
Focus on the Contribution of Oxidative Stress in Skin Aging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
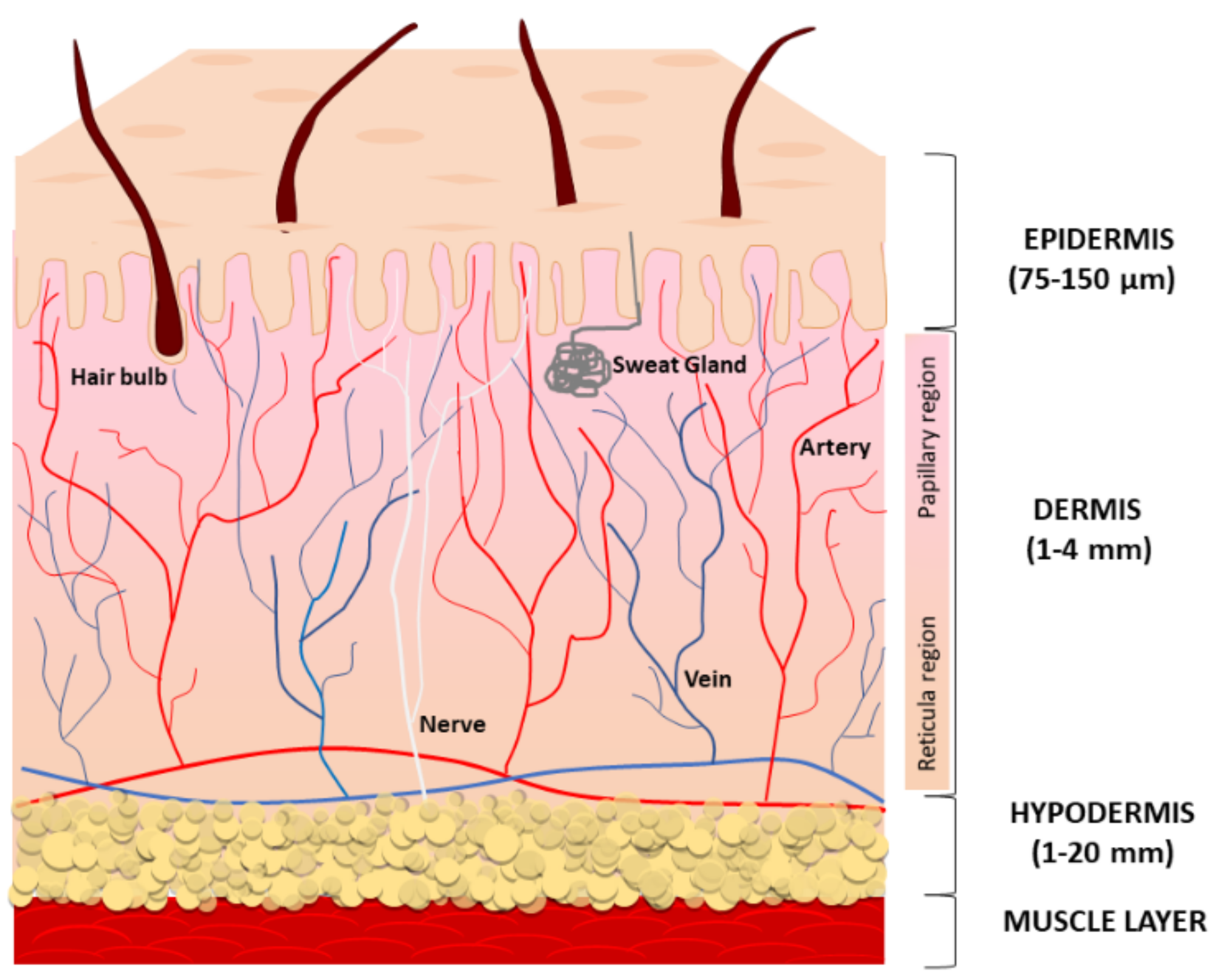
1.1. Human Skin Tissue Structure and Biology
1.2. Skin Antioxidant Defense System
1.3. A Brief Introduction to Skin Aging
2. The Role of Oxidative Stress in Chronological Senescence of the Skin
3. The Role of Oxidative Stress in Extrinsic Aging of the Skin
4. ROS and Hair Greying
5. Contribution of ROS in Common Age-Related Skin Diseases
5.1. Evidence of the Impact of ROS and Age-Related Skin Alteration on Tissue Vulnerability
5.2. Contribution of ROS in Age-Related Skin Cancers
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nielsen, K.P.; Zhao, L.; Stamnes, J.J.; Stamnes, K.; Moan, J. The importance of the depth distribution of melanin in skin for DNA protection and other photobiological processes. J. Photochem. Photobiol. B 2006, 82, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Boulais, N.; Misery, L. Merkel cells. J. Am. Acad. Dermatol. 2007, 57, 147–165. [Google Scholar] [CrossRef]
- Matsui, T.; Amagai, M. Dissecting the formation, structure and barrier function of the stratum corneum. Int. Immunol. 2015, 27, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Eckhart, L.; Tschachler, E.; Gruber, F. Autophagic Control of Skin Aging. Front. Cell Dev. Biol. 2019, 7, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halprin, K.M. Epidermal “turnover time”—A re-examination. Br. J. Dermatol. 1972, 86, 14–19. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Gunn, D.A.; Adams, P.D.; Pawlikowski, J.S.; Griffiths, C.E.; van Heemst, D.; Slagboom, P.E.; Westendorp, R.G.; Maier, A.B. P16INK4a Positive Cells in Human Skin Are Indicative of Local Elastic Fiber Morphology, Facial Wrinkling, and Perceived Age. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin melanocytes: Biology and development. Postepy Derm. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.C. The migratory pathway of neural crest cells into the skin of mouse embryos. Dev. Biol. 1973, 34, 39–46. [Google Scholar] [CrossRef]
- Birlea, S.A.; Costin, G.E.; Roop, D.R.; Norris, D.A. Trends in Regenerative Medicine: Repigmentation in Vitiligo Through Melanocyte Stem Cell Mobilization. Med. Res. Rev. 2017, 37, 907–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Fisher, D.E. Melanocyte stem cells as potential therapeutics in skin disorders. Expert Opin. Biol. Ther. 2014, 14, 1569–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Fukunaga-Kalabis, M.; Yu, H.; Xu, X.; Kong, J.; Lee, J.T.; Herlyn, M. Human dermal stem cells differentiate into functional epidermal melanocytes. J. Cell Sci. 2010, 123, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoerter, J.D.; Bradley, P.; Casillas, A.; Chambers, D.; Denholm, C.; Johnson, K.; Weiswasser, B. Extrafollicular dermal melanocyte stem cells and melanoma. Stem Cells Int. 2012, 2012, 407079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsepkolenko, A.; Tsepkolenko, V.; Dash, S.; Mishra, A.; Bader, A.; Melerzanov, A.; Giri, S. The regenerative potential of skin and the immune system. Clin. Cosmet. Investig. Dermatol. 2019, 12, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. FASEB J. 1997, 11, 51–59. [Google Scholar] [CrossRef]
- De Laporte, L.; Rice, J.J.; Tortelli, F.; Hubbell, J.A. Tenascin C promiscuously binds growth factors via its fifth fibronectin type III-like domain. PLoS ONE 2013, 8, e62076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briquez, P.S.; Hubbell, J.A.; Martino, M.M. Extracellular Matrix-Inspired Growth Factor Delivery Systems for Skin Wound Healing. Adv. Wound Care 2015, 4, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, K.; Pei, M. Age associated communication between cells and matrix: A potential impact on stem cell-based tissue regeneration strategies. Organogenesis 2014, 10, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, A.; Oh, S.J. Age related changes of the extracellular matrix and stem cell maintenance. Prev. Med. 2012, 54, S50–S56. [Google Scholar] [CrossRef] [PubMed]
- Asumda, F.Z. Age-associated changes in the ecological niche: Implications for mesenchymal stem cell aging. Stem. Cell Res. Ther. 2013, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihermann, A.C.; Lorencini, M.; Brohem, C.A.; de Carvalho, C.M. Elastin structure and its involvement in skin photoageing. Int. J. Cosmet. Sci. 2017, 39, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Juarez, C.F.; Plikus, M.V. Emerging nonmetabolic functions of skin fat. Nat. Rev. Endocrinol. 2018, 14, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Alam, W.; Hasson, J.; Reed, M. Clinical approach to chronic wound management in older adults. J. Am. Geriatr. Soc. 2021, 69, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Guerrero-Juarez, C.F.; Hata, T.; Bapat, S.P.; Ramos, R.; Plikus, M.V.; Gallo, R.L. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015, 347, 67–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellei, B.; Migliano, E.; Tedesco, M.; Caputo, S.; Papaccio, F.; Lopez, G.; Picardo, M. Adipose tissue-derived extracellular fraction characterization: Biological and clinical considerations in regenerative medicine. Stem Cell Res. Ther. 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Mazini, L.; Malka, G.; Zeller, M.; Cottin, Y.; Vergely, C. The Crosstalk of Adipose-Derived Stem Cells (ADSC), Oxidative Stress, and Inflammation in Protective and Adaptive Responses. Int. J. Mol. Sci. 2020, 21, 9262. [Google Scholar] [CrossRef] [PubMed]
- Bliznakov, E.G. Aging, mitochondria, and coenzyme Q (10): The neglected relationship. Biochimie 1999, 81, 1131–1132. [Google Scholar] [PubMed]
- Krol, E.S.; Kramer-Stickland, K.A.; Liebler, D.C. Photoprotective actions of topically applied vitamin E. Drug Metab. Rev. 2000, 32, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.; Valacchi, G. Antioxidants and the response of skin to oxidative stress: Vitamin E as a key indicator. Skin Pharmacol. Appl. Skin Physiol. 2002, 15, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.J.; Traber, M.G.; Tsang, K.; Cross, C.E.; Packer, L. In vivo exposure to ozone depletes vitamins C and E and induces lipid peroxidation in epidermal layers of murine skin. Free Radic. Biol. Med. 1997, 23, 385–391. [Google Scholar] [CrossRef]
- Rhie, G.; Shin, M.H.; Seo, J.Y.; Choi, W.W.; Cho, K.H.; Kim, K.H.; Park, K.C.; Eun, H.C.; Chung, J.H. Aging- and photoaging-dependent changes of enzymic and nonenzymic antioxidants in the epidermis and dermis of human skin in vivo. J. Investig. Dermatol. 2001, 117, 1212–1217. [Google Scholar] [CrossRef] [Green Version]
- McArdle, F.; Rhodes, L.E.; Parslew, R.; Jack, C.I.; Friedmann, P.S.; Jackson, M.J. UVR-induced oxidative stress in human skin in vivo: Effects of oral vitamin C supplementation. Free Radic. Biol. Med. 2002, 33, 1355–1362. [Google Scholar] [CrossRef]
- Geesin, J.C.; Darr, D.; Kaufman, R.; Murad, S.; Pinnell, S.R. Ascorbic acid specifically increases type I and type III procollagen messenger RNA levels in human skin fibroblast. J. Investig. Dermatol. 1988, 90, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.M.; LuValle, P.A.; Zoia, O.; Quaglino, D., Jr.; Giro, M. Ascorbate differentially regulates elastin and collagen biosynthesis in vascular smooth muscle cells and skin fibroblasts by pretranslational mechanisms. J. Biol. Chem. 1997, 272, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Burke, K.E. Interaction of vitamins C and E as better cosmeceuticals. Dermatol. Ther. 2007, 20, 314–321. [Google Scholar] [CrossRef]
- Landis, G.N.; Tower, J. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 2005, 126, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Saxena, P.; Selvaraj, K.; Khare, S.K.; Chaudhary, N. Superoxide dismutase as multipotent therapeutic antioxidant enzyme: Role in human diseases. Biotechnol. Lett. 2022, 44, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Inagaki, J.; Saito, M.; Ikeda, Y.; Tsuda, C.; Noda, Y.; Kawakami, S.; Shirasawa, T.; Shimizu, T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun. 2009, 382, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, N.; Maity, P.; Singh, K.; Ferchiu, F.; Wlaschek, M.; Scharffetter-Kochanek, K. The role of manganese superoxide dismutase in skin aging. Dermatoendocrinology 2012, 4, 232–235. [Google Scholar] [CrossRef] [Green Version]
- Treiber, N.; Maity, P.; Singh, K.; Kohn, M.; Keist, A.F.; Ferchiu, F.; Sante, L.; Frese, S.; Bloch, W.; Kreppel, F.; et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 2011, 10, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Demaria, M.; Melov, S.; Campisi, J. Pleiotropic age-dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10407–10412. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.H.; Rhie, G.E.; Kim, Y.K.; Park, C.H.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. H2O2 accumulation by catalase reduction changes MAP kinase signaling in aged human skin in vivo. J. Investig. Dermatol. 2005, 125, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.Y.; Lee, H.C.; Fahn, H.J.; Wei, Y.H. Oxidative damage elicited by imbalance of free radical scavenging enzymes is associated with large-scale mtDNA deletions in aging human skin. Mutat. Res. 1999, 423, 11–21. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [Green Version]
- Hellemans, L.; Corstjens, H.; Neven, A.; Declercq, L.; Maes, D. Antioxidant enzyme activity in human stratum corneum shows seasonal variation with an age-dependent recovery. J. Investig. Dermatol. 2003, 120, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung-Htut, M.T.; Ayer, A.; Breitenbach, M.; Dawes, I.W. Oxidative stresses and ageing. Subcell. Biochem. 2012, 57, 13–54. [Google Scholar]
- Lopez-Torres, M.; Shindo, Y.; Packer, L. Effect of age on antioxidants and molecular markers of oxidative damage in murine epidermis and dermis. J. Investig. Dermatol. 1994, 102, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Poljšak, B.; Dahmane, R. Free radicals and extrinsic skin aging. Dermatol. Res. Pract. 2012, 2012, 135206. [Google Scholar] [CrossRef] [Green Version]
- Vermeij, W.P.; Alia, A.; Backendorf, C. ROS quenching potential of the epidermal cornified cell envelope. J. Investig. Dermatol. 2011, 131, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Investig. Dermatol. 1994, 102, 122–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S.U.; Thiele, J.J.; Cross, C.E.; Packer, L. Vitamin C, uric acid, and glutathione gradients in murine stratum corneum and their susceptibility to ozone exposure. J. Investig. Dermatol. 1999, 113, 1128–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodby, B.; Penta, K.; Pecorelli, A.; Lila, M.A.; Valacchi, G. Skin Health from the Inside Out. Annu. Rev. Food Sci. Technol. 2020, 11, 235–254. [Google Scholar] [CrossRef] [Green Version]
- Parrado, C.; Mercado-Saenz, S.; Perez-Davo, A.; Gilaberte, Y.; Gonzalez, S.; Juarranz, A. Environmental Stressors on Skin Aging. Mechanistic Insights. Front. Pharmacol. 2019, 10, 759. [Google Scholar] [CrossRef]
- Sarangarajan, R.; Apte, S.P. The polymerization of melanin: A poorly understood phenomenon with egregious biological implications. Melanoma Res. 2006, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Edwards, C.; Gaskell, S.; Pearse, A.; Marks, R. Melanin content and distribution in the surface corneocyte with skin phototypes. Br. J. Dermatol. 1996, 135, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Wendt, J.; Rauscher, S.; Burgstaller-Muehlbacher, S.; Fae, I.; Fischer, G.; Pehamberger, H.; Okamoto, I. Human Determinants and the Role of Melanocortin-1 Receptor Variants in Melanoma Risk Independent of UV Radiation Exposure. JAMA Dermatol. 2016, 152, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Duan, E. Fighting against Skin Aging: The Way from Bench to Bedside. Cell Transplant. 2018, 27, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Zouboulis, C.C.; Makrantonaki, E.; Nikolakis, G. When the skin is in the center of interest: An aging issue. Clin. Dermatol. 2019, 37, 296–305. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Bekou, V.; Zouboulis, C.C. Genetics and skin aging. Dermatoendocrinology 2012, 4, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, S.H.; Pappas, A. Differentiation and characterization of human facial subcutaneous adipocytes. Adipocyte 2014, 4, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.B. Physiology of sweat gland function: The roles of sweating and sweat composition in human health. Temperature 2019, 6, 211–259. [Google Scholar] [CrossRef] [Green Version]
- Buffoli, B.; Rinaldi, F.; Labanca, M.; Sorbellini, E.; Trink, A.; Guanziroli, E.; Rezzani, R.; Rodella, L.F. The human hair: From anatomy to physiology. Int. J. Dermatol. 2014, 53, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Velarde, M.C.; Demaria, M. Targeting Senescent Cells: Possible Implications for Delaying Skin Aging: A Mini-Review. Gerontology 2016, 62, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathi, E.; Charoudeh, H.N.; Sanaat, Z.; Farahzadi, R. Telomere shortening as a hallmark of stem cell senescence. Stem Cell Investig. 2019, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kornicka, K.; Marycz, K.; Tomaszewski, K.A.; Marędziak, M.; Śmieszek, A. The Effect of Age on Osteogenic and Adipogenic Differentiation Potential of Human Adipose Derived Stromal Stem Cells (hASCs) and the Impact of Stress Factors in the Course of the Differentiation Process. Oxid. Med. Cell. Longev. 2015, 2015, 309169. [Google Scholar] [CrossRef] [PubMed]
- Orciani, M.; Gorbi, S.; Benedetti, M.; Di Benedetto, G.; Mattioli-Belmonte, M.; Regoli, F.; Di Primio, R. Oxidative stress defense in human-skin-derived mesenchymal stem cells versus human keratinocytes: Different mechanisms of protection and cell selection. Free Radic. Biol. Med. 2010, 49, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.C.; Hsieh, T.Y.; Lai, H.S.; Young, T.H. High glucose-induced reactive oxygen species generation promotes stemness in human adipose-derived stem cells. Cytotherapy 2016, 18, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Saretzki, G.; Döcke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Saretzki, G.; Murphy, M.P.; von Zglinicki, T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell 2003, 2, 141–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.J.; Hwang, Y.C.; Kang, C.M.; Choy, H.E.; Park, S.C. Reduction of UV-induced cell death in the human senescent fibroblasts. Mol. Cells 2000, 10, 415–422. [Google Scholar] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Melo Pereira, S.; Ribeiro, R.; Logarinho, E. Approaches towards Longevity: Reprogramming, Senolysis, and Improved Mitotic Competence as Anti-Aging Therapies. Int. J. Mol. Sci. 2019, 20, 938. [Google Scholar] [CrossRef] [Green Version]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 2019, 18, e12841. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Disease or not, aging is easily treatable. Aging 2018, 10, 3067–3078. [Google Scholar] [CrossRef]
- Tuttle, C.S.L.; Waaijer, M.E.C.; Slee-Valentijn, M.S.; Stijnen, T.; Westendorp, R.; Maier, A.B. Cellular senescence and chronological age in various human tissues: A systematic review and meta-analysis. Aging Cell 2020, 19, e13083. [Google Scholar] [CrossRef] [Green Version]
- Bellei, B.; Picardo, M. Premature cell senescence in human skin: Dual face in chronic acquired pigmentary disorders. Ageing Res. Rev. 2020, 57, 100981. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Sahin, E.; Depinho, R.A. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 2010, 464, 520–528. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef]
- Krutmann, J.; Schikowski, T.; Morita, A.; Berneburg, M. Environmentally-Induced (Extrinsic) Skin Aging: Exposomal Factors and Underlying Mechanisms. J. Investig. Dermatol. 2021, 141, 1096–1103. [Google Scholar] [CrossRef]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Bellei, B.; Migliano, E.; Picardo, M. Therapeutic potential of adipose tissue-derivatives in modern dermatology. Exp. Dermatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Garcovich, S. Adipose-Derived Mesenchymal Stem Cells (AD-MSCs) against Ultraviolet (UV) Radiation Effects and the Skin Photoaging. Biomedicines 2021, 9, 532. [Google Scholar] [CrossRef]
- Passeron, T.; Krutmann, J.; Andersen, M.L.; Katta, R.; Zouboulis, C.C. Clinical and biological impact of the exposome on the skin. J. Eur. Acad. Dermatol. Venereol. 2020, 34 (Suppl. S4), 4–25. [Google Scholar] [CrossRef]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef] [PubMed]
- Friedman, O. Changes associated with the aging face. Facial Plast. Surg. Clin. N. Am. 2005, 13, 371–380. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Blog, F.B.; Szabo, G. Effects of aging and chronic sun exposure on melanocytes in human skin. J. Investig. Dermatol. 1979, 73, 141–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordlund, J.J. The lives of pigment cells. Dermatol. Clin. 1986, 4, 407–418. [Google Scholar] [CrossRef]
- Kim, M.; Kim, S.M.; Kwon, S.; Park, T.J.; Kang, H.Y. Senescent fibroblasts in melasma pathophysiology. Exp. Dermatol. 2019, 28, 719–722. [Google Scholar] [CrossRef]
- Kang, H.Y.; Lee, J.W.; Papaccio, F.; Bellei, B.; Picardo, M. Alterations of the pigmentation system in the aging process. Pigment Cell Melanoma Res. 2021, 34, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Rittié, L.; Fisher, G.J. Natural and sun-induced aging of human skin. Cold Spring Harb. Perspect. Med. 2015, 5, a015370. [Google Scholar] [CrossRef]
- El-Domyati, M.; Medhat, W.; Abdel-Wahab, H.M.; Moftah, N.H.; Nasif, G.A.; Hosam, W. Forehead wrinkles: A histological and immunohistochemical evaluation. J. Cosmet. Dermatol. 2014, 13, 188–194. [Google Scholar] [CrossRef]
- Labat-Robert, J.; Fourtanier, A.; Boyer-Lafargue, B.; Robert, L. Age dependent increase of elastase type protease activity in mouse skin. Effect of UV-irradiation. J. Photochem. Photobiol. B 2000, 57, 113–118. [Google Scholar] [CrossRef]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, T.; Li, S.; Jiang, X.; Oberley, L.W. Altered levels of primary antioxidant enzymes in progeria skin fibroblasts. Biochem. Biophys. Res. Commun. 1999, 257, 163–167. [Google Scholar] [CrossRef]
- Chung, J.H.; Kang, S.; Varani, J.; Lin, J.; Fisher, G.J.; Voorhees, J.J. Decreased extracellular-signal-regulated kinase and increased stress-activated MAP kinase activities in aged human skin in vivo. J. Investig. Dermatol. 2000, 115, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef] [Green Version]
- Boo, Y.C. Natural Nrf2 Modulators for Skin Protection. Antioxidants 2020, 9, 812. [Google Scholar] [CrossRef]
- Fisher, G.J.; Datta, S.C.; Talwar, H.S.; Wang, Z.Q.; Varani, J.; Kang, S.; Voorhees, J.J. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature 1996, 379, 335–339. [Google Scholar] [CrossRef]
- Kohl, E.; Steinbauer, J.; Landthaler, M.; Szeimies, R.M. Skin ageing. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 873–884. [Google Scholar] [CrossRef]
- Quan, T.; He, T.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces Smad7 via induction of transcription factor AP-1 in human skin fibroblasts. J. Biol. Chem. 2005, 280, 8079–8085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.; Qin, Z.; Xu, Y.; He, T.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation induces CYR61/CCN1, a mediator of collagen homeostasis, through activation of transcription factor AP-1 in human skin fibroblasts. J. Investig. Dermatol. 2010, 130, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Haustead, D.J.; Stevenson, A.; Saxena, V.; Marriage, F.; Firth, M.; Silla, R.; Martin, L.; Adcroft, K.F.; Rea, S.; Day, P.J.; et al. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-κB. Sci. Rep. 2016, 6, 26846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef] [PubMed]
- Meinke, M.C.; Busch, L.; Lohan, S.B. Wavelength, dose, skin type and skin model related radical formation in skin. Biophys. Rev. 2021, 13, 1091–1100. [Google Scholar] [CrossRef]
- Okayama, Y. Oxidative stress in allergic and inflammatory skin diseases. Curr. Drug Targets Inflamm. Allergy 2005, 4, 517–519. [Google Scholar] [CrossRef]
- Kandola, K.; Bowman, A.; Birch-Machin, M.A. Oxidative stress—A key emerging impact factor in health, ageing, lifestyle and aesthetics. Int. J. Cosmet. Sci. 2015, 37 (Suppl. S2), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cedikova, M.; Pitule, P.; Kripnerova, M.; Markova, M.; Kuncova, J. Multiple roles of mitochondria in aging processes. Physiol. Res. 2016, 65, S519–S531. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, K.; Hanna, R.; Birch-Machin, M.A. What is the role of mitochondrial dysfunction in skin photoaging? Exp. Dermatol. 2018, 27, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995, 27, 647–653. [Google Scholar] [CrossRef]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Yang, J.H.; Lee, H.C.; Wei, Y.H. Photoageing-associated mitochondrial DNA length mutations in human skin. Arch. Dermatol. Res. 1995, 287, 641–648. [Google Scholar] [PubMed]
- Kaneko, N.; Vierkoetter, A.; Kraemer, U.; Sugiri, D.; Matsui, M.; Yamamoto, A.; Krutmann, J.; Morita, A. Mitochondrial common deletion mutation and extrinsic skin ageing in German and Japanese women. Exp. Dermatol. 2012, 21 (Suppl. S1), 26–30. [Google Scholar] [CrossRef] [PubMed]
- Mellem, D.; Sattler, M.; Pagel-Wolff, S.; Jaspers, S.; Wenck, H.; Rübhausen, M.A.; Fischer, F. Fragmentation of the mitochondrial network in skin in vivo. PLoS ONE 2017, 12, e0174469. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Albensi, B.C. The nuclear factor kappa B (NF-κB) signaling pathway is involved in ammonia-induced mitochondrial dysfunction. Mitochondrion 2021, 57, 63–75. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell senescence, rapamycin and hyperfunction theory of aging. Cell Cycle 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. The role of cellular senescence in skin aging. J. Investig. Dermatol. Symp. Proc. 1998, 3, 1–5. [Google Scholar]
- Blagosklonny, M.V. Aging and immortality: Quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle 2006, 5, 2087–2102. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.; Bates, P.; Fisher, R.; Richardson, L.; Graham, C.F. Disproportionate growth in mice with Igf-2 transgenes. Proc. Natl. Acad. Sci. USA 1994, 91, 10365–10369. [Google Scholar] [CrossRef] [Green Version]
- Makrantonaki, E.; Zouboulis, C.C. German National Genome Research Network 2 The skin as a mirror of the aging process in the human organism—State of the art and results of the aging research in the German National Genome Research Network 2 (NGFN-2). Exp. Gerontol. 2007, 42, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luebberding, S.; Krueger, N.; Kerscher, M. Age-related changes in skin barrier function–Quantitative evaluation of 150 female subjects. Int. J. Cosmet. Sci. 2013, 35, 183–190. [Google Scholar] [CrossRef]
- Feng, J.; Luo, J.; Yang, P.; Du, J.; Kim, B.S.; Hu, H. Piezo2 channel-Merkel cell signaling modulates the conversion of touch to itch. Science 2018, 360, 530–533. [Google Scholar] [CrossRef] [Green Version]
- Ale, I.S.; Maibach, H.I. Irritant contact dermatitis. Rev. Environ. Health 2014, 29, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.J. Introduction to skin aging. J. Tissue Viability 2017, 26, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Eun, H.C. Angiogenesis in skin aging and photoaging. J. Dermatol. 2007, 34, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Pierzga, J.M.; Frymoyer, A.; Kenney, W.L. Delayed distribution of active vasodilation and altered vascular conductance in aged skin. J. Appl. Physiol. 2003, 94, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Bonta, M.; Daina, L.; Muţiu, G. The process of ageing reflected by histological changes in the skin. Rom. J. Morphol. Embryol. 2013, 54, 797–804. [Google Scholar]
- Dufour, A.; Candas, V. Ageing and thermal responses during passive heat exposure: Sweating and sensory aspects. Eur. J. Appl. Physiol. 2007, 100, 19–26. [Google Scholar] [CrossRef]
- Fernandez-Flores, A.; Saeb-Lima, M.; Cassarino, D.S. Histopathology of aging of the hair follicle. J. Cutan. Pathol. 2019, 46, 508–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flood, K.S.; Houston, N.A.; Savage, K.T.; Kimball, A.B. Genetic basis for skin youthfulness. Clin. Dermatol. 2019, 37, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Naval, J.; Alonso, V.; Herranz, M.A. Genetic polymorphisms and skin aging: The identification of population genotypic groups holds potential for personalized treatments. Clin. Cosmet. Investig. Dermatol. 2014, 7, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.C.; Wakamatsu, K.; Ito, S.; Kadekaro, A.L.; Kobayashi, N.; Groden, J.; Kavanagh, R.; Takakuwa, T.; Virador, V.; Hearing, V.J.; et al. Human melanocortin 1 receptor variants, receptor function and melanocyte response to UV radiation. J. Cell Sci. 2002, 115, 2349–2355. [Google Scholar] [CrossRef]
- Castejón-Griñán, M.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Chen, J.; Yang, J.; Chen, S.; Jameson, J.; Swope, V.B.; Cheng, T.; Kadakia, M.; Abdel-Malek, Z. Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol. Cancer Res. 2012, 10, 778–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henri, P.; Beaumel, S.; Guezennec, A.; Poumès, C.; Stoebner, P.E.; Stasia, M.J.; Guesnet, J.; Martinez, J.; Meunier, L. MC1R expression in HaCaT keratinocytes inhibits UVA-induced ROS production via NADPH oxidase- and cAMP-dependent mechanisms. J. Cell Physiol. 2012, 227, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Mosby, N.; Yang, J.; Xu, A.; Abdel-Malek, Z.; Kadekaro, A.L. alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment. Cell Melanoma Res. 2009, 22, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Fargnoli, M.C.; Spica, T.; Sera, F.; Pellacani, G.; Chiarugi, A.; Seidenari, S.; Carli, P.; Chimenti, S.; Peris, K. Re: MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J. Natl. Cancer Inst. 2006, 98, 144–145. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Mao, W.; Chen, J.; Goding, C.R.; Cui, R.; Xu, Z.X.; Miao, X. The protective role of MC1R in chromosome stability and centromeric integrity in melanocytes. Cell Death Discov. 2021, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Fargnoli, M.C.; Gandini, S.; Maisonneuve, P.; Liu, F.; Kayser, M.; Nijsten, T.; Han, J.; Kumar, R.; Gruis, N.A.; et al. M-SKIP Study Group MC1R gene variants and non-melanoma skin cancer: A pooled-analysis from the M-SKIP project. Br. J. Cancer 2015, 113, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Elfakir, A.; Ezzedine, K.; Latreille, J.; Ambroisine, L.; Jdid, R.; Galan, P.; Hercberg, S.; Gruber, F.; Malvy, D.; Tschachler, E.; et al. Functional MC1R-gene variants are associated with increased risk for severe photoaging of facial skin. J. Investig. Dermatol. 2010, 130, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Kokot, A.; Sindrilaru, A.; Schiller, M.; Sunderkötter, C.; Kerkhoff, C.; Eckes, B.; Scharffetter-Kochanek, K.; Luger, T.A.; Böhm, M. Alpha-Melanocyte-Stimulating Hormone Suppresses Bleomycin-Induced Collagen Synthesis and Reduces Tissue Fibrosis in a Mouse Model of Scleroderma: Melanocortin Peptides as a Novel Treatment Strategy for Scleroderma? Arthritis Rheum. 2009, 60, 592–603. [Google Scholar] [CrossRef]
- Böhm, M.; Luger, T.A. Melanocortins in fibroblast biology--current update and future perspective for dermatology. Exp. Dermatol. 2004, 13 (Suppl. S4), 16–21. [Google Scholar] [CrossRef]
- De Souza, K.S.; Cantaruti, T.A.; Azevedo, G.M., Jr.; Galdino, D.A.; Rodrigues, C.M.; Costa, R.A.; Vaz, N.M.; Carvalho, C.R. Improved cutaneous wound healing after intraperitoneal injection of alpha-melanocyte-stimulating hormone. Exp. Dermatol. 2015, 24, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Dinparastisaleh, R.; Mirsaeidi, M. Antifibrotic and Anti-Inflammatory Actions of α-Melanocytic Hormone: New Roles for an Old Player. Pharmaceuticals 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Guida, S.; Ciardo, S.; De Pace, B.; De Carvalho, N.; Peccerillo, F.; Manfredini, M.; Farnetani, F.; Chester, J.; Kaleci, S.; Manganelli, M.; et al. The influence of MC1R on dermal morphological features of photo-exposed skin in women revealed by reflectance confocal microscopy and optical coherence tomography. Exp. Dermatol. 2019, 28, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Pain, S.; Dezutter, C.; Reymermier, C.; Vogelgesang, B.; Delay, E.; André, V. Age-related changes in pro-opiomelanocortin (POMC) and related receptors in human epidermis. Int. J. Cosmet. Sci. 2010, 32, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.C.; Hamer, M.A.; Gunn, D.A.; Deelen, J.; Lall, J.S.; van Heemst, D.; Uh, H.W.; Hofman, A.; Uitterlinden, A.G.; Griffiths, C.E.M.; et al. A Genome-Wide Association Study Identifies the Skin Color Genes IRF4, MC1R, ASIP, and BNC2 Influencing Facial Pigmented Spots. J. Investig. Dermatol. 2015, 135, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Tan, J.; Hüls, A.; Ding, A.; Liu, Y.; Matsui, M.S.; Vierkötter, A.; Krutmann, J.; Schikowski, T.; Jin, L.; et al. Genetic variants associated with skin aging in the Chinese Han population. J. Dermatol. Sci. 2017, 86, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kang, S.; Lee, W.J. Menopause, Ultraviolet Exposure, and Low Water Intake Potentially Interact with the Genetic Variants Related to Collagen Metabolism Involved in Skin Wrinkle Risk in Middle-Aged Women. Int. J. Environ. Res. Public Health 2021, 18, 2044. [Google Scholar] [CrossRef] [PubMed]
- Orioli, D.; Dellambra, E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells 2018, 7, 268. [Google Scholar] [CrossRef] [Green Version]
- Bormann, F.; Rodríguez-Paredes, M.; Hagemann, S.; Manchanda, H.; Kristof, B.; Gutekunst, J.; Raddatz, G.; Haas, R.; Terstegen, L.; Wenck, H.; et al. Reduced DNA methylation patterning and transcriptional connectivity define human skin aging. Aging Cell 2016, 15, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, G.; Hagemann, S.; Aran, D.; Söhle, J.; Kulkarni, P.P.; Kaderali, L.; Hellman, A.; Winnefeld, M.; Lyko, F. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenet. Chromatin 2013, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Baumann, L. Skin ageing and its treatment. J. Pathol. 2007, 211, 241–251. [Google Scholar] [CrossRef]
- Szabo, G. The number of melanocytes in human epidermis. Br. Med. J. 1954, 1, 1016–1017. [Google Scholar] [CrossRef]
- Fitzpatrick, T.B. Hypomelanosis. South. Med. J. 1964, 57, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.P. Pigmentary changes of the ageing skin. Br. J. Dermatol. 1990, 122 (Suppl. S35), 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shlivko, I.L.; Petrova, G.A.; Zor’kina, M.V.; Tchekalkina, O.E.; Firsova, M.S.; Ellinsky, D.O.; Agrba, P.D.; Kamensky, V.A.; Donchenko, E.V. Complex assessment of age-specific morphofunctional features of skin of different anatomic localizations. Skin Res. Technol. 2013, 19, e85–e92. [Google Scholar] [CrossRef]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Maize, J.C.; Foster, G. Age-related changes in melanocytic naevi. Clin Exp. Dermatol. 1979, 4, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Chikeka, I.; Hornyak, T.J. Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence. Dermatol. Clin. 2017, 35, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef]
- Waaijer, M.E.; Parish, W.E.; Strongitharm, B.H.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.; Sedivy, J.M.; Westendorp, R.G.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Adamus, J.; Aho, S.; Meldrum, H.; Bosko, C.; Lee, J.M. p16INK4A influences the aging phenotype in the living skin equivalent. J. Investig. Dermatol. 2014, 134, 1131–1133. [Google Scholar] [CrossRef] [Green Version]
- Gilhar, A.; Ullmann, Y.; Karry, R.; Shalaginov, R.; Assy, B.; Serafimovich, S.; Kalish, R.S. Ageing of human epidermis: The role of apoptosis, Fas and telomerase. Br. J. Dermatol. 2004, 150, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Giangreco, A.; Qin, M.; Pintar, J.E.; Watt, F.M. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 2008, 7, 250–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, S.; Ray, S.; Tarone, R.E.; Kraemer, K.H.; Grossman, L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: Reduced DNA repair capacity and increased DNA mutability. Mutat. Res. 1996, 364, 117–123. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, J.; Dame, M.K.; Rittie, L.; Fligiel, S.E.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Pathol. 2006, 168, 1861–1868. [Google Scholar] [CrossRef] [Green Version]
- Le Varlet, B.; Chaudagne, C.; Saunois, A.; Barré, P.; Sauvage, C.; Berthouloux, B.; Meybeck, A.; Dumas, M.; Bonté, F. Age-related functional and structural changes in human dermo-epidermal junction components. J. Investig. Dermatol. Symp. Proc. 1998, 3, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langton, A.K.; Halai, P.; Griffiths, C.E.; Sherratt, M.J.; Watson, R.E. The impact of intrinsic ageing on the protein composition of the dermal-epidermal junction. Mech. Ageing Dev. 2016, 156, 14–16. [Google Scholar] [CrossRef]
- Waldera Lupa, D.M.; Kalfalah, F.; Safferling, K.; Boukamp, P.; Poschmann, G.; Volpi, E.; Götz-Rösch, C.; Bernerd, F.; Haag, L.; Huebenthal, U.; et al. Characterization of Skin Aging-Associated Secreted Proteins (SAASP) Produced by Dermal Fibroblasts Isolated from Intrinsically Aged Human Skin. J. Investig. Dermatol. 2015, 135, 1954–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, G.J.; Shao, Y.; He, T.; Qin, Z.; Perry, D.; Voorhees, J.J.; Quan, T. Reduction of fibroblast size/mechanical force down-regulates TGF-β type II receptor: Implications for human skin aging. Aging Cell 2016, 15, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Smith, M.J.; Siow, R.C.M.; Liu, K.K. Ageing modulates human dermal fibroblast contractility: Quantification using nano-biomechanical testing. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118972. [Google Scholar] [CrossRef]
- Brun, C.; Jean-Louis, F.; Oddos, T.; Bagot, M.; Bensussan, A.; Michel, L. Phenotypic and functional changes in dermal primary fibroblasts isolated from intrinsically aged human skin. Exp. Dermatol. 2016, 25, 113–119. [Google Scholar] [CrossRef]
- Yuan, W.; Varga, J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad. J. Biol. Chem. 2001, 276, 38502–38510. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Quan, T.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative exposure impairs TGF-β pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age 2014, 36, 9623. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashcroft, G.S.; Herrick, S.E.; Tarnuzzer, R.W.; Horan, M.A.; Schultz, G.S.; Ferguson, M.W. Human ageing impairs injury-induced in vivo expression of tissue inhibitor of matrix metalloproteinases (TIMP)-1 and -2 proteins and mRNA. J. Pathol. 1997, 183, 169–176. [Google Scholar] [CrossRef]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Stephan-Otto Attolini, C.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef] [Green Version]
- Bayreuther, K.; Rodemann, H.P.; Hommel, R.; Dittmann, K.; Albiez, M.; Francz, P.I. Human skin fibroblasts in vitro differentiate along a terminal cell lineage. Proc. Natl. Acad Sci. USA 1988, 85, 5112–5116. [Google Scholar] [CrossRef] [Green Version]
- Mine, S.; Fortunel, N.O.; Pageon, H.; Asselineau, D. Aging alters functionally human dermal papillary fibroblasts but not reticular fibroblasts: A new view of skin morphogenesis and aging. PLoS ONE 2008, 3, e4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorteau, J.; Chevalier, F.P.; Bonnet, S.; Barthélemy, T.; Lopez-Gaydon, A.; Martin, L.S.; Bechetoille, N.; Lamartine, J. Maintenance of Chronological Aging Features in Culture of Normal Human Dermal Fibroblasts from Old Donors. Cells 2022, 11, 858. [Google Scholar] [CrossRef]
- Solé-Boldo, L.; Raddatz, G.; Schütz, S.; Mallm, J.P.; Rippe, K.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef] [Green Version]
- Janson, D.; Rietveld, M.; Mahé, C.; Saintigny, G.; El Ghalbzouri, A. Differential effect of extracellular matrix derived from papillary and reticular fibroblasts on epidermal development in vitro. Eur. J. Dermatol. 2017, 27, 237–246. [Google Scholar] [CrossRef]
- Haydont, V.; Neiveyans, V.; Perez, P.; Busson, É.; Lataillade, J.; Asselineau, D.; Fortunel, N.O. Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling. Cells 2020, 9, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Characteristics of the Aging Skin. Adv. Wound Care 2013, 2, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvioli, S.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Bacalini, M.G.; Garagnani, P.; Giuliani, C.; Fontanesi, E.; Ostan, R.; et al. Immune system, cell senescence, aging and longevity--inflamm-aging reappraised. Curr. Pharm. Des. 2013, 19, 1675–1679. [Google Scholar]
- Chen, B.; Yang, J.; Song, Y.; Zhang, D.; Hao, F. Skin Immunosenescence and Type 2 Inflammation: A Mini-Review With an Inflammaging Perspective. Front. Cell Dev. Biol. 2022, 10, 835675. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, T.G.; Kim, S.H.; Park, J.Y.; Lee, M.; Lee, J.W.; Lee, S.H.; Lee, M.G. Epidermal Barrier Function Is Impaired in Langerhans Cell-Depleted Mice. J. Investig. Dermatol. 2019, 139, 1182–1185. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Dearman, R.J.; Kimber, I.; Griffiths, C.E.M. Langerhans cells express human β-defensin 3: Relevance for immunity during skin ageing. Br. J. Dermatol. 2018, 179, 1170–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, M.G.; Markofski, M.M.; Carrillo, A.E. Elevated Inflammatory Status and Increased Risk of Chronic Disease in Chronological Aging: Inflamm-aging or Inflamm-inactivity? Aging Dis. 2019, 10, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Szántó, M.; Dózsa, A.; Antal, D.; Szabó, K.; Kemény, L.; Bai, P. Targeting the gut-skin axis-Probiotics as new tools for skin disorder management? Exp. Dermatol. 2019, 28, 1210–1218. [Google Scholar] [CrossRef] [Green Version]
- Boyajian, J.L.; Ghebretatios, M.; Schaly, S.; Islam, P.; Prakash, S. Microbiome and Human Aging: Probiotic and Prebiotic Potentials in Longevity, Skin Health and Cellular Senescence. Nutrients 2021, 13, 4550. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 16748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavker, R.M.; Gerberick, G.F.; Veres, D.; Irwin, C.J.; Kaidbey, K.H. Cumulative effects from repeated exposures to suberythemal doses of UVB and UVA in human skin. J. Am. Acad. Dermatol. 1995, 32, 53–62. [Google Scholar] [CrossRef]
- Oikarinen, A. The aging of skin: Chronoaging versus photoaging. Photodermatol. Photoimmunol. Photomed. 1990, 7, 3–4. [Google Scholar]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-degrading metalloproteinases in photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contet-Audonneau, J.L.; Jeanmaire, C.; Pauly, G. A histological study of human wrinkle structures: Comparison between sun-exposed areas of the face, with or without wrinkles, and sun-protected areas. Br. J. Dermatol. 1999, 140, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Gilchrest, B.A. Photoaging. J. Investig. Dermatol. 2013, 133, E2–E6. [Google Scholar] [CrossRef] [Green Version]
- Oresajo, C.; Pillai, S.; Manco, M.; Yatskayer, M.; McDaniel, D. Antioxidants and the skin: Understanding formulation and efficacy. Dermatol. Ther. 2012, 25, 252–259. [Google Scholar] [CrossRef]
- Georgakopoulou, E.; Evangelou, K.; Havaki, S.; Townsend, P.; Kanavaros, P.; Gorgoulis, V.G. Apoptosis or senescence? Which exit route do epithelial cells and fibroblasts preferentially follow? Mech. Ageing Dev. 2016, 156, 17–24. [Google Scholar] [CrossRef]
- Gupta, V.; Sharma, V.K. Skin typing: Fitzpatrick grading and others. Clin. Dermatol. 2019, 37, 430–436. [Google Scholar] [CrossRef]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef]
- Schwarz, T.; Luger, T.A. Effect of UV irradiation on epidermal cell cytokine production. J. Photochem. Photobiol. B 1989, 4, 1–13. [Google Scholar] [CrossRef]
- Li, Y.; Lei, D.; Swindell, W.R.; Xia, W.; Weng, S.; Fu, J.; Worthen, C.A.; Okubo, T.; Johnston, A.; Gudjonsson, J.E.; et al. Age-Associated Increase in Skin Fibroblast-Derived Prostaglandin E2 Contributes to Reduced Collagen Levels in Elderly Human Skin. J. Investig. Dermatol. 2015, 135, 2181–2188. [Google Scholar] [CrossRef] [Green Version]
- Habib, M.A.; Salem, S.A.; Hakim, S.A.; Shalan, Y.A. Comparative immunohistochemical assessment of cutaneous cyclooxygenase-2 enzyme expression in chronological aging and photoaging. Photodermatol. Photoimmunol. Photomed. 2014, 30, 43–51. [Google Scholar] [CrossRef]
- Bosset, S.; Bonnet-Duquennoy, M.; Barré, P.; Chalon, A.; Kurfurst, R.; Bonté, F.; Schnébert, S.; Le Varlet, B.; Nicolas, J.F. Photoageing shows histological features of chronic skin inflammation without clinical and molecular abnormalities. Br. J. Dermatol. 2003, 149, 826–835. [Google Scholar] [CrossRef]
- Hossain, M.R.; Ansary, T.M.; Komine, M.; Ohtsuki, M. Diversified Stimuli-Induced Inflammatory Pathways Cause Skin Pigmentation. Int. J. Mol. Sci. 2021, 22, 3970. [Google Scholar] [CrossRef]
- Ikehata, H. Mechanistic considerations on the wavelength-dependent variations of UVR genotoxicity and mutagenesis in skin: The discrimination of UVA-signature from UV-signature mutation. Photochem. Photobiol. Sci. 2018, 17, 1861–1871. [Google Scholar] [CrossRef]
- Sarkar, S.; Gaddameedhi, S. Solar ultraviolet-induced DNA damage response: Melanocytes story in transformation to environmental melanomagenesis. Environ. Mol. Mutagen. 2020, 61, 736–751. [Google Scholar] [CrossRef]
- Imokawa, G. Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment. Cell Res. 2004, 17, 96–110. [Google Scholar] [CrossRef]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key regulatory role of dermal fibroblasts in pigmentation as demonstrated using a reconstructed skin model: Impact of photo-aging. PLoS ONE 2014, 9, e114182. [Google Scholar] [CrossRef]
- Bastonini, E.; Bellei, B.; Filoni, A.; Kovacs, D.; Iacovelli, P.; Picardo, M. Involvement of non-melanocytic skin cells in vitiligo. Exp. Dermatol. 2019, 28, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Brenner, M.; Degitz, K.; Besch, R.; Berking, C. Differential expression of melanoma-associated growth factors in keratinocytes and fibroblasts by ultraviolet A and ultraviolet B radiation. Br. J. Dermatol. 2005, 153, 733–739. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.H.; Kim, E.K. Repeated exposure of human fibroblasts to UVR induces secretion of stem cell factor and senescence. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1577–1580. [Google Scholar] [CrossRef]
- Kovacs, D.; Cardinali, G.; Aspite, N.; Cota, C.; Luzi, F.; Bellei, B.; Briganti, S.; Amantea, A.; Torrisi, M.R.; Picardo, M. Role of fibroblast-derived growth factors in regulating hyperpigmentation of solar lentigo. Br. J. Dermatol. 2010, 163, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Monestier, S.; Gaudy, C.; Gouvernet, J.; Richard, M.A.; Grob, J.J. Multiple senile lentigos of the face, a skin ageing pattern resulting from a life excess of intermittent sun exposure in dark-skinned caucasians: A case-control study. Br. J. Dermatol. 2006, 154, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Bastiaens, M.; Hoefnagel, J.; Westendorp, R.; Vermeer, B.J.; Bouwes Bavinck, J.N. Solar lentigines are strongly related to sun exposure in contrast to ephelides. Pigment Cell Res. 2004, 17, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Hearing, V.J. Modifying skin pigmentation—Approaches through intrinsic biochemistry and exogenous agents. Drug Discov. Today Dis. Mech. 2008, 5, e189–e199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Park, J.Y.; Kim, S.J.; Kang, H.Y. Characteristics of keratinocytes in facial solar lentigo with flattened rete ridges: Comparison with melasma. Clin. Exp. Dermatol. 2015, 40, 489–494. [Google Scholar] [CrossRef]
- Morgan, A.M.; Lo, J.; Fisher, D.E. How does pheomelanin synthesis contribute to melanomagenesis?: Two distinct mechanisms could explain the carcinogenicity of pheomelanin synthesis. Bioessays 2013, 35, 672–676. [Google Scholar] [CrossRef] [Green Version]
- Egambaram, O.P.; Kesavan Pillai, S.; Ray, S.S. Materials Science Challenges in Skin UV Protection: A Review. Photochem. Photobiol. 2020, 96, 779–797. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, J.Y.; Martinez, R.M.; Morocho-Jácome, A.L.; Castillo-Gómez, T.S.; Pereda-Contreras, V.J.; Rosado, C.; Velasco, M.V.R.; Baby, A.R. Skin impacts from exposure to ultraviolet, visible, infrared, and artificial lights—A review. J. Cosmet. Laser Ther. 2021, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.; Bastiaens, M.T.; Bajdik, C.D.; Willemze, R.; Westendorp, R.G.; Bouwes Bavinck, J.N. Leiden Skin Cancer Study Effect of smoking and sun on the aging skin. J. Investig. Dermatol. 2003, 120, 548–554. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, D.; Farris, P.; Valacchi, G. Atmospheric skin aging-Contributors and inhibitors. J. Cosmet. Dermatol. 2018, 17, 124–137. [Google Scholar] [CrossRef]
- Ortiz, A.; Grando, S.A. Smoking and the skin. Int. J. Dermatol. 2012, 51, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Leow, Y.H.; Maibach, H.I. Cigarette smoking, cutaneous vasculature, and tissue oxygen. Clin. Dermatol. 1998, 16, 579–584. [Google Scholar] [CrossRef]
- Ono, Y.; Torii, K.; Fritsche, E.; Shintani, Y.; Nishida, E.; Nakamura, M.; Shirakata, Y.; Haarmann-Stemmann, T.; Abel, J.; Krutmann, J.; et al. Role of the aryl hydrocarbon receptor in tobacco smoke extract-induced matrix metalloproteinase-1 expression. Exp. Dermatol. 2013, 22, 349–353. [Google Scholar] [CrossRef]
- Lahmann, C.; Bergemann, J.; Harrison, G.; Young, A.R. Matrix metalloproteinase-1 and skin ageing in smokers. Lancet 2001, 357, 935–936. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Yang, Y.; Ares, I.; Martínez, M.; Lopez-Torres, B.; Martínez-Larrañaga, M.R.; Wang, X.; Anadón, A.; Martinez, M.A. Neonicotinoids: Mechanisms of systemic toxicity based on oxidative stress-mitochondrial damage. Arch. Toxicol. 2022, 96, 1493–1520. [Google Scholar] [CrossRef]
- Sule, R.O.; Condon, L.; Gomes, A.V. A Common Feature of Pesticides: Oxidative Stress-The Role of Oxidative Stress in Pesticide-Induced Toxicity. Oxid. Med. Cell. Longev. 2022, 2022, 5563759. [Google Scholar] [CrossRef] [PubMed]
- Berge, U.; Behrens, J.; Rattan, S.I. Sugar-induced premature aging and altered differentiation in human epidermal keratinocytes. Ann. N. Y. Acad. Sci. 2007, 1100, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Sejersen, H.; Rattan, S.I. Dicarbonyl-induced accelerated aging in vitro in human skin fibroblasts. Biogerontology 2009, 10, 203–211. [Google Scholar] [CrossRef]
- Halkoum, R.; Salnot, V.; Capallere, C.; Plaza, C.; L’honoré, A.; Pays, K.; Friguet, B.; Nizard, C.; Petropoulos, I. Glyoxal Induces Senescence in Human Keratinocytes through Oxidative Stress and Activation of the Protein Kinase B/FOXO3a/p27(KIP1) Pathway. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, M.; Gensberger-Reigl, S.; Henle, T.; Pischetsrieder, M. Food-derived 1,2-dicarbonyl compounds and their role in diseases. Semin. Cancer Biol. 2018, 49, 1–8. [Google Scholar] [CrossRef]
- Radjei, S.; Friguet, B.; Nizard, C.; Petropoulos, I. Prevention of dicarbonyl-mediated advanced glycation by glyoxalases: Implication in skin aging. Biochem. Soc. Trans. 2014, 42, 518–522. [Google Scholar] [CrossRef]
- Saewan, N.; Jimtaisong, A. Natural products as photoprotection. J. Cosmet. Dermatol. 2015, 14, 47–63. [Google Scholar] [CrossRef]
- Michalak, M. Plant-Derived Antioxidants: Sig.gnificance in Skin Health and the Ageing Process. Int. J. Mol. Sci. 2022, 23, 585. [Google Scholar] [CrossRef] [PubMed]
- Pinnell, S.R. Regulation of collagen biosynthesis by ascorbic acid: A review. Yale J. Biol. Med. 1985, 58, 553–559. [Google Scholar]
- Petruk, G.; Del Giudice, R.; Rigano, M.M.; Monti, D.M. Antioxidants from Plants Protect against Skin Photoaging. Oxid. Med. Cell. Longev. 2018, 2018, 1454936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forni, C.; Facchiano, F.; Bartoli, M.; Pieretti, S.; Facchiano, A.; D’Arcangelo, D.; Norelli, S.; Valle, G.; Nisini, R.; Beninati, S.; et al. Beneficial Role of Phytochemicals on Oxidative Stress and Age-Related Diseases. Biomed. Res. Int. 2019, 2019, 8748253. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Gao, Q.; Ma, C.; Ge, Y.; You, L.; Liu, R.H.; Fu, X.; Liu, D. Effect of polysaccharides from Tremella fuciformis on UV-induced photoaging. J. Funct. Foods 2016, 20, 400–410. [Google Scholar] [CrossRef]
- Ye, Y.; Ji, D.; You, L.; Zhou, L.; Zhao, Z.; Brennan, C. Structural properties and protective effect of Sargassum fusiforme polysaccharides against ultraviolet B radiation in hairless Kun Ming mice. J. Funct. Foods 2018, 43, 8–16. [Google Scholar] [CrossRef]
- Hinek, A.; Kim, H.J.; Wang, Y.; Wang, A.; Mitts, T.F. Sodium L-ascorbate enhances elastic fibers deposition by fibroblasts from normal and pathologic human skin. J. Dermatol. Sci. 2014, 75, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Choi, G.I.; Kim, E.K.; Choi, Y.J.; Sohn, K.C.; Lee, Y.; Kim, C.D.; Yoon, T.J.; Sohn, H.J.; Han, S.H.; et al. Hair greying is associated with active hair growth. Br. J. Dermatol. 2011, 165, 1183–1189. [Google Scholar] [CrossRef]
- Jadkauskaite, L.; Coulombe, P.A.; Schäfer, M.; Dinkova-Kostova, A.T.; Paus, R.; Haslam, I.S. Oxidative stress management in the hair follicle: Could targeting NRF2 counter age-related hair disorders and beyond? Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Arck, P.C.; Overall, R.; Spatz, K.; Liezman, C.; Handjiski, B.; Klapp, B.F.; Birch-Machin, M.A.; Peters, E.M. Towards a “free radical theory of graying”: Melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. FASEB J. 2006, 20, 1567–1569. [Google Scholar] [CrossRef]
- Sato, S.; Kukita, A.; Jimbow, K. Electron microscopic studies of dendritic cells in the human gray and white matrix during anagen. In Pigment Cell Mechanisms in Pigmentation; McGovern, V.J., Russel, P., Eds.; Karger: Basel, Switzerland, 1973; Volume 1, pp. 20–26. [Google Scholar]
- Trüeb, R.M. The impact of oxidative stress on hair. Int. J. Cosmet. Sci. 2015, 37 (Suppl. S2), 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Luo, L.F.; Liu, X.M.; Zhou, Q.; Xu, S.Z.; Lei, T.C. Premature graysing as a consequence of compromised antioxidant activity in hair bulb melanocytes and their precursors. PLoS ONE 2014, 9, e93589. [Google Scholar]
- Kauser, S.; Westgate, G.E.; Green, M.R.; Tobin, D.J. Human hair follicle and epidermal melanocytes exhibit striking differences in their aging profile which involves catalase. J. Investig. Dermatol. 2011, 131, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.M.; Decker, H.; Hartmann, H.; Chavan, B.; Rokos, H.; Spencer, J.D.; Hasse, S.; Thornton, M.J.; Shalbaf, M.; Paus, R.; et al. Senile hair graying: H2O2-mediated oxidative stress affects human hair color by blunting methionine sulfoxide repair. FASEB J. 2009, 23, 2065–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallreuter, K.U.; Gibbons, N.C.; Zothner, C.; Abou Elloof, M.M.; Wood, J.M. Hydrogen peroxide-mediated oxidative stress disrupts calcium binding on calmodulin: More evidence for oxidative stress in vitiligo. Biochem. Biophys. Res. Commun. 2007, 360, 70–75. [Google Scholar] [CrossRef]
- Maresca, V.; Roccella, M.; Roccella, F.; Camera, E.; Del Porto, G.; Passi, S.; Grammatico, P.; Picardo, M. Increased sensitivity to peroxidative agents as a possible pathogenic factor of melanocyte damage in vitiligo. J. Investig. Dermatol. 1997, 109, 310–313. [Google Scholar] [CrossRef] [Green Version]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.J.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc. 1999, 4, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Bellei, B.; Pitisci, A.; Ottaviani, M.; Ludovici, M.; Cota, C.; Luzi, F.; Dell’Anna, M.L.; Picardo, M. Vitiligo: A possible model of degenerative diseases. PLoS ONE 2013, 8, e59782. [Google Scholar] [CrossRef] [Green Version]
- Gan, E.Y.; Cario-André, M.; Pain, C.; Goussot, J.F.; Taïeb, A.; Seneschal, J.; Ezzedine, K. Follicular vitiligo: A report of 8 cases. J. Am. Acad. Dermatol. 2016, 74, 1178–1184. [Google Scholar] [CrossRef]
- Wood, J.M.; Chavan, B.; Hafeez, I.; Schallreuter, K.U. Regulation of tyrosinase by tetrahydropteridines and H2O. Biochem. Biophys. Res. Commun. 2004, 325, 1412–1417. [Google Scholar] [CrossRef]
- Koga, S.; Nakano, M.; Tero-Kubota, S. Generation of superoxide during the enzymatic action of tyrosinase. Arch. Biochem. Biophys. 1992, 292, 570–575. [Google Scholar] [CrossRef]
- Seiberg, M. Age-induced hair greying—The multiple effects of oxidative stress. Int. J. Cosmet. Sci. 2013, 35, 532–538. [Google Scholar] [CrossRef]
- Paus, R. A neuroendocrinological perspective on human hair follicle pigmentation. Pigment Cell Melanoma Res. 2011, 24, 89–106. [Google Scholar] [CrossRef]
- O′Sullivan, J.D.B.; Nicu, C.; Picard, M.; Chéret, J.; Bedogni, B.; Tobin, D.J.; Paus, R. The biology of human hair greying. Biol. Rev. Camb. Philos. Soc. 2021, 96, 107–128. [Google Scholar] [CrossRef]
- Lu, Z.; Fischer, T.W.; Hasse, S.; Sugawara, K.; Kamenisch, Y.; Krengel, S.; Funk, W.; Berneburg, M.; Paus, R. Profiling the response of human hair follicles to ultraviolet radiation. J. Investig. Dermatol. 2009, 129, 1790–1804. [Google Scholar] [CrossRef]
- Haslam, I.S.; Jadkauskaite, L.; Szabó, I.L.; Staege, S.; Hesebeck-Brinckmann, J.; Jenkins, G.; Bhogal, R.K.; Lim, F.L.; Farjo, N.; Farjo, B.; et al. Oxidative Damage Control in a Human (Mini-) Organ: Nrf2 Activation Protects against Oxidative Stress-Induced Hair Growth Inhibition. J. Investig. Dermatol. 2017, 137, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Van Neste, D.; Tobin, D.J. Hair cycle and hair pigmentation: Dynamic interactions and changes associated with aging. Micron 2004, 35, 193–200. [Google Scholar] [CrossRef]
- Zhou, C.; Li, X.; Wang, C.; Zhang, J. Alopecia Areata: An Update on Etiopathogenesis, Diagnosis, and Management. Clin. Rev. Allergy Immunol. 2021, 61, 403–423. [Google Scholar] [CrossRef]
- Upton, J.H.; Hannen, R.F.; Bahta, A.W.; Farjo, N.; Farjo, B.; Philpott, M.P. Oxidative stress-associated senescence in dermal papilla cells of men with androgenetic alopecia. J. Investig. Dermatol. 2015, 135, 1244–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloepper, J.E.; Baris, O.R.; Reuter, K.; Kobayashi, K.; Weiland, D.; Vidali, S.; Tobin, D.J.; Niemann, C.; Wiesner, R.J.; Paus, R. Mitochondrial function in murine skin epithelium is crucial for hair follicle morphogenesis and epithelial-mesenchymal interactions. J. Investig. Dermatol. 2015, 135, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Zou, Z.; Chang, H.; Shen, Q.; Liu, L.; Xing, D. Photobiomodulation therapy for hair regeneration: A synergetic activation of β-CATENIN in hair follicle stem cells by ROS and paracrine WNTs. Stem Cell Rep. 2021, 16, 1568–1583. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Dupuis, G.; Witkowski, J.M.; Larbi, A. The Role of Immunosenescence in the Development of Age-Related Diseases. Rev. Investig. Clin. 2016, 68, 84–91. [Google Scholar]
- Laube, S. Skin infections and ageing. Ageing Res. Rev. 2004, 3, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin barrier immunity and ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, J.M.; Vafaie, J.; Scheinfeld, N.S. Skin infections in the elderly. Dermatol. Clin. 2004, 22, 51–61. [Google Scholar] [CrossRef]
- Stevens, N.E.; Cowin, A.J.; Kopecki, Z. Skin Barrier and Autoimmunity-Mechanisms and Novel Therapeutic Approaches for Autoimmune Blistering Diseases of the Skin. Front. Immunol. 2019, 10, 1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, F.; Fania, L.; Sinagra, J.L.M.; Salemme, A.; Di Zenzo, G. Bullous Pemphigoid: Trigger and Predisposing Factors. Biomolecules 2020, 10, 1432. [Google Scholar] [CrossRef] [PubMed]
- Deotto, M.L.; Spiller, A.; Sernicola, A.; Alaibac, M. Bullous pemphigoid: An immune disorder related to aging (Review). Exp. Ther. Med. 2022, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Dey-Rao, R.; Seiffert-Sinha, K.; Sinha, A.A. Increased oxidative stress in pemphigus vulgaris is related to disease activity and HLA-association. Autoimmunity 2016, 49, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Perl, S.; Rappersberger, K.; Födinger, D.; Anegg, B.; Hönigsmann, H.; Ortel, B. Bullous pemphigoid induced by PUVA therapy. Dermatology 1996, 193, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Pfau, A.; Hohenleutner, U.; Hohenleutner, S.; Eckert, F.; Landthaler, M. UV-A-provoked localized bullous pemphigoid. Acta Derm. Venereol. 1994, 74, 314–316. [Google Scholar] [PubMed]
- Farage, M.A.; Miller, K.W.; Berardesca, E.; Maibach, H.I. Clinical implications of aging skin: Cutaneous disorders in the elderly. Am. J. Clin. Dermatol. 2009, 10, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Y.; Ucmak, D.; Selek, S.; Dertlioğlu, S.B.; Sula, B.; Bozkus, F.; Turan, E. Oxidative stress index may play a key role in patients with pemphigus vulgaris. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, M.H.; Djalali, M.; Daneshpazhooh, M.; Zarei, M.; Eshraghian, M.R.; Derakhshanian, H.; Chams-Davatchi, C. Evaluation of antioxidant enzyme activity and antioxidant capacity in patients with newly diagnosed pemphigus vulgaris. Clin. Exp. Dermatol. 2015, 40, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jedličková, H.; Cai, Y.; Rehman, A.; Gammon, L.; Ahmad, U.S.; Uttagomol, J.; Parkinson, E.K.; Fortune, F.; Wan, H. Oxidative Stress-Mediated YAP Dysregulation Contributes to the Pathogenesis of Pemphigus Vulgaris. Front. Immunol. 2021, 12, 649502. [Google Scholar] [CrossRef] [PubMed]
- Tétart, F.; Joly, P. Eczema in elderly people. Eur. J. Dermatol. 2020, 30, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Katoh, N.; Tennstedt, D.; Abellan van Kan, G.; Saint Aroman, M.; Loir, A.; Bacqueville, D.; Duprat, L.; Guiraud, B.; Bessou-Touya, S.; Duplan, H. Gerontodermatology: The fragility of the epidermis in older adults. J. Eur. Acad. Dermatol. Venereol. 2018, 32 (Suppl. S4), 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, C.; Vogt, T. Seborrheic keratosis. J. Dtsch. Dermatol. Ges. 2008, 6, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Pambianchi, E.; Pecorelli, A.; Woodby, B.; Messano, N.; Therrien, J.P.; Lila, M.A.; Valacchi, G. Redox regulation of cutaneous inflammasome by ozone exposure. Free Radic. Biol. Med. 2020, 152, 561–570. [Google Scholar] [CrossRef]
- Corsini, E.; Galbiati, V.; Nikitovic, D.; Tsatsakis, A.M. Role of oxidative stress in chemical allergens induced skin cells activation. Food Chem. Toxicol. 2013, 61, 74–81. [Google Scholar] [CrossRef]
- Nakashima, H.; Fujimoto, M.; Asashima, N.; Watanabe, R.; Kuwano, Y.; Yazawa, N.; Maruyama, N.; Okochi, H.; Kumanogoh, A.; Tamaki, K. Serum chemokine profile in patients with bullous pemphigoid. Br. J. Dermatol. 2007, 156, 454–459. [Google Scholar] [CrossRef]
- Chen, D.; Hao, H.; Fu, X.; Han, W. Insight into Reepithelialization: How Do Mesenchymal Stem Cells Perform? Stem. Cells Int. 2016, 2016, 6120173. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Kakanj, P.; Leptin, M.; Eming, S.A. Regulation of the Wound Healing Response during Aging. J. Investig. Dermatol. 2021, 141, 1063–1070. [Google Scholar] [CrossRef]
- Kamenisch, Y.; Berneburg, M. Progeroid syndromes and UV-induced oxidative DNA damage. J. Investig. Dermatol. Symp. Proc. 2009, 14, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Goorochurn, R.; Viennet, C.; Tissot, M.; Locatelli, F.; Granger, C.; Varin-Blank, N.; Humbert, P.; Le Roy, C. Differential morphological and functional features of fibroblasts explanted from solar lentigo. Br. J. Dermatol. 2017, 177, e109–e111. [Google Scholar] [CrossRef]
- Kovacs, D.; Bastonini, E.; Ottaviani, M.; Cota, C.; Migliano, E.; Dell′Anna, M.L.; Picardo, M. Vitiligo Skin: Exploring the Dermal Compartment. J. Investig. Dermatol. 2018, 138, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Dell′Anna, M.L.; Ottaviani, M.; Albanesi, V.; Vidolin, A.P.; Leone, G.; Ferraro, C.; Cossarizza, A.; Rossi, L.; Picardo, M. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J. Investig. Dermatol. 2007, 127, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Dell′Anna, M.L.; Ottaviani, M.; Kovacs, D.; Mirabilii, S.; Brown, D.A.; Cota, C.; Migliano, E.; Bastonini, E.; Bellei, B.; Cardinali, G.; et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci. Rep. 2017, 7, 13663. [Google Scholar] [CrossRef] [Green Version]
- Seçkin, H.Y.; Kalkan, G.; Baş, Y.; Akbaş, A.; Önder, Y.; Özyurt, H.; Sahin, M. Oxidative stress status in patients with melasma. Cutan. Ocul. Toxicol. 2014, 33, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Arámbula, A.; Torres-Álvarez, B.; Cortés-García, D.; Fuentes-Ahumada, C.; Castanedo-Cázares, J.P. CD4, IL-17, and COX-2 Are Associated With Subclinical Inflammation in Malar Melasma. Am. J. Dermatopathol. 2015, 37, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, T.H.; Park, T.J.; Kang, H.Y. p16(ink4a) Positivity of Melanocytes in Non-Segmental Vitiligo. Diagnostics 2020, 10, 878. [Google Scholar] [CrossRef]
- Spritz, R.A.; Santorico, S.A. The Genetic Basis of Vitiligo. J. Investig. Dermatol. 2021, 141, 265–273. [Google Scholar] [CrossRef]
- Patel, P.; Hussain, K. Merkel cell carcinoma. Clin. Exp. Dermatol. 2021, 46, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Tsatsou, F.; Trakatelli, M.; Patsatsi, A.; Kalokasidis, K.; Sotiriadis, D. Extrinsic aging: UV-mediated skin carcinogenesis. Dermatoendocrinology 2012, 4, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcovich, S.; Colloca, G.; Sollena, P.; Andrea, B.; Balducci, L.; Cho, W.C.; Bernabei, R.; Peris, K. Skin Cancer Epidemics in the Elderly as An Emerging Issue in Geriatric Oncology. Aging Dis. 2017, 8, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribero, S.; Stucci, L.S.; Marra, E.; Marconcini, R.; Spagnolo, F.; Orgiano, L.; Picasso, V.; Queirolo, P.; Palmieri, G.; Quaglino, P.; et al. Effect of Age on Melanoma Risk, Prognosis and Treatment Response. Acta Derm. Venereol. 2018, 98, 624–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balch, C.M.; Buzaid, A.C.; Soong, S.J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A., Jr.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar] [CrossRef]
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 2001, 63, 88–102. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, L.L.; Lim, H.W.; Mohammad, T.F. Sunscreens and Photoaging: A Review of Current Literature. Am. J. Clin. Dermatol. 2021, 22, 819–828. [Google Scholar] [CrossRef] [PubMed]
- González, S.; Aguilera, J.; Berman, B.; Calzavara-Pinton, P.; Gilaberte, Y.; Goh, C.L.; Lim, H.W.; Schalka, S.; Stengel, F.; Wolf, P.; et al. Expert Recommendations on the Evaluation of Sunscreen Efficacy and the Beneficial Role of Non-filtering Ingredients. Front. Med. 2022, 9, 790207. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Dixon, K.M.; Deo, S.S.; Holliday, C.J.; Slater, M.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J. Investig. Dermatol. 2007, 127, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.J.; Gordon-Thomson, C.; Cole, L.; Stern, H.; Halliday, G.M.; Damian, D.L.; Reeve, V.E.; Mason, R.S. 1α,25-Dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. J. Steroid Biochem. Mol. Biol. 2013, 136, 131–138. [Google Scholar] [CrossRef]
- Gordon-Thomson, C.; Gupta, R.; Tongkao-on, W.; Ryan, A.; Halliday, G.M.; Mason, R.S. 1α,25 dihydroxyvitamin D3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photochem. Photobiol. Sci. 2012, 11, 1837–1847. [Google Scholar] [CrossRef]
- Snaidr, V.A.; Damian, D.L.; Halliday, G.M. Nicotinamide for photoprotection and skin cancer chemoprevention: A review of efficacy and safety. Exp. Dermatol. 2019, 28 (Suppl. S1), 15–22. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.C.; Halliday, G.M.; Damian, D.L. Nicotinamide enhances repair of arsenic and ultraviolet radiation-induced DNA damage in HaCaT keratinocytes and ex vivo human skin. PLoS ONE 2015, 10, e0117491. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.E.; Sheaff, M.T. The pathology of ageing: Concepts and mechanisms. J. Pathol. 2007, 211, 111–113. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Margherita De Santi, M.; Schürfeld, K.; Santopietro, R.; Lalinga, A.V.; Fimiani, M.; Biagioli, M.; Brogi, M.; De Felice, C.; Luzi, P.; et al. Quantitative in situ evaluation of telomeres in fluorescence in situ hybridization-processed sections of cutaneous melanocytic lesions and correlation with telomerase activity. Br. J. Dermatol. 2002, 146, 399–408. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence--halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Reis-Filho, J.S.; da Rocha Dias, S.; Hayward, R.; Savage, K.; Delmas, V.; Larue, L.; Pritchard, C.; Marais, R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Goel, V.K.; Ibrahim, N.; Jiang, G.; Singhal, M.; Fee, S.; Flotte, T.; Westmoreland, S.; Haluska, F.S.; Hinds, P.W.; Haluska, F.G. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 2009, 28, 2289–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF mutations are common somatic events in melanocytic nevi. J. Investig. Dermatol. 2004, 122, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, N.C.; Jung, J.; Liu, T.; Wilde, M.; Holmen, S.L.; Grossman, D. Familial melanoma-associated mutations in p16 uncouple its tumor-suppressor functions. J. Investig. Dermatol. 2013, 133, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.; Ally, D.S.; Sheahan, M.D.; Clark, W.H., Jr.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef]
- Kuwata, T.; Kitagawa, M.; Kasuga, T. Proliferative activity of primary cutaneous melanocytic tumours. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 359–364. [Google Scholar] [CrossRef]
- Maldonado, J.L.; Timmerman, L.; Fridlyand, J.; Bastian, B.C. Mechanisms of cell-cycle arrest in Spitz nevi with constitutive activation of the MAP-kinase pathway. Am. J. Pathol. 2004, 164, 1783–1787. [Google Scholar] [CrossRef] [Green Version]
- Vredeveld, L.C.; Possik, P.A.; Smit, M.A.; Meissl, K.; Michaloglou, C.; Horlings, H.M.; Ajouaou, A.; Kortman, P.C.; Dankort, D.; McMahon, M.; et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012, 26, 1055–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Kajiya, H.; Ozeki, S.; Okabe, K.; Ikebe, T. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16(INK4a). Biochem. Biophys. Res. Commun. 2014, 452, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nishioka, K. Enhanced expression of p16 in seborrhoeic keratosis; a lesion of accumulated senescent epidermal cells in G1 arrest. Br. J. Dermatol. 2003, 149, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Vaddavalli, P.L.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022, 38, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.M. The Role of p53 in Progression of Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef]
- Loureiro, J.B.; Abrantes, M.; Oliveira, P.A.; Saraiva, L. P53 in skin cancer: From a master player to a privileged target for prevention and therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188438. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; English, D.; Randell, P.L.; Nakkazawa, K.; Martel, N.; Armstrong, B.K.; Yamasaki, H. UV and skin cancer: Specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc. Natl. Acad. Sci. USA 1994, 91, 360–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouhtit, A.; Ueda, M.; Nakazawa, H.; Ichihashi, M.; Dumaz, N.; Sarasin, A.; Yamasaki, H. Quantitative detection of ultraviolet-specific p53 mutations in normal skin from Japanese patients. Cancer Epidemiol. Biomarkers Prev. 1997, 6, 433–438. [Google Scholar] [PubMed]
- Ouhtit, A.; Nakazawa, H.; Armstrong, B.K.; Kricker, A.; Tan, E.; Yamasaki, H.; English, D.R. UV-radiation-specific p53 mutation frequency in normal skin as a predictor of risk of basal cell carcinoma. J. Natl. Cancer Inst. 1998, 90, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Alspach, E.; Fu, Y.; Stewart, S.A. Senescence and the pro-tumorigenic stroma. Crit. Rev. Oncog. 2013, 18, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worrede, A.; Douglass, S.M.; Weeraratna, A.T. The dark side of daylight: Photoaging and the tumor microenvironment in melanoma progression. J. Clin. Investig. 2021, 131, e143763. [Google Scholar] [CrossRef]
- Vogel, V. Unraveling the Mechanobiology of Extracellular Matrix. Annu. Rev. Physiol. 2018, 80, 353–387. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E.; Cheri, S.; Fanti, M.; Marongiu, F. Aging and Cancer: The Waning of Community Bonds. Cells 2021, 10, 2269. [Google Scholar] [CrossRef]
- Ingber, D.E. Can cancer be reversed by engineering the tumor microenvironment? Semin. Cancer Biol. 2008, 18, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, S.J.; Wang, R.K.; Duncan, D.D.; Kulesz-Martin, M.; Lee, K. Imaging the mechanical stiffness of skin lesions by in vivo acousto-optical elastography. Opt. Express 2006, 14, 9770–9779. [Google Scholar] [CrossRef] [Green Version]
- Berking, C.; Takemoto, R.; Schaider, H.; Showe, L.; Satyamoorthy, K.; Robbins, P.; Herlyn, M. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001, 61, 8306–8316. [Google Scholar]
- Godic, A.; Poljšak, B.; Adamic, M.; Dahmane, R. The role of antioxidants in skin cancer prevention and treatment. Oxid. Med. Cell. Longev. 2014, 2014, 860479. [Google Scholar] [CrossRef]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol. 2003, 148, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Duryee, M.J.; Clemens, D.L.; Opperman, P.J.; Thiele, G.M.; Duryee, L.M.; Garvin, R.P.; Anderson, D.R. Malondialdehyde-Acetaldehyde Modified (MAA) Proteins Differentially Effect the Inflammatory Response in Macrophage, Endothelial Cells and Animal Models of Cardiovascular Disease. Int. J. Mol. Sci. 2021, 22, 12948. [Google Scholar] [CrossRef]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Gadjeva, V.; Dimov, A.; Georgieva, N. Influence of therapy on the antioxidant status in patients with melanoma. J. Clin. Pharm. Ther. 2008, 33, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Bellei, B.; Migliano, E.; Picardo, M. A Framework of Major Tumor-Promoting Signal Transduction Pathways Implicated in Melanoma-Fibroblast Dialogue. Cancers 2020, 12, 3400. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Rebecca, V.; Fedorenko, I.V.; Messina, J.L.; Mathew, R.; Maria-Engler, S.S.; Basanta, D.; Smalley, K.S.; Anderson, A.R. Senescent fibroblasts in melanoma initiation and progression: An integrated theoretical, experimental, and clinical approach. Cancer Res. 2013, 73, 6874–6885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moinfar, F.; Beham, A.; Friedrich, G.; Deutsch, A.; Hrzenjak, A.; Luschin, G.; Tavassoli, F.A. Macro-environment of breast carcinoma: Frequent genetic alterations in the normal appearing skins of patients with breast cancer. Mod. Pathol. 2008, 21, 639–646. [Google Scholar] [CrossRef]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alili, L.; Sack, M.; Puschmann, K.; Brenneisen, P. Fibroblast-to-myofibroblast switch is mediated by NAD(P)H oxidase generated reactive oxygen species. Biosci. Rep. 2014, 34, e00089. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [Green Version]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: A new genetically tractable model for human cancer associated fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef]


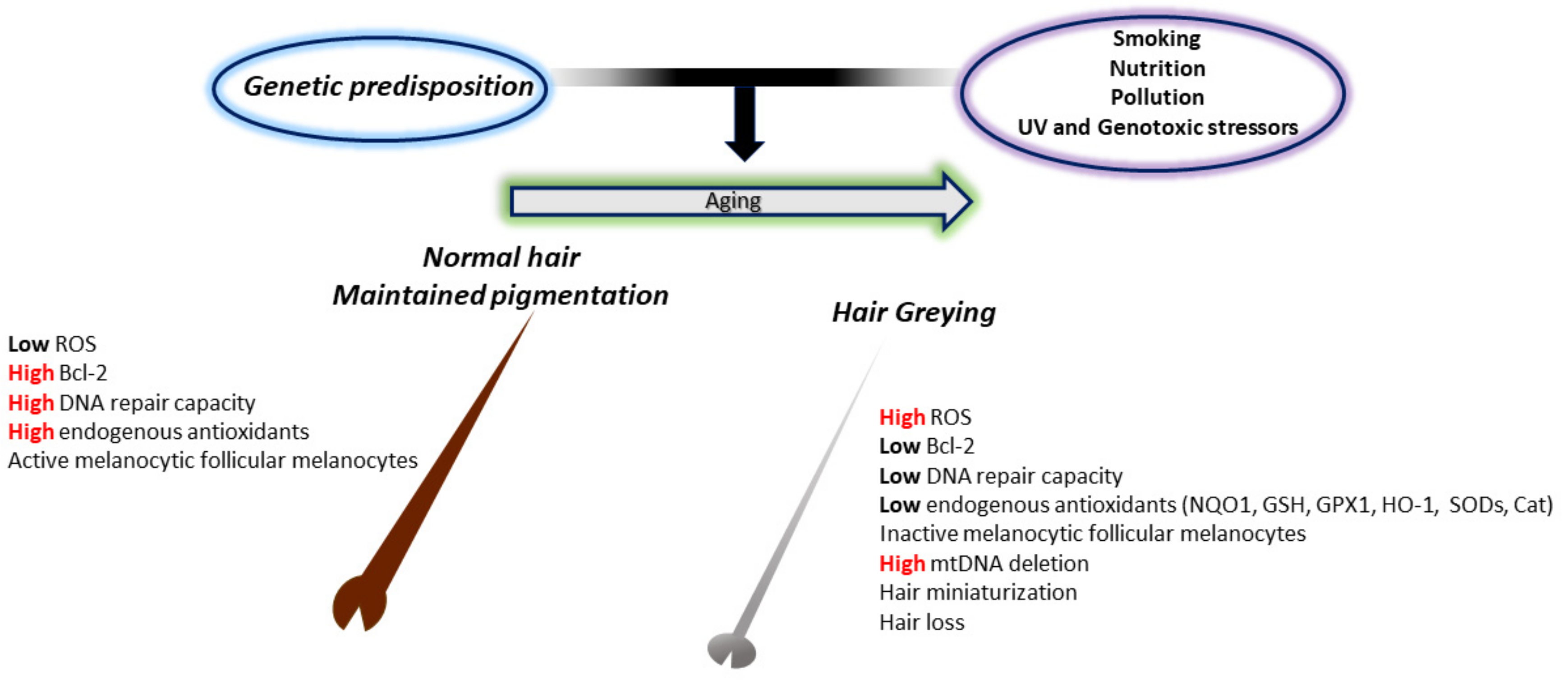

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papaccio, F.; D′Arino, A.; Caputo, S.; Bellei, B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants 2022, 11, 1121. https://doi.org/10.3390/antiox11061121
Papaccio F, D′Arino A, Caputo S, Bellei B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants. 2022; 11(6):1121. https://doi.org/10.3390/antiox11061121
Chicago/Turabian StylePapaccio, Federica, Andrea D′Arino, Silvia Caputo, and Barbara Bellei. 2022. "Focus on the Contribution of Oxidative Stress in Skin Aging" Antioxidants 11, no. 6: 1121. https://doi.org/10.3390/antiox11061121
APA StylePapaccio, F., D′Arino, A., Caputo, S., & Bellei, B. (2022). Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants, 11(6), 1121. https://doi.org/10.3390/antiox11061121