Eduardo, J.C.; Mafra, D. Effects of Resveratrol Supplementation in Nrf2 and NF-κB Expressions in Nondialyzed Chronic Kidney Disease Patients: A Randomized, Double-Blind, Placebo-Controlled, Crossover Clinical Trial. J. Ren. Nutr. 2016, 26, 401–406. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Kujawski, K.; Sacewicz-Hofman, I.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. Are Nutraceuticals Beneficial in Chronic Kidney Disease? Pharmaceutics 2021, 13, 231. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, W.; Zhou, Z.; Liu, J.; Li, X.; Chen, L.; Weng, J.; Yu, Z. Curcumin attenuates urinary excretion of albumin in type II diabetic patients with enhancing nuclear factor erythroid-derived 2-like 2 (Nrf2) system and repressing inflammatory signaling efficacies. Exp. Clin. Endocrinol. Diabetes 2015, 123, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, L.; Salarolli, R.; Cardozo, L.F.M.F.; Santos, R.S.; de Brito, J.S.; Kemp, J.A.; Reis, D.; de Paiva, B.R.; Stenvinkel, P.; Lindholm, B.; et al. Impact of curcumin supplementation on expression of inflammatory transcription factors in hemodialysis patients: A pilot randomized, double-blind, controlled study. Clin. Nutr. 2020, 39, 3594–3600. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Osorio, A.S.; García-Niño, W.R.; González-Reyes, S.; Álvarez-Mejía, A.E.; Guerra-León, S.; Salazar-Segovia, J.; Falcón, I.; Montes de Oca-Solano, H.; Madero, M.; Pedraza-Chaverri, J. The Effect of Dietary Supplementation with Curcumin on Redox Status and Nrf2 Activation in Patients With Nondiabetic or Diabetic Proteinuric Chronic Kidney Disease: A Pilot Study. J. Ren. Nutr. 2016, 26, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Pakfetrat, M.; Basiri, F.; Malekmakan, L.; Roozbeh, J. Effects of turmeric on uremic pruritus in end stage renal disease patients: A double-blind randomized clinical trial. J. Nephrol. 2014, 27, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Khajehdehi, P.; Pakfetrat, M.; Javidnia, K.; Azad, F.; Malekmakan, L.; Nasab, M.H.; Dehghanzadeh, G. Oral supplementation of turmeric attenuates proteinuria, transforming growth factor-β and interleukin-8 levels in patients with overt type 2 diabetic nephropathy: A randomized, double-blind and placebo-controlled study. Scand. J. Urol. Nephrol. 2011, 45, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.X.; Devereaux, P.J.; Hill, A.; Sood, M.; Aggarwal, B.; Dubois, L.; Hiremath, S.; Guzman, R.; Iyer, V.; James, M.; et al. Oral curcumin in elective abdominal aortic aneurysm repair: A multicentre randomized controlled trial. CMAJ 2018, 190, E1273–E1280. [Google Scholar] [CrossRef]
- Lewandowska, U.; Szewczyk, K.; Hrabec, E.; Janecka, A.; Gorlach, S. Overview of metabolism and bioavailability enhancement of polyphenols. J. Agric. Food Chem. 2013, 61, 12183–12199. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. Curcumin May (Not) Defy Science. ACS Med. Chem. Lett. 2017, 8, 467–470. [Google Scholar] [CrossRef]
- Tang, M.; Larson-Meyer, D.E.; Liebman, M. Effect of cinnamon and turmeric on urinary oxalate excretion, plasma lipids, and plasma glucose in healthy subjects. Am. J. Clin. Nutr. 2008, 87, 1262–1267. [Google Scholar] [CrossRef]
- Pfau, A.; Ermer, T.; Coca, S.G.; Tio, M.C.; Genser, B.; Reichel, M.; Finkelstein, F.O.; März, W.; Wanner, C.; Waikar, S.S.; et al. High Oxalate Concentrations Correlate with Increased Risk for Sudden Cardiac Death in Dialysis Patients. J. Am. Soc. Nephrol. 2021, 32, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Hajirahimkhan, A.; Simmler, C.; Dong, H.; Lantvit, D.D.; Li, G.; Chen, S.N.; Nikolić, D.; Pauli, G.F.; van Breemen, R.B.; Dietz, B.M.; et al. Induction of NAD(P)H:Quinone Oxidoreductase 1 (NQO1) by Glycyrrhiza Species Used for Women’s Health: Differential Effects of the Michael Acceptors Isoliquiritigenin and Licochalcone A. Chem. Res. Toxicol. 2015, 28, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jiang, H.; Shi, Y. Upregulation of heme oxygenase-1 expression by curcumin conferring protection from hydrogen peroxide-induced apoptosis in H9c2 cardiomyoblasts. Cell Biosci. 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Santel, T.; Pflug, G.; Hemdan, N.Y.; Schäfer, A.; Hollenbach, M.; Buchold, M.; Hintersdorf, A.; Lindner, I.; Otto, A.; Bigl, M.; et al. Curcumin inhibits glyoxalase 1: A possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE 2008, 3, e3508. [Google Scholar] [CrossRef]
- Shen, C.; Cheng, W.; Yu, P.; Wang, L.; Zhou, L.; Zeng, L.; Yang, Q. Resveratrol pretreatment attenuates injury and promotes proliferation of neural stem cells following oxygen-glucose deprivation/reoxygenation by upregulating the expression of Nrf2, HO-1 and NQO1 in vitro. Mol. Med. Rep. 2016, 14, 3646–3654. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Reversal of Insulin Resistance in Overweight and Obese Subjects by trans-Resveratrol and Hesperetin Combination-Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef]
- Jeffery, E.H.; Stewart, K.E. Upregulation of quinone reductase by glucosinolate hydrolysis products from dietary broccoli. Methods Enzymol. 2004, 382, 457–469. [Google Scholar] [CrossRef]
- Schachtele, S.J.; Hu, S.; Lokensgard, J.R. Modulation of experimental herpes encephalitis-associated neurotoxicity through sulforaphane treatment. PLoS ONE 2012, 7, e36216. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective effect of sulforaphane against methylglyoxal cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef]
- Fujiki, T.; Ando, F.; Murakami, K.; Isobe, K.; Mori, T.; Susa, K.; Nomura, N.; Sohara, E.; Rai, T.; Uchida, S. Tolvaptan activates the Nrf2/HO-1 antioxidant pathway through PERK phosphorylation. Sci. Rep. 2019, 9, 9245. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Saar-Kovrov, V.; Zidek, W.; Orth-Alampour, S.; Fliser, D.; Jankowski, V.; Biessen, E.A.L.; Jankowski, J. Reduction of protein-bound uraemic toxins in plasma of chronic renal failure patients: A systematic review. J. Intern. Med. 2021, 290, 499–526. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, T.; Eloot, S.; Vanholder, R.; Glorieux, G.; van der Sande, F.M.; Scheijen, J.L.; Leunissen, K.M.; Kooman, J.P.; Schalkwijk, C.G. Protein-bound uraemic toxins, dicarbonyl stress and advanced glycation end products in conventional and extended haemodialysis and haemodiafiltration. Nephrol. Dial. Transplant. 2015, 30, 1395–1402. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Possible Mechanism | Observed Effects in Patients with CKD | Reference for the Patient Data |
|---|---|---|---|
| Oxidative stress | Oxidants and thiol-reactive electrophiles modify Keap1 → increase in Nrf2 translocation to the nucleus → effect on transcription of Nrf2 targets | DKD: oxidative damage of renal glomeruli, | [54] |
| Nrf2 protein ↑ | [116] | ||
| NQO1 protein ↑ | |||
| LN: oxidative damage of renal glomeruli, | |||
| Nrf2 protein ↑ | |||
| NQO1 protein ↑ | |||
| CKD5-HD: synovial tissue | [117] | ||
| MDA ↑ | |||
| HO-1 protein ↑ | |||
| Uremic toxins Indoxyl sulfate | (a) induction of ROS → effect on transcription of Nrf2 targets as above (b) activation of AhR → induction of Nrf2 gene transcription | CKD3/4:PBMCs, positive correlation between plasma indoxyl sulfate (1–11 mg/L) and Nrf2 gene expression | [121] |
| Methylglyoxal | Keap1 cross-linking → Keap1 dimers → Nrf2 accumulation and Nrf2-target induction | T2D with and without DKD: Lymphocytes, HO-1 protein ↑ Plasma, NQO1 protein ↑ Renal cells, Nrf2, NQO1, HO-1 protein ↑ | [125] |
| [126] | |||
| [37,54,127,128] | |||
| NF-κB | NF-κB binding sites in Nrf2 gene → Nrf2 induction | CKD3/4: PBMCs, positive correlation between NF-κB and Nrf2 gene expression ↑ | [131] |
| Factor | Possible Mechanism | Observed Effects in Patients with CKD | Reference for the Patient Data |
|---|---|---|---|
| Increased GSK-3β | GSK-3β → phosphorylation of Nrf2 → ubiquitinylation by β-TrCP → Nrf2 degradation ↑ | ADPKD_CKD1–3b: kidney tissue, Nrf2 protein ↓ | [144] * |
| Uremic toxins Indoxyl sulfate | ↓ Nrf2 gene and protein expression with high indoxyl-sulfate concentrations | CKD-5HD: PBMCs, plasma indoxyl sulfate (mean ~29 mg/L) correlated negatively with Nrf2 gene expression | [156] |
| Substance | Nrf2-System Response | Reference |
|---|---|---|
| Bardoxolone methyl | CKD4 and T2D: serum GGT ↑ | [161] |
| Resveratrol | CKD3/4: no effect on PBMC Nrf2 gene expression | [169] |
| Curcumin | CKD1–3a and T2D: PBMC Nrf2 and NQO1 protein ↑ | [172] |
| Curcumin | CKD5-HD, diabetes, hypertension: no effect on PBMC Nrf2 gene expression | [173] |
| Curcumin | CKD, proteinuria, diabetes: no effect on PBMC Nrf2 binding activity | [174] |
| Tin proto-porphyrin | CKD3/4, diabetes, hypertension: plasma NQO1, HO-1, and ferritin ↑ | [115] |
| Substance | Response | Reference |
|---|---|---|
| Curcumin | NQO1 activity↑ | [182] |
| Curcumin | HO-1 protein↑ | [183] |
| Curcumin | GLO1 activity↓ | [184] |
| Resveratrol | NQO1 protein↑ | [185] |
| Resveratrol | HO-1 protein↑ | [185] |
| Resveratrol | GLO1 gene expression and activity↑ | [186] |
| Sulforaphane | NQO1 activity↑ | [187] |
| Sulforaphane | HO-1 protein↑ | [188] |
| Sulforaphane | GLO1 protein and activity↑ | [189] |
| Bardoxolone methyl | NQO1 gene expression↑ | [34] |
| Bardoxolone methyl | HO-1 gene expression and protein↑ | [190] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Scholze, A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants 2022, 11, 1112. https://doi.org/10.3390/antiox11061112
Aranda-Rivera AK, Cruz-Gregorio A, Pedraza-Chaverri J, Scholze A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants. 2022; 11(6):1112. https://doi.org/10.3390/antiox11061112
Chicago/Turabian StyleAranda-Rivera, Ana Karina, Alfredo Cruz-Gregorio, José Pedraza-Chaverri, and Alexandra Scholze. 2022. "Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls" Antioxidants 11, no. 6: 1112. https://doi.org/10.3390/antiox11061112
APA StyleAranda-Rivera, A. K., Cruz-Gregorio, A., Pedraza-Chaverri, J., & Scholze, A. (2022). Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants, 11(6), 1112. https://doi.org/10.3390/antiox11061112